
Abstract
Activates the pituitary gland, leading to increased secretion of growth hormone. This activation plays a crucial role in growth, metabolism, and tissue repair. Bovine serum albumin, a well-studied protein, demonstrates the interaction between proteins-ligands and transports various compounds in the bloodstream. This study elucidates the molecular interactions between L-ornithine and BSA through spectroscopic analysis, identifying binding modes, constants, and structural changes. Molecular docking techniques are employed to correlate with experimental data from fluorescence and UV spectroscopy analyses.The study uses Fourier transform infrared spectroscopy, UV–Visible spectroscopy, fluorescence spectroscopy, circular dichroism spectroscopy and molecular docking to analyze the interaction between bovine serum albumin and L-ornithine, providing thermodynamic parameters for understanding the protein structure and its binding.The interactions between L-ornithine and bovine serum albumin (BSA) were characterized using UV–visible spectroscopy, which demonstrated a hyperchromic shift. Fluorescence spectroscopy revealed that L-ornithine quenches the intrinsic fluorescence of both BSA and human serum albumin (HSA) via a static quenching mechanism, resulting in the formation of stable BSA-L-ornithine and HSA-L-ornithine complexes. Furthermore, Fourier-transform infrared (FTIR) and circular dichroism (CD) spectroscopy indicated a decrease in the secondary structure of the proteins, as evidenced by shifts in the amide II band and a concomitant reduction in α-helix content. Molecular docking studies suggested that L-ornithine binds to BSA within subdomain II. Collectively, these findings provide valuable insights into the metabolic pathways of L-ornithine and its potential pharmacological relevance. The study involves more precise information which revealing the insight of L-ornithine bioavailability. The finding enhances our understanding of interaction with potential implications for drug delivery.
Similar content being viewed by others
Introduction
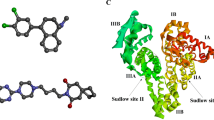
A drug’s pharmacokinetic and pharmacodynamic properties are substantially influenced by how it interacts with serumalbumin1. Drugs and alternative small compounds attach amendable to serum and albumin components, acting as bearers. Serum albumin raises the solvability of non-polar medications in blood plasma, influencing the drug’s transportation to tissues both in vivo and in vitro. It is crucial to acknowledge how a drug interacts with serum albumin. Serum albumin, a key circulatory protein, maintains blood pH, contributes to colloid osmotic hypertension, and facilitates the movement and elimination of endogenic and foreign ligands, influencing their metabolism2,3,4,5. These investigations can shed light on the structural characteristics of drugs that impact their therapeutic efficacy. In light of this, it is an important field of research for those studying clinical medicine, biology, and chemistry.The binding leaning of ligands to proteins, such as bovine serum albumin (BSA) (Fig. 1), significantly impacts their distribution, metabolism, excretion, and toxicity. Accurately estimating the binding affinity is essential for comprehending the characteristics of the binding process in biological systems6. Blood serum contains a well-known protein called BSA, and it is crucial for controlling osmotic pressures and facilitating the transportation of numerous endogenous compounds within the body7. BSA is usually used as an exemplary protein for assessing the binding interactions between drugs and proteins. This is due to its cost-effectiveness, ready availability, and some unique ligand-binding characteristics8. BSA consists of 583 amino acid remnants, 17 disulfide linkages, and a free cysteine remnant in its basic structure. BSA subdomain IA contains amino acids 6–105, while subdomain IB consists of amino acids 119–196. Amino acids 197–291, 314–481, 482–510, and 511–583 are present in subdomains IIA, IIB, IIIA, and IIIB, respectively. It also contains two tryptophan residues: Trp 134 in subdomain IB and Trp 213 in subdomain IIA which is important for interaction study9,10,11,12. BSA shares 76% of its similarities with human serum albumin (HSA), which makes it an exemplary protein in clinical medicine13.

3D Structure of bovine serum albumin (BSA) (PDB ID-4F5S).
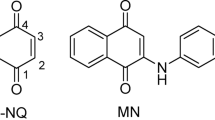
L-Ornithine (LO) (Fig. 2) is an unencoded free amino acid not engaged in the process of protein synthesis. Its primary function is to stimulate the pituitary gland, promoting the release of growth hormone14. It may exert a direct influence on the central nervous system, potentially alleviating stress and enhancing sleep quality, as well as reducing fatigue symptoms in humans (Fig. 3)15. It effectively reduces blood ammonia levels, offering potential benefits to individuals with hepatic encephalopathy or serving as a preventative measure against its development16. It serves as an intermediate in the urea cycle. It is predominantly generated in the cytosol through the arginase pathway, primarily derived from arginine, and subsequently transported into the mitochondrion via a specialized transport system17.

