
Abstract
Multiple sclerosis (MS) is characterized as an immune-mediated central nervous system disease marked by chronic inflammation, demyelination, and progressive neurodegeneration. In this study, we evaluated the contribution of low-frequency and rare genetic variants to MS susceptibility within one of the largest family-based MS cohorts to date, comprising 215 individuals from 59 Turkish multiplex MS families. Whole exome sequencing was conducted on all samples including affected and unaffected members, followed by investigation of the effect of well-established human leukocyte antigen loci for MS on the elevated MS risk observed in our families. Subsequently, a gene-based burden analysis was performed on candidate genes identified through both our segregation analysis and existing literature. To prioritize the genes and pathways that are potentially associated with MS, a segregation-based analysis of the variants was conducted and complemented by gene-based pathway enrichment analysis. Our results highlighted the significance of the extracellular matrix in MS pathogenesis, as we identified laminin-related genes including LAMA5 and LAMB1 from both the segregation analysis and gene-based burden test. Hemidesmosome assembly emerged as a key pathway in our analysis, primarily driven by the identification of DST and PLEC as significant genes in the gene-based segregation analysis. Finally, we identified two rare coding variants passing our allele frequency and deleteriousness score-based filters, rs41266745 (C> T) in the CD109 gene with CADD phred score 24 and rs143093165 (T> G) in the ITPR1 gene with CADD phred score 22 and LOEUF 0.325, segregating within more than one family. Overall, this is one of the first and largest family-based MS studies from Turkey that features a unique cohort from an admixed population that enabled the detection of novel low-frequency and rare variants associated with MS. The findings from this study offer valuable insights that could guide future research aimed at further exploring and understanding the factors contributing to MS risk.
Similar content being viewed by others

Introduction
Multiple sclerosis (MS) is an immune-mediated disease of the central nervous system characterized by chronic inflammation, demyelination, and progressive neurodegeneration. Even though the exact cause of MS is still unknown, studies over the years have shown that both genetic and environmental factors contribute to disease susceptibility and progression. Many different loci contribute to MS susceptibility and a large genome-wide association study (GWAS) performed by the International Multiple Sclerosis Genetic Consortium (IMSGC) has uncovered more than 232 genetic loci including common human leukocyte antigens(HLA) variants1. However, these variants lack sufficient explanation for MS susceptibility in other populations2. HLA alleles are well-recognized as key genetic determinants of MS susceptibility, with common HLA gene variants contributing approximately 20-30% to the overall heritability of the disease3. The HLA-DRB1*15:01 allele is the most significant genetic risk factor, with carriers having over three times the likelihood of developing MS compared to non-carriers, contributing to approximately 10.5% of the genetic variance associated with the disease4. Other HLA variants, including HLA-DR3, HLA-DR4, and various DQ alleles, also contribute to MS risk, though their impact varies across populations, reflecting the genetic diversity in disease risk. The association with HLA-DRB115:01 is particularly prominent in Northern European populations, whereas different alleles may have stronger associations in other ethnic groups5.
Discovered MS loci can only explain about half of disease heritability where a large part of disease heritability remains unexplained. One potential source of the missing heritability is the presence of low-frequency and rare variants that exhibit moderate to large effect sizes. Standard methods for variant detection lack sufficient power to identify associations with rare variants due to their low frequency in the population6.
The most extensive effort to date in detecting low-frequency and rare variants associated with MS involved a large-scale meta-analysis conducted by the IMSGC, revealing that coding low-frequency variants account for 5% of MS heritability3. Individuals with a first-degree relative diagnosed with MS exhibit a markedly elevated risk, estimated to be seven times higher than that of the general population, underscoring the substantial genetic contribution in familial MS cases7. These analyses reveal that while common variants significantly contribute to disease risk, rare functional variants may also play an essential role, particularly in specific familial contexts where they might interact with common risk variants8,9.