Structure of L-Ornithine (LO).

Mode of actions of LO.
Understanding the interaction between BSA and medicinal drugs, particularly LO, is vital due to BSA’s physiological importance and widespread presence in plasma. Given its abundance, BSA may act as a reservoir or carrier for BSA, potentially influencing the drug’s distribution and bioavailability. Conversely, BSA might serve as an off-target molecule, leading to unintended interactions that could disrupt its normal biological functions. Exploring the LO-BSA interaction is crucial for analysing the drug’s pharmacological profile. This study employs a combination of spectroscopic and computational methods to unravel the molecular basis of this interaction, assess its effects on BSA’s structural and functional properties, and determine its implications for drug safety and therapeutic efficacy. These findings are significant not only for optimizing LO’s clinical use but also for advancing drug design strategies by reducing off-target effects and enhancing therapeutic outcomes.
This paper aimed to elucidate the interaction mechanism between L-Ornithine (LO) and bovine serum albumin (BSA), as well as to examine the impact of LO binding on BSA’s structural changes.
Materials and methods
Chemical and preparation of sample
BSA and LO were bought from Sigma Chemical Co. and HIMEDIA. Each experiment, employing methodologies such as molecular docking, Fourier transform infrared spectroscopy (FTIR), UV–visible (UV–Vis) spectroscopy, fluorescence spectroscopy, and circular dichroism (CD) spectroscopy, utilized exclusively deionized water. A phosphate buffer solution with a concentration of 0.01 M (pH 7.4) was utilized. This buffer was prepared by combining sodium phosphate dibasic anhydrous and sodium dihydrogen orthophosphate monohydrate in deionized water to ensure the solution remained free from impurities. The prepared solution of BSA and LO was formulated in a phosphate buffer to achieve final concentrations of 10 μM and 0.5 mM, respectively, at a pH of 7.4. All data obtained from different instruments can be analysed using Origin 2024 software.
UV–Vis. spectra measurement
By operating Shimadzu’s UV-1900 UV–Vis, all UV–Vis. absorbance spectra were obtained. The investigations were conducted at room temperature. The apparatus introduced an error of ± 1 nm for the wavelength (λ) and ± 0.00001 for the absorbance (A). Spectra within the range of 200 to 400 nm were considered, and the maximum absorption point was determined based on the data obtained from spectra.
Fluorescence spectra measurement
The fluorescence intensity was quantified using a Shimadzu 5301PC fluorescence spectrophotometer equipped with a tungsten lamp and a spectral width of 10 nm. A quartz cell was filled with a precisely calibrated 3.0 mL aliquot of BSA and HSA solution at a concentration of 10 × 10−6 mol/L. The various concentration of LO (0 to 50 μM for BSA and 0 to 25 μM for HSA) was titrated to a stable quantity of BSA and HSA. After excitation with BSA and HSA at 278 nm, steady-state fluorescence was observed. The fluorescence intensities of each LO in buffer solutions were evaluated to eliminate the fluorescence background. The emission spectrum was collected in the 260–575 nm region. Various ratios of LO:BSA and LO: HSA solution was mixed in separate 10 mL volumetric flasks. Afterward, these mixtures were authorized to stand at room temperature for 5 min and then stimulate for 30 min at identified temperatures. Quenching spectra were documented at temperature 310 K using an excitation wavelength of 278 nm. During the fluorescence measurements, the slit widths for both emission and excitation were set to 5 nm, and the scan speed was adjusted to 600 nm/min.
FT-IR spectra measurement
The FT-IR spectra were listed utilizing an attenuated total reflection (ATR) sampling appliance attached to an FT-IR (PerkinElmer) spectrophotometer 2 by using the detector DTGS. The final concentrations of BSA and LO in all these mixtures were 0.3 mM each and analysed the change in secondary structure when drug LO added to BSA. To analyse these solutions, IR spectra were collected spanning the transmittance range of 4000–400 cm−1.
CD spectroscopy measurement
A JASCO-j-715 spectropolarimeter was employed to meticulously analyse the CD spectra of BSA solutions both in the absence and presence of LO. CD spectra of BSA were recorded in the range of 190–300 nm using a 0.1 cm cell length with 0.2 nm intervals. A 0.01 M phosphate buffer was incorporated into a prepared solution containing BSA (10 μM). To acquire CD spectra, the molar ratio of BSA to LO was adjusted within the range of 1:5 to 1:10.Each spectrum was analysed by BeStSel software (https://bestsel.elte.hu/), to evaluate the percentage of the α-helix.
Molecular docking studies
Molecular docking investigations of LO-BSA were done by operating the Glide module of the Schrodinger software. The receptor grid was built with default parameters and a van der Waals radius of 1.0A. Using Glide’s Extra Precision (XP) ligand docking, these grids were used to ascertain the interlinkage between the ligand and the receptor18.
Results and discussion
UV–Vis. spectroscopy study for the interaction of LO-BSA
To study drug-protein interactions, UV–Vis. spectra can be employed to monitor alterations in the absorption properties of either the drug or the protein molecules. This method offers pertinent insights into the binding and behaviour of drugs in the presence of proteins19. The intensity of a LO-BSA interaction is determined by observing the shift in the maximum band from ligand-free to binding-BSA structure. BSA’s electronic absorption spectrum has a significant absorption band at around 278 nm, is caused by the aromatic Trp and Tyr29 remnants. The molar ratio of the BSA-LO complex is determined through spectroscopic titration, where the BSA concentration remains constant while varying amounts of LO are introduced. Absorbance experiments were conducted at room temperature, covering the wavelength range of 200–400 nm. A cuvette with a 1 mm path length and a 1000 nm/min scanning speed was utilized. While the concentration of LO varied from 5 µM to 50 µM, the BSA concentration was maintained at 10 µM throughout the experiments.The insertion of LO gradually increased the absorption intensity of BSA and caused a hyperchromic shift, as depicted in Fig. 4. This hyperchromic effect, characterized by a rise in the intensity of absorption, suggests the binding of LO to the protein surface. Furthermore, the observed hyperchromic reaction may indicate an enhanced exposure of tryptophan residues to the solvent.