Family-based genetic association studies can provide an advantage over case-control studies in detecting rare variant associations due to enrichment and multiple sampling of rare variants with moderate to large effect sizes in families with multiple individuals with the phenotype of interest10. Furthermore, depending on the structure of the pedigrees, family-based studies can offer greater robustness against genotyping errors, thereby enhancing the overall accuracy of the findings. In addition to using a family-based approach to explore the contribution of low-frequency variants to disease risk, analyzing samples from diverse populations holds significant value for identifying novel common and rare variant associations, as the majority of association studies in the past have predominantly utilized samples of European ancestry. Investigating multi-ethnic and admixed populations is particularly crucial for detecting low-frequency variants, as these variants are more recent compared to common variants and thus exhibit greater population specificity11.
The past decade, several studies have identified rare, potentially pathogenic genetic variants in patients with MS (pwMS) from multi-incident families using whole-exome sequencing (WES)10,12,13,14,15,16,17,18,19,20,21. In a study involving nine multi-incident MS families, WES revealed novel candidate genes, specifically, genetic variants influencing endothelial function, such as those in ICAM1, can affect the transcellular migration of autoreactive T-cells across the blood-brain barrier (BBB), contributing to neuroinflammation and subsequent demyelination21. Laminins and collagens are essential components of the extracellular matrix (ECM) that play critical roles in the pathology of MS, influencing processes such as inflammation, demyelination, and remyelination22.
In this study, we analyzed one of the largest cohorts of multiplex MS families to this date consisting of 59 multiply affected Turkish families (Turkish Familial MS - TuFAMS Consortium), aiming to discover novel low-frequency and rare variants (LFRV) associated with MS risk. For this purpose, we first assessed the segregation pattern of known HLA variants associated with MS in our families. Then we employed a segregation-based analysis of variants. For the prioritization of variants and genes, metrics such as the Combined Annotation Dependent Depletion (CADD) Phred score, probability of loss-of-function intolerance (pLI), and loss-of-function observed/expected upper bound fraction (LOEUF) were used. Further pathway enrichment analysis was utilized using gene-based results derived from the segregation analysis, where variants were collapsed into gene units. This approach led us to search for the low likelihood of observing the same variants across different families due to their low frequencies. By considering pathways and biological processes, this analysis allowed for a broader exploration of disease pathogenesis, providing a more comprehensive understanding of the underlying mechanisms rather than solely investigating individual variants or genes. The results show that the ECM-related pathways were significantly associated with incompletely segregated genes in multiple sclerosis families. Findings from previously conducted large population-based association studies, as well as other family-based studies, were thoroughly examined and compared with our results to determine whether any previously identified MS-associated variants and genes were detected by our analysis.
Results
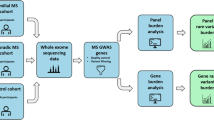
Exome sequencing of 215 individuals from 59 families, including 138 pwMS and 77 non-MS relatives, resulted in 589,587 variants, of which 40% were rare (minor allele frequency (MAF) \(\le\) 0.01). After stringent filtering, including removing common variants, Mendelian inconsistencies, and Hardy-Weinberg equilibrium failures, the dataset was refined to 38,851 variants across 14,036 genes. Segregation analysis under autosomal dominant and recessive models revealed 12 variants completely segregating with MS in more than two families. Genes such as LAMA5 and DST were identified with the highest number of segregating families, and pathway enrichment analysis highlighted ECM organization and collagen formation as key processes associated with MS. HLA-DRB1*15:01, a well-established MS susceptibility allele, exhibited complete segregation in one family and incomplete segregation in five others, supporting complex inheritance patterns within MS families. These findings support the involvement of both common and rare variants, particularly in ECM-related pathways, in MS pathogenesis (Fig. 1).

Workflow of whole exome sequencing and genetic analysis for multiple sclerosis families. This workflow outlines the stepwise approach for identifying and prioritizing genetic variants contributing to MS heritability in a cohort of 215 subjects from 59 families (138 MS cases, 77 unaffected). WES was performed, followed by HLA haplotyping using HLA-HD and variant calling, yielding 589,587 variants. Subsequent filtering removed variants on sex chromosomes and those failing Hardy-Weinberg Equilibrium (HWE), reducing the dataset to 510,102 variants. A low-frequency and rare variants (LFRV) filter was applied based on Genome Aggregation Database (gnomAD) MAF, and predicted impacts, considering variants with potentially deleterious effects. Segregation analyses under complete and incomplete penetrance models were conducted alongside burden analysis of genes harboring multiple variants. Enrichment analysis using g:Profiler identified significant pathways associated with MS. The results were further filtered based on gene and variant segregation patterns.