UV–Vis. spectra of BSA-LO complex having pH7.4 at room temperature.
Fluorescence spectroscopy study for interaction of LO-BSA
Fluorescence quenching is a sensitive method for investigating the interaction of tiny compounds with proteins. In BSA, two Trp residues (Trp-213 and Trp-134) exhibit relatively high fluorescence intensities and are responsible for changes in the microenvironment20. Two independent quenching mechanisms are employed to investigate the binding relationship between a quencher and macromolecules: static quenching and dynamic (or collision) quenching. A non-fluorescent fluorophore-quencher combination is described as the result of static quenching, while dynamic quenching involves the quencher diffusing to the fluorophore while it is stimulated. Without emitting any photons, the fluorophore rapidly returns to its ground state21. Based on the observed data, depicted in Fig. 5, the emission peak of BSA and HSA emerged at334 and 338 nm, and its intensity progressively diminished with increasing concentrations of LO shown in Figs. 5 and 6. In addition, the hypochromic shift (decrease in intensity) indicates the binding of LO’s with BSA, alters the micro-environment around Trpresidue in BSA and HSAchanged due to its interaction with LO. Consequently, fluorescence spectra of the BSA-LO complex were recorded at310K to examine the alternation in the Stern–Volmer constant (Ksv).

Fluorescence spectroscopy of BSA with and without LO at 310 K and pH 7.4.

Fluorescence spectroscopy of HSA with and without LO at 310 K and pH 7.4.
To assess the BSA-LO interaction by a quenching mechanism and compute the quenching parameters of BSA caused by LO, the Stern–Volmer equation was employed22.
In Eq. (1), F0 and F illustrate the fluorescence intensity of BSA and HSA with and without LO, [CQ] stands for the drug concentration,\({K}_{q}\) denotes the biomolecular quenching rate constant, τ0 corresponds to the moderate lifespan of the molecule without drugwith a fixed value of 10−8 s, and \({K}_{sv}\) signifies the Stern–Volmer quenching constant23. Figures 7 and 8 illustrates the Stern–Volmer plots of \(\frac{{F}_{0}}{F}\) against [Q] at 310 K.

The Stern–Volmer plot for the Fluorescence quenching of BSA by LO at 310 K.

The Stern–Volmer plot for the Fluorescence quenching of HSA by LO at 310 K.
As it is widely recognized, considering that the protein contains independent binding sites, the consecutive Eq. (2) can be utilized to obtain the assessment of the binding constant (Kb) and the number of binding sites (n).
where F and F0, respectively, represent the protein’s fluorescence intensity with and without a quencher24. The initial concentration is denoted by [Q], log [(F0-F)/F] v/s log [Q] may thus be plotted to regulate the values of Kb and n for the interaction between BSA and LO quencher.Using Eqs. (1) and (2) calculate the important parameters to determine the different types of fluorescence quenching. Figures 9 and 10, the resulting plots of \(\frac{{F}_{o}}{F}\) versus [C] of the interaction between BSA-LO, and HSA-LO at temperature 310 K were constructed, and their values of \({K}_{q}\) and \({K}_{SV}\) were evaluated.The computed Kq value, displayed in Tables 1 and 2 was higher than the maximum scattering collision quenching constant of 2 × 1010Lmol−1 s, indicating that quenching was facilitated by a static mechanism.

Linear plot of log (F0-F)/F versus log [Q] of BSA:LO complex.

Linear plot of log (F0-F)/F versus log [Q] of HSA:LO complex.
Gibbs free energy change (ΔG) is obtained from the following equation:
In Eq. (3) R and T respectively, represent the universal gas constant (1.987calmol−1 K−1) and temperature (310 K)25. Tables 1 and 2, representa summary of the computed outcomes of the LO- BSA and LO-HSA combination. Conversely, the data n (n ≈ 1) suggests LO’s binding position on BSA and HSA belonged to a single unique class.
FT-IR spectroscopy study for the interaction of LO-BSA
FT-IR spectra provided additional evidence for the interaction between LO and BSA. Protein’s infrared spectra revealed many amide bands caused by peptide vibrations. The amide I mode of the peptide group is commonly employed in protein subsidiary structure investigations26. The amideI and IIbands appeared at 1600–1700 cm−1 and 1500–1600 cm−1. Compared to amide II, amide I is more responsiblefor changes in the secondary structures of proteins27,28. Hydrogen bonding and transition dipole coupling significantly impact the conformational subtlety of amide bands. The amide I band (C=O stretch)appears at 1653 cm−1 and the II band (C-N stretch with N–H bending)appears at 1548 cm−1 is analogous to the protein’s subsidiary structure29,30.The BSA-LO spectral data were obtained at 298 K using the attenuated total reflectance (ATR) approach with a resolution of 4.0 cm−1 and 10 scans. The solution of LO was gradually added to the BSA, yielding combinations with various ratios (1:1, 1:3, and 1:10) of BSA. Thechanges observed in Fig. 11 the FT-IR spectrum of BSA-LO is due tothe multiple vibrational bands caused by various functional groups such as N–H, C=O, and amide groups. It was evident from Figs. 11 and 12 that the addition of LO had an impact on the amide I and II bands. The amide I was amended from 1644 cm−1 to 1639 cm−1 and the amide II was gone from 1539 to 1535 cm−1, with an increase in the concentration of LO. And it concludes that decrease in the helical shape of protein through BSA-LO complexes.

FT-IR spectra of LO-BSA at 298 K and pH 7.4

Changes in the Amide I and Amide II region of BSA with LO.
CD spectroscopy study for the interaction of LO-BSA
CD spectroscopy, a delicate method for examining protein conformational changes, was used in this investigation31. CD spectroscopy’s extraordinary sensitivity to sample perturbations allows proteins to be investigated qualitatively and quantitatively in terms of the effect of temperature, chemical agents, solvent components, pH, detergents, and ligand-combining interactivity32. The far-UV–Vis. region (190-250 nm) can reveal subsidiary structure composition in proteins. Peptide bonds are the predominant absorbing groups in this area. The asymmetric protein environment and innate chirality create induced asymmetry in some achiral ligand molecules during reversible binding interactions. CD spectroscopy measures optical activity in the visible region of light absorption33. This is a highly effective approach to investigating protein secondary conformations and gaining a thorough knowledge of the structural behaviour of the BSA-LO interaction. The CD spectroscopy studies were conducted to interrogate the variation in the secondary structure or composition of BSA produced by LO induction. At room temperature, the BSA and the molecular ratio of BSA-LO was managed at 1:0, 1:5, and 1:10, respectively (Fig. 13). To investigate variations in the secondary structure of BSA, CD spectra were collected in the absence and occupation of various immersion of LO. The CD spectroscopy of pure BSA showed two negative peaks at 208 and 222 nm, indicating its α-helical structure. The α-helical structure’s π-π* transition is represented by the 208 nm band, whereas the α-helix’s n-π* transition is responsible for the 222 nm band20,34.