Demographic and clinical characteristics of multiple sclerosis patients and non-affected family members
The demographic and known clinical characteristics of 138 individuals diagnosed with MS and 77 non-affected family members reveal key differences across sex, age, and disease progression. Among affected family members, 58.69% were female and 41.30% were male, while the non-affected group had a higher proportion of males (54.54%). We compared the gender distribution between pwMS and healthy individuals to assess sex-based differences in MS susceptibility. Using generalized estimating equation (GEE) to account for familial clustering, we found that females had higher odds of MS (OR = 1.68, 95% CI: [1.01, 2.8], p = 4.4e−02). The mean age of affected individuals was 40.5 years, with a range of 18 to 69 years, whereas non-affected members have a mean age of 47.3 years (17–83 years). The distribution of MS subtypes shows that the majority of patients had relapsing-remitting MS (RRMS, 81 cases), followed by secondary progressive MS (SPMS, 17 cases), with smaller numbers of radiologically isolated syndrome (RIS), single attack MS (SAMS), primary progressive MS (PPMS), and progressive-relapsing MS (PRMS). The average Expanded Disability Status Scale (EDSS) score among affected individuals was 2.3, with a broad range of disease severity from minimal to more advanced disability (0 to 7.5) (Fig. 2). Regarding kinship, 61.17% of affected family members were first-degree relatives, 10.68% were second-degree, 12.62% were third-degree, and 15.53% were fourth-degree or more distant relatives (Supp. Table S1).

The demographic landscape of TuFaMS Consortium. (A) Comparison of the proportion of pwMS and healthy individuals by gender. The OR calculated using GEE to account for familial clustering. (B) Distribution of the number of patients with MS (pwMS) in families. (C) Clinical subtypes of MS in the TuFaMS cohort.
Exome sequencing, quality control, and variant filtering
Exome sequencing (40\(\times\) mean coverage in the exonic region), quality control, and variant calling produced a dataset of 589,587 variants of which approximately 40% were rare (MAF \(\le\) 0.01), 15% were low frequency( 0.01 < MAF \(\le\) 0.05 ) and 45% were common (MAF > 0.05). Removing X and Y chromosome variants, Mendelian inconsistencies, variants failing HWE at p < 10\(^{-4}\), and common variants led to a set consisting of 289,024 low-frequency and rare variants. In silico scoring and annotation tools were used to determine predicted deleterious variants which further reduced the number of variants to a final set of 38,851 variants across 14,036 genes. Before proceeding to segregation analysis, relatedness within and between families was assessed using kinship-based inference for GWAS (KING)23. This evaluation aimed to identify both intra-family inconsistencies and unreported inter-family relationships. No significant relationship was detected between any two families, as the relatedness value exceeded 0.05, suggesting cryptic relatedness as a significant factor.
Segregation patterns of HLA variants and their contribution to MS heritability
Twenty-nine different HLA types were identified in each individual. HLA haplotypes were typed and segregation patterns were examined in our families. We investigated HLA alleles with both risk and protective effects in MS.
Complete segregation of HLA-DRB1*15:01 was observed in only one family whereas incomplete segregation was observed in five families with both MS and non-MS participants (Table 1). The transmission of HLA-DRB1*15:01 with MS in our families was tested using transmission disequilibrium test (TDT)24. TDT statistics were calculated using our families and we detected an association between HLA-DRB1*15:01 and MS with odds ratio (OR) of 4.66 (95% confidence interval [CI]: 1.34–16.24) with combined p-value = 0.01. The strong OR and significant p-value indicate an association; however, the potentially low statistical power of the TDT test in our family cohort may have increased the risk of type I error, as indicated by the wide CI. We also used mixed-effects logistic regression to assess the association between the HLA-DRB1*15:01 haplotype and MS risk, using full extent of our dataset and accounting for complex pedigree structures and missing parental data. The haplotype was associated with an increased MS risk (OR=2.46, 95% CI: [1.32, 4.57], p-value=4.5e-03). Although the results point to a possible association, the small sample size means the strength of this finding should be viewed with some caution (Fig. 3).

Percentage of all MS and non-MS samples across our families carrying HLA-DRB1*15:01 as heterozygous (1 copy), homozygous (2 copies), and none (0 copy). Displayed OR and p-value statistics are from TDT analysis.
Burden analysis of low-frequency and rare variants
The aggregate burden of LFRVs was evaluated for the associated genes from the literature as well as from our segregation analysis, to which we refer as our “discovery” set. Incorporating the kinship matrix and gender as covariates in the mixed linear model revealed that the phenotypic variance explained by gender was 0.0388104, indicating a minimal contribution. Additionally, no significant differences were observed in OR/Beta values at the variant level when gender was included as a covariate. These findings suggest that gender has a limited impact on the burden analyses in this study. After the p-value correction for the total number of genes tested (\(\alpha\) = 0.05/8360 = 5.9e−06), no genes were found to be statistically significant. However, three genes from the literature with positive burden statistics (b.stat > 0) showed a different LFRV variant burden between the cases and controls for the “damaging” set of variants (p-value < 0.05). Additionally, eight genes were found to show a similar LFRV burden for the genes from our segregation filter. Lastly, the burden of LFRVs for the “control” variants was examined in these 11 genes. As expected, this analysis did not reveal any significant associations for these genes (Table 2).
Identification of variants exhibiting complete segregation across multiple families
The final set of variants was filtered down primarily according to autosomal dominant segregation models based on the cohort’s segregation patterns under both incomplete and complete penetrance assumptions. Variants segregating completely according to the autosomal dominant model were further assessed to determine if they also segregate fully in more than one family. We also investigated whether these variants were present in the families without non-MS samples to validate our findings.
We investigate genes with variants completely segregating for autosomal dominant inheritance mode in our families (Table 3). The top genes with the most families carrying at least one variant within a gene and showing complete segregation patterns were DST, FBN3 and AHNAK (Full list available in Supp. Table S3).
Gene-based segregation and pathway enrichment analysis
Gene-based analysis was done by collapsing segregating variants to genes and checking the number of families with observed segregation for at least one variant in a given gene. Gene level analysis is useful for accounting for low variant MAFs resulting in low probabilities of observing the same variants segregating in more than one family. As a result, we detected 584 genes with variants segregating incompletely with MS for more than one family. TTN is the top gene with the 12 families that carry at least one variant in this gene segregating with MS according to the given model followed by PLEC, KIAA1522, DST, and ABCA4 genes (full list available in Supp. Table S2). We also reviewed previous MS studies, including large-scale GWAS and other family-based studies, to see if any genes identified from our incomplete filter had been previously associated with MS. Using a compiled list of genes previously associated with MS8, we found an overlap of 25 genes including PLEC, SOX8, CUX2, USP34, and ILF3 from previous IMSGC studies1,3.
In the following step, we performed pathway enrichment analysis using g:Profiler on the genes from the incomplete segregation filter to reveal if we are detecting enrichment from MS-specific pathways25. To achieve a comprehensive analysis, multiple pathway databases, including the Human Phenotype Ontology (HPO), Reactome, Wikipathways, and KEGG were utilized.
We observed an enrichment pattern mainly revolving around ECM organization and collagen formation which included some of the top genes from our gene-based segregation analysis such as DST and PLEC as well as LAMA5, LAMA1, and LAMB1 laminin-related genes playing a regulatory role in the BBB structure26 (Table 4).

Enrichment analysis results of (A) incomplete dominant genes in gene-based segregation across HPO, Reactome, WikiPathways, and KEGG Pathway databases. Q-value (false discovery rate [FDR]) cutoff is 0.01 for significance. The gradient color indicates Q-values, where darker red represents higher statistical significance (Q = 0). Node size corresponds to gene set size. Larger nodes represent larger gene sets and edge thickness indicates shared genes between pathways. (B) Protein-protein interaction network of genes from segregation filter enriching for ECM organization and their clustering using k-means clustering within STRING. The edge thickness represents the significance of the associations, while the dotted edges indicate gene interactions between distinct clusters.