The BSA and BSA-LO CD spectra at 298 K in a 0.01 M phosphate buffer with a pH of 7.4.
BESTSEL, a tool designed for secondary structure analysis and folding recognition from protein CD spectra, can be utilized to determine secondary structures. Figure 14 depicts the CD spectra of pure BSA and BSA with increasing concentrations of LO.The α-helical matter of BSA decreased from 33.2 to 14.9% after changing the ratios of BSA: LO (1:0, 1:1, and 1:5, respectively).The addition of molar ratios ([BSA]/[LO]) of 1:0, 1:1, and 1:5 caused noticeable changes in the shapes and positions of these peaks, suggesting the initiation of protein denaturation.The structural modifications of proteins, including α-helices and β-sheets, are mainly located in the core and are held together by hydrogen bonds. When positively charged coating agents attach to or penetrate negatively charged plasma proteins, they can increase the spacing between these hydrogen bonds, reducing the proportion of α-helical structures. This may further lead to a decrease in α-helices when BSA interacts with LO stabilised by positively charged coatings.This observed decrease can be attributed to the potential disruption or alteration of the secondary structure of the protein induced by the linkages with the molecules of LO35,36,37.

Helicity content for pure BSA and BSA-LO systems using Bestsel software.
Molecular docking study for interaction of LO-BSA
Preparation of protein
The crystallinemake-up of BSA (4F5S) was taken from the Protein Data Bank database (www.rcsb.org)38. Before proceeding with the docking technique, we optimized the structure of protein in Schrodinger by using the protein preparation wizard module (Prepwiz). In the first phase, the pretreated option was used to allocate bond ordering and join hydrogen. Epic ionizes heteroatoms at biological pH to assess protein absorbency along with medication solvability. H-bonds are then optimized to minimize steric confrontations. Applying the force field of OPLS3, the protein’s structurewas minimized to the lowest energy state.
Preparation of ligand
The ligprep tool was used to transform 2-dimensional ligand structures into 3-dimensional. Furthermore, the structure of the ligand was enhanced by reducing its energy by applying the OPLS3 force field. Docking investigations was conducted using the optimized low-energy ligand conformations39,40. A computational technique called “molecular docking” typically provides results that are consistent with experimental data, particularly when examining interactions between tiny compounds and biological macromolecules. According to crystal structural studies, Domain I (1–195), ___domain II (196–383), and ___domain III (384–583) are the three homologous domains that make up BSA. Each ___domain has two sub-domains, A and B. Sub-domains IIA and IIIA distribute the primary binding sites for small compounds corresponding to sites I and II. Our site probe competitive tests suggest that LO is bound to BSA at sub-___domain IIA. The following BSA residue interacts through hydrogen bond and hydrophobically with LO: ASP 118, GLU-125 and LYS-116, THR 121, LEU 122 as shown in Fig. 15, and a 3D view of the docking is shown in Fig. 16.

2-Dimensional view of LO binds with BSA.