The pathway enrichment analysis of the incompletely segregated genes in multiple sclerosis families reveals significant associations with ECM-receptor interaction, focal adhesion, laminin interactions, and collagen chain trimerization among other pathways (Fig. 4 A). To further break down and group the ECM organization pathway to gain novel insights for genes such as LAMA5, LAMB1 based on their specific molecular interactions with ECM, which is involved in BBB structure27, we analyzed the genes enriching for this pathway and their interactions with each other using k-means clustering of protein-protein network of enriched genes with STRING28 (Fig. 4 B, Table 5). Moreover, pathway enrichment analysis of fully segregated genes reveals associations (FDR < 0.08) with processes involved in cell-cell communication, cell junction organization, and abnormal circulating alpha-fetoprotein concentrations (Supp. Table S4) .
Discussion
To our knowledge, this is one of the largest studies of MS with a family-based design to this date where we evaluated the role of novel LFRVs associated with MS risk by analyzing WES data from 59 Turkish families with mostly multiple affected members. Familial studies are crucial in uncovering inherited genetic factors that may not be detectable in unrelated populations, providing unique insights into the genetic architecture of MS. First-degree relatives of patients with MS have a sevenfold increased risk of developing the disease compared to the general population29. Our results, showing 61.17% of affected family members as first-degree relatives and decreasing prevalence across second, third, and more distant degrees, align with the global prevalence of familial MS supporting the hypothesis that closer genetic relationships increase the MS risk. Although this study represents one of the largest cohorts of multiplex MS families and offers certain advantages over large-scale case-control studies, particularly due to the enrichment of low-frequency variants through inheritance, the sample size remains statistically underpowered to detect individual variants associated with MS3. To address these limitations in statistical power, we employed a multi-faceted approach that combined gene prioritization with a gene-based burden analysis and segregation-based variant followed by pathway enrichment. Moreover, we examined the role of well-known HLA loci in MS and assessed how consanguinity might influence the increased MS risk in these families. From the known HLA loci that are associated with MS, we analyzed 29 HLA types including HLA-DRB1*15:01 which is the most significantly MS-associated allele identified to this day3. Across our families, we observed only one family with complete segregation of this risk allele with MS and an additional five families with a high segregation pattern. We observed that the majority of patients across our families do not carry the HLA-DRB1*15:01 allele overall. However, we did not observe any notable or significant patterns of segregation or distribution for other HLA variants previously associated with MS or reported as protective variants for MS from the literature within our dataset.
After that, we filtered variants that exhibited two penetration models, which are complete penetrance where all individuals with MS in a family possess the variant while none of the healthy individuals do, and incomplete penetrance, where a specific proportion of healthy individuals may carry the segregating variant, with autosomal dominant inheritance. Given the genetic heterogeneity of MS, identifying the exact set of single rare variants across different families presents a significant challenge. To address this challenge, we employed a gene-based approach, aggregating the variants identified from the segregation analysis into genes. The genes identified through the segregation analysis were then further examined using over-representation analysis to uncover significant pathways.
Our enrichment results highlight the significance of the ECM-related pathways, particularly the LAMA genes involved, including LAMA5 and LAMB1. LAMB1 was also detected by the rare variant burden analysis. Pathway enrichment results identified ECM organization, collagen formation, and laminin interactions as key processes associated with MS, with notable genes such as LAMA5, LAMB1, and PLEC playing critical roles. These proteins contribute to BBB integrity, which is essential in regulating immune cell infiltration into the central nervous system27. In a study, gene expression analysis revealed that the expression of LAMA5 significantly increased in the cerebral cortex of both male and female EAE mice compared to healthy controls. This increase was more pronounced in female EAE mice, with LAMA5 expression levels rising 3.85-fold compared to a 3.04-fold increase in males. These findings suggest that ECM components, such as laminins encoded by LAMA5, play a crucial role in the pathophysiology of MS by contributing to the structural changes in the brain associated with neuroinflammation and demyelination30. Moreover, studies have demonstrated that laminin-\(\alpha\)4 plays a pivotal role in facilitating T lymphocyte infiltration into the central nervous system. Laminin-\(\alpha\)4 null mice exhibit significantly reduced disease susceptibility and severity due to dramatically decreased T lymphocyte migration into the brain. This reduction is attributed to the compensatory and ubiquitous expression of laminin-\(\alpha\)5, which selectively inhibits integrin \(\alpha\)6\(\beta\)1-mediated migration of T lymphocytes through laminin-\(\alpha\)4. Consequently, the inhibitory role of laminin-\(\alpha\)5 in these null mice contributes to a less severe MS phenotype. These findings suggest targeting the interaction between T lymphocytes and endothelial basement membrane laminins, particularly inhibiting integrin \(\alpha\)6\(\beta\)1 interactions with laminin-\(\alpha\)431.