3-Dimensional view of LO binds with BSA.
Conclusion
The interactions between LO and BSA were examined through UV–Vis spectroscopy, fluorescence spectroscopy, CD spectroscopy, and molecular docking techniques. UV–Vis spectroscopy revealed that LO binding to BSA results in a hyperchromic shift, indicating increased absorbance. Fluorescence spectroscopy showed a quenching effect on BSA fluorescence intensity, with parameters Kb = 5.183 × 103 Lmol−1, n = 0.8941, Ksv = 1.1 × 104 Lmol−1, Kq = 1.1 × 1012 Lmol−1 s−1, and ΔG = -5.269 kcal/mol, suggesting strong binding and dynamic quenching mechanisms.FT-IR spectroscopy and CD spectroscopy further illustrate that LO interaction induces significant alterations in the secondary structure of BSA, notably shifts in the amide I and amide II bands. These spectral changes indicate a decrease in α-helical content and an overall perturbation of the protein’s secondary structure. Molecular docking confirmed that LO predominantly binds to subdomain II of BSA. This comprehensive analysis elucidates the molecular basis of LO’s influence on the BSA microenvironment, providing insights into the potential modification of BSA for enhanced drug delivery and efficacy. These findings contribute to a deeper understanding of LO’s pharmacological interactions and its impact on protein function.
Data availability
The data will be available on request to the corresponding author.
Abbreviations
- UV–Vis.:
-
UV–Visible
- FT-IR:
-
Fourier transform infrared spectroscopy
- CD:
-
Circular dichroism
- LO:
-
L-Ornithine
- BSA:
-
Bovine serum albumin
- HSA:
-
Human serum albumin
- Trp:
-
Tryptophan
- Tyr:
-
Tyrosine
References
Jiang, S. L., Wang, W. J., Hu, Z. Y., Zhang, R. J. & Shi, J. H. Comprehending the intermolecular interaction of JAK inhibitor fedratinib with bovine serum albumin (BSA)/human alpha-1-acid glycoprotein (HAG): Multispectral methodologies and molecular simulation. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 304, 123277 (2024).
Kaumbekova, S., Sugita, M., Sakaguchi, N., Takahashi, Y., Sadakane, A. & Umezawa, M. Effect of Acetonitrile on the Conformation of Bovine Serum Albumin. ACS omega (2024).
Mavani, A., Ray, D., Aswal, V. K. & Bhattacharyya, J. Understanding the molecular interaction of BSA protein with antibiotic sulfa molecule (s) for novel drug development. J. Mol. Struct. 1287, 135697 (2023).
Sun, X. et al. Multispectral and molecular docking investigations on the interaction of primethamine/trimethoprim with BSA/HSA. J. Biomol. Struct. Dyn. 38(3), 934–942 (2020).
Abdelaziz, M. A., Shaldam, M., El-Domany, R. A. & Belal, F. Multi-Spectroscopic, thermodynamic and molecular dynamic simulation studies for investigation of interaction of dapagliflozin with bovine serum albumin. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 264, 120298 (2022).
Zhou, K. L., Pan, D. Q., Lou, Y. Y. & Shi, J. H. Intermolecular interaction of fosinopril with bovine serum albumin (BSA): The multi-spectroscopic and computational investigation. J. Mol. Recognit. 31(8), e2716 (2018).
Afsharan, H., Hasanzadeh, M., Shadjou, N. & Jouyban, A. Interaction of some cardiovascular drugs with bovine serum albumin at physiological conditions using glassy carbon electrode: A new approach. Mater. Sci. Eng. C 65, 97–108. https://doi.org/10.1016/j.msec.2016.03.112 (2016).
Shi, J. H., Pan, D. Q., Jiang, M., Liu, T. T. & Wang, Q. Binding interaction of ramipril with bovine serum albumin (BSA): Insights from multi-spectroscopy and molecular docking methods. J. Photochem. Photobiol. B Biol. 164, 103–111. https://doi.org/10.1016/j.jphotobiol.2016.09.025 (2016).
Zhang, R. J. et al. Exploring binding interaction of baricitinib with bovine serum albumin (BSA): Multi-spectroscopic approaches combined with theoretical calculation. J. Mol. Liq. 354, 118831. https://doi.org/10.1016/j.molliq.2022.118831 (2022).
Chaves, O. A., Cesarin-Sobrinho, D., Serpa, C., da Silva, M. B., de Lima, M. E. F. & Netto-Ferreira, J. C. The presence of NSAIDs may affect the binding capacity of serum albumin to the natural products hymecromone and umbelliferone. Int. J. Biol. Macromol. 137981 (2024).