In insight of segregation results, the hemidesmosome assembly-related genes are also significantly associated with MS, emphasizing key genes such as DST, ITGB4, and PLEC. Notably, DST was also identified as a top candidate gene in an Italian family-based study by Mascia et al., which employed a methodology similar to that of our study. DST encodes the dystonin protein, a key component in maintaining cellular structural integrity. While this gene has been implicated in other neurodegenerative disorders, its association with MS has not been previously reported8. The replication of DST as a top candidate in our analysis, along with its involvement in relevant biological pathways and its established role in neurodegenerative diseases, suggests that it may be a promising candidate for further investigation in the context of MS pathogenesis. Recent research highlights that PLEC is a vitamin D receptor super-enhancer, which influences MS activity32. Our segregation analysis provides further evidence supporting PLEC as a compelling candidate gene for MS, consistent with its mapping by a significant association identified (rs3923387-T) in the 2019 IMSGC GWAS1. Interestingly, PLEC is also implicated in vascular dementia, contributing to neuronal loss through carotid artery occlusion33. PLEC interacts with AHNAK, which is also a candidate gene in our segregation analysis-, a scaffold protein involved in the formation of the BBB and calcium-dependent membrane processes in hematopoietic cells. Their established interaction in previous studies suggests that they may play a significant role in the MS etiology34. In MS, aberrant migration of immune cells across the BBB is a key event in the disease’s progression. ITGB4 is implicated in this process, facilitating interactions that might promote the entry of immune cells into the central nervous system, exacerbating inflammation. A recent study also suggests that ITGB4 might interact with ECM components, influencing demyelination and axonal damage, hallmarks of MS pathology35.
The fully segregating variants within CD109 and ITPR1 emerged as promising candidates for further investigation. We propose that CD109, which acts as a TGF-\(\beta\) co-receptor, could be a promising candidate gene for MS. CD109 is known to antagonize TGF-\(\beta\) signaling and ECM expression in fibroblasts and keratinocytes. In our study, we identified a C to T substitution in the CD109 (rs4126674) in two MS families (FMS17 and FMS18). The link between CD109 and MS may be mediated through TGF-\(\beta\), which plays a role in endothelial-mesenchymal transition, a crucial tissue modeling mechanism in neuroinflammation in MS36. ITPR1 has been associated with autoimmunity in the central nervous system, particularly in cases of autoimmune cerebellar ataxia, encephalitis, and peripheral neuropathy. ITPR1 was also mapped by a significant GWAS hit, rs4560284-A, in IMSGC GWAS from 20191. Autoantibodies against ITPR1 have been found in patients with neurological autoimmunity, which may indicate overlapping immune-mediated mechanisms that could contribute to MS-like neuroinflammatory conditions37.
To further capitalize on the unique dataset of multiply affected MS families, we analyzed the impact of the rate of consanguineous marriages within these families on the risk of developing MS. However, we did not observe an increased rate of consanguinity within our families that would be associated with an increased MS risk, contrary to findings reported in some previous studies, which were in line with a recently conducted national registry-based study38,39
Methods
Multi-incident families recruitment
Families were recruited through a large national collaborative effort known as the Turkish Familial MS Consortium, coordinated by the University of Istanbul-Cerrahpasa and initiated in 2017. Centers across Türkiye were asked to identify familial MS cases with multiple affected individuals having a first-degree relationship, along with their first and second-degree relatives. The participants were provided with patient information forms, consent documents, and family relation details. Clinical data were collected from 59 families, comprising a total of 215 samples, including 138 affected and 77 unaffected individuals, across 48 provinces and 26 centers in Türkiye. Of these 215 samples, 116 were from females and 99 from males. All samples were subsequently analyzed using exome sequencing. Among them, 138 were obtained from affected individuals (81 females and 57 males), while the remaining 77 samples were from unaffected individuals (35 females and 42 males). All methods in this study were carried out in accordance with relevant guidelines and regulations. The experimental protocols were reviewed and approved by the Clinical Research Ethics Committee of Istanbul University-Cerrahpaşa Medical Faculty (Approval Number: 83045809-604.01.02 / A-45). Informed consent was obtained from all participants or their legal guardians prior to their inclusion in the study.
DNA isolation and whole exome sequencing
Genomic DNA was isolated from peripheral blood using a standard salting-out procedure40. WES was performed on three different sets of individuals using different kits. 154 individuals were sequenced using Illumina’s PCR-free WES approach to ensure high-depth coverage and reduced amplification bias. 52 individuals underwent sequencing using the Illumina DNA Prep kit with exome enrichment through the Illumina Exome 2.5 Enrichment Kit in combination with Twist Bioscience reagents, optimizing exome capture. 9 individuals were sequenced by Macrogen using the Twist Human Core Exome kit with additional RefSeq enrichment.
HLA haplotyping
HLA typing was performed by HLA-HD41 which performs mapping of reads to the alleles in the HLA dictionary and calculates a score to identify the HLA types. Twenty-nine different HLA types were identified for each individual separately with the read length parameter (-m) selected as 90. Segregation of each HLA type was checked for each family. Transmission of HLA haplotypes in the families was tested using PLINK’s -tdt function to calculate the TDT statistics24. We applied mixed-effects logistic regression using the R lme4 package to evaluate the association between the HLA haplotypes and MS risk42. This model was selected to account for familial relationships and missing parental data in our cohort by incorporating random effects to control for shared genetic background. The odds ratio (OR) and 95% confidence interval (CI) were computed to estimate the effect size. Statistical significance was determined at a p-value threshold of 0.05.
Variant calling and annotation
Fastqc was used to assess the quality of the paired-end reads. Trim-Galore was used for removing adapters and low-quality sequences. Reads were mapped to human reference genome hg38 using Burrows-Wheeler Aligner-v0.7.17 (BWA)43. Pre-processing (MarkDuplicates, FixMateInformation and BaseRecalibrator) and variant calling (HaplotypeCaller) were done using Genome Analysis Toolkit’s (GATK)44 best practices45,46. All VCF files were merged and multiallelic sites were split into biallelic records using bcftools47. The merged VCF file was annotated using vcfanno48, Ensembl Variant Effect Predictor (VEP, version 109)49. ANNOVAR50 was used for annotation of in silico prediction scores using dbNSFP version 4.2. Mendelian inconsistencies were detected from 34 trios in our families using vcftools and removed from the main variant set51. Hardy-Weinberg p-values were calculated using vcftools for each site using founder samples from families if available and if not, one sample from each family included. Variant sites failing HWE at p < 10\(^{-4}\) were excluded.
Family relatedness and sex analysis
KING23 software was employed to identify any unknown relatedness between families. Pedigree error checking with default settings and inconsistent results with at least 2 degrees of inconsistency in the prediction were investigated. Peddy52 tool investigates sex discrepancies by evaluating the heterozygosity ratio of the X chromosome. Sex is determined by calculating the heterozygosity ratio, defined as the number of heterozygous variants divided by the number of homozygous alternate variants.
Burden testing of rare variants
The contribution of aggregated LFRVs was evaluated using the PedGene gene-based association tool, which incorporated pedigree-based kinship information to account for the relatedness among family members53. A burden test was conducted to evaluate the contribution of LFRVs with the same effect direction in the genes with non-zero variance and that had at least two variants from the “damaging” variant list used for the segregation analysis. In addition, a “control” variant list was created for the same genes, consisting of rare synonymous variants. The burden tests were carried out separately for each group and the rare variant burden was compared between the case and control groups8.