Siddiqui, S., Ameen, F., ur Rehman, S., Sarwar, T. & Tabish, M. Studying the interaction of drug/ligand with serum albumin. Journal of Molecular Liquids 336, 116200. https://doi.org/10.1016/j.molliq.2021.116200 (2021).
Qi, H. et al. The different interactions of two anticancer drugs with bovine serum albumin based on multi-spectrum method combined with molecular dynamics simulations. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 259, 119809. https://doi.org/10.1016/j.saa.2021.119809 (2021).
Shi, J. H., Pan, D. Q., Jiang, M., Liu, T. T. & Wang, Q. In vitro study on binding interaction of quinapril with bovine serum albumin (BSA) using multi-spectroscopic and molecular docking methods. J. Biomol. Struct. Dyn. 35(10), 2211–2223. https://doi.org/10.1080/07391102.2016.1213663 (2017).
Canbay, A. et al. l-Ornithine-l-Aspartate (LOLA) normalizes metabolic parameters in models of steatosis, insulin resistance and metabolic syndrome. Pharmaceutics 16(4), 506 (2024).
Miyake, M. et al. Randomised controlled trial of the effects of L-ornithine on stress markers and sleep quality in healthy workers. Nutr. J. 13, 1–8. https://doi.org/10.1186/1475-2891-13-53 (2014).
Goh, E. T. et al. L-ornithine L-aspartate for prevention and treatment of hepatic encephalopathy in people with cirrhosis. Cochrane Database Syst. Rev. https://doi.org/10.1002/14651858.CD012410.pub2 (2018).
Shi, H. P. et al. Effect of supplemental ornithine on wound healing. J. Surg. Res. 106(2), 299–302. https://doi.org/10.1006/jsre.2002.6471 (2002).
Saravanan, K., Karthikeyan, S., Sugarthi, S. & Stephen, A. D. Binding studies of known molecules with acetylcholinesterase and bovine serum albumin: A comparative view. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 259, 119856. https://doi.org/10.1016/j.saa.2021.119856 (2021).
Wani, T. A., Bakheit, A. H., Abounassif, M. A. & Zargar, S. Study of interactions of an anticancer drug neratinib with bovine serum albumin: Spectroscopic and molecular docking approach. Front. Chem. 6, 47 (2018).
Liu, X., Ling, Z., Zhou, X., Ahmad, F. & Zhou, Y. Comprehensive spectroscopic probing the interaction and conformation impairment of bovine serum albumin (BSA) by herbicide butachlor. J. Photochem. Photobiol. B Biol. 162, 332–339. https://doi.org/10.1016/j.jphotobiol.2016.07.005 (2016).
Salam, M. A., Rokonujjaman, M., Rahman, A., Sultana, U. N. & Sultan, M. Z. Study of in vitro interaction of sildenafil citrate with bovine serum albumin by fluorescence spectroscopy. Pharmacol. Pharm. 6(2), 94–101 (2015).
Rashid, M. A., Rabbi, S. I., Sultana, T., Sultan, M. Z. & Sultan, M. Z. Fluorescence spectroscopic study of interaction between olanzapine and bovine serum albumin. Pharm. Anal. Acta 6, 408 (2015).
Nan, Z., Hao, C., Ye, X., Feng, Y. & Sun, R. Interaction of graphene oxide with bovine serum albumin: A fluorescence quenching study. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 210, 348–354 (2019).
Jiang, M. et al. Combined spectroscopies and molecular docking approach to characterizing the binding interaction between lisinopril and bovine serum albumin. Luminescence 31(2), 468–477. https://doi.org/10.1002/bio.2984 (2016).
Cui, F.-L., Fan, J., Li, J.-P. & Zhi-De, Hu. Interactions between 1-benzoyl-4-p-chlorophenyl thiosemicarbazide and serum albumin: Investigation by fluorescence spectroscopy. Bioorg. Med. Chem. 12(1), 151–157. https://doi.org/10.1016/j.bmc.2003.10.018 (2004).
Raghav, D., Mahanty, S. & Rathinasamy, K. Characterizing the interactions of the antipsychotic drug trifluoperazine with bovine serum albumin: Probing the drug-protein and drug-drug interactions using multi-spectroscopic approaches. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 226, 117584. https://doi.org/10.1016/j.saa.2019.117584 (2020).
Lin, S.-Y., Wei, Y.-S., Li, M.-J. & Wang, S.-L. Effect of ethanol or/and captopril on the secondary structure of human serum albumin before and after protein binding. Eur. J. Pharm. Biopharm. 57(3), 457–464. https://doi.org/10.1016/j.ejpb.2004.02.005 (2004).
Brauner, J. W., Flach, C. R. & Mendelsohn, R. A quantitative reconstruction of the amide I contour in the IR spectra of globular proteins: From structure to spectrum. J. Am. Chem. Soc. 127(1), 100–109. https://doi.org/10.1021/ja0400685 (2005).
Bakkialakshmi, S. & Chandrakala, D. A spectroscopic investigations of anticancer drugs binding to bovine serum albumin. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 88, 2–9. https://doi.org/10.1016/j.saa.2011.10.076 (2012).
Katrahalli, U., Jaldappagari, S. & Kalanur, S. S. Study of the interaction between fluoxetine hydrochloride and bovine serum albumin in the imitated physiological conditions by multi-spectroscopic methods. J. Lumin. 130(2), 211–216. https://doi.org/10.1016/j.jlumin.2009.07.033 (2010).
Jattinagoudar, L., Meti, M., Nandibewoor, S. & Chimatadar, S. Evaluation of the binding interaction between bovine serum albumin and dimethyl fumarate, an anti-inflammatory drug by multispectroscopic methods. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 156, 164–171 (2016).
Wang, Q. et al. Binding interaction of atorvastatin with bovine serum albumin: Spectroscopic methods and molecular docking. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 156, 155–163 (2016).
Mohammadi, F., Bordbar, A. K., Divsalar, A., Mohammadi, K. & Saboury, A. A. Analysis of binding interaction of curcumin and diacetylcurcumin with human and bovine serum albumin using fluorescence and circular dichroism spectroscopy. Protein J. 28, 189–196. https://doi.org/10.1007/s10930-009-9184-1 (2009).
Yasmeen, S., Khatun, S. & Qais, F. A. Characterization of interactions between cromolyn sodium and bovine serum albumin by spectroscopic, calorimetric and computational methods. J. Biomol. Struct. Dyn. https://doi.org/10.1080/07391102.2019.1586588 (2019).
Buddanavar, A. T. & Nandibewoor, S. T. Multi-spectroscopic characterization of bovine serum albumin upon interaction with atomoxetine. J. Pharm. Anal. 7(3), 148–155. https://doi.org/10.1016/j.jpha.2016.10.001 (2017).
Rahman, Y., Afrin, S., Alhaji Isa, M., Ahmed, S. & Tabish, M. Elucidating the molecular interaction of serum albumin with nizatidine and the role of β-cyclodextrin: Multi-spectroscopic and computational approach. J. Biomol. Struct. Dyn. https://doi.org/10.1080/07391102.2019.1604265 (2019).
Chowdhury, T. et al. Unveiling the binding interaction of zinc (II) complexes of homologous Schiff-base ligands on the surface of BSA protein: A combined experimental and theoretical approach. Appl. Organomet. Chem. 34(4), e5556. https://doi.org/10.1002/aoc.5556 (2020).
Bujacz, A. Structures of bovine, equine and leporine serum albumin. Acta Crystallogr. Sect. D Biol. Crystallogr. 68(10), 1278–1289. https://doi.org/10.1107/S0907444912027047 (2012).
Pawar, S. K. & Jaldappagari, S. Interaction of repaglinide with bovine serum albumin: Spectroscopic and molecular docking approaches. J. Pharm. Anal. 9(4), 274–283. https://doi.org/10.1016/j.jpha.2019.03.007 (2019).
Gulati, P., Solanki, P., Verma, A. K. & Kumar, A. Interaction of 4-ethyl phenyl sulfate with bovine serum albumin: Experimental and molecular docking studies. Plos One 19(10), e0309057. https://doi.org/10.1371/journal.pone.0309057 (2024).
Acknowledgements
The research was primarily supported by Chandigarh University, for which the authors are grateful.
Author information
Authors and Affiliations
Contributions
S.K.: Conceptualization, Writing; A.P.: Methodology, Data analysis; H.D.K.: Software, writing; A.R.P.: Project management, Figure preparation; J.K.: Supervision, Methodology; B.J.: Data analysis, literature survey; G.C.S.: Software, writing; A.B.: Conceptualization, Writing; A.J.S.: Supervision, Funding.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Koundal, S., Pathania, A., Kour, H.D. et al. Molecular interaction study of L-Ornithine with bovine serum albumin using spectroscopic and molecular docking methods. Sci Rep 15, 11997 (2025). https://doi.org/10.1038/s41598-025-93108-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-93108-z