Variant filtering, segregation, and enrichment analyses
For the segregation analysis, we mainly investigated a dominant mode of inheritance due to the structure of our pedigrees, and two different filters were applied to identify the variants with complete and incomplete penetrance. For the complete penetrance filter, 34 families, containing both MS and non-MS individuals, were used. Filter was applied to each family separately to identify variants found in pwMS but not in unaffected family members. If any variant was found in any non-MS sample or if the genotype quality was less than 20, it was filtered out. MAF < 0.05 (gnomAD all populations-exome) and CADD Phred score \(\le\) 15 filters were applied to the remaining variants, and variants occurring in more than one family were selected. In addition to single nucleotide changes, frameshift insertion and deletions with HIGH impact score annotated in VEP were selected. For the incomplete penetrance filter, the variants found in at least 66% of pwMS within a family with both MS and non-MS participants were chosen. We allowed 33% of non-MS participants within a family and 33% of all non-MS participants across our sample set to carry the variant of interest for this filter. Families with no non-MS participants were used as a form of validation to check variants and genes from the segregation analysis.
Pathway and ontology over-representation analyses were performed using g:profiler and Enrichr on genes identified through incomplete gene-based segregation. These analyses encompassed the Reactome, KEGG, Wikipathway, and HPO databases54,55,56,57,58. Pathways and terms with FDR below 0.01, along with their shared genes, were visualized using the Enrichmap app in Cytoscape version 19.059,60.
Limitations
Although this study represents one of the largest family-based designs for MS to date, the sample size remains modest and still underpowered for the detection of rare variants. To overcome these power limitations, we employed an aggregation-based approach and utilized multiple methods to compare results, thereby increasing our confidence in the findings. Furthermore, the top results from the analyses were subjected to additional scrutiny using pathway enrichment methods.
A literature-based approach was also employed to characterize and prioritize our findings, ultimately generating a list of candidate variants and genes with the strongest associations with MS risk within our families.
Another significant limitation of this study is the lack of an independent replication cohort of multiplex MS families; from the same or similar population in terms of ancestry, however, obtaining a similar cohort with available whole exome sequencing data presents a considerable challenge.
Data availability
All data and scripts generated during this study are available from the corresponding author upon reasonable request. A comprehensive list of prioritized variants and genes, along with the relevant information, is provided in the supplementary files. Additionally, these prioritized variants have been uploaded to ClinVar for public access and further validation. The following list of ClinVar accession identifiers contains these variants; VCV000289396, VCV001027084, VCV000809956, VCV000809958, VCV000357584, VCV000357589, VCV003600999, VCV000357622, VCV003257589, VCV003601001, VCV000252602, VCV000436328, VCV000196854, VCV002063149, VCV000287621, VCV000196717, VCV000471582, VCV003600998, VCV003601002, VCV000714023, VCV001442403, VCV003601000.
References
I. M. S. G. Consortium*\(\dagger\) et al. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 365, eaav7188. https://doi.org/10.1126/science.aav7188 (2019).
Breedon, J. R. et al. Polygenic risk score prediction of multiple sclerosis in individuals of south Asian ancestry. Brain Commun. 5, fcad041 (2023).
Mitrovič, M. et al. Low-frequency and rare-coding variation contributes to multiple sclerosis risk. Cell 175, 1679–1687 (2018).
Hollenbach, J. A. & Oksenberg, J. R. The immunogenetics of multiple sclerosis: A comprehensive review. J. Autoimmun. 64, 13–25 (2015).
Dashti, M., Ateyah, K., Alroughani, R. & Al-Temaimi, R. Replication analysis of variants associated with multiple sclerosis risk. Sci. Rep. 10, 7327 (2020).
Lill, C. M. Recent advances and future challenges in the genetics of multiple sclerosis. Front. Neurol. 5, 130 (2014).
Steenhof, M. et al. Distribution of disease courses in familial vs sporadic multiple sclerosis. Acta Neurol. Scand. 139, 231–237 (2019).
Mascia, E. et al. Burden of rare coding variants in an Italian cohort of familial multiple sclerosis. J. Neuroimmunol. 362, 577760 (2022).
Clarelli, F. et al. Contribution of rare and low-frequency variants to multiple sclerosis susceptibility in the Italian continental population. Front. Genet. 12, 800262 (2022).
Zrzavy, T. et al. Exome-sequence analyses of four multi-incident multiple sclerosis families. Genes 11, 988 (2020).
Peterson, R. E. et al. Genome-wide association studies in ancestrally diverse populations: Opportunities, methods, pitfalls, and recommendations. Cell 179, 589–603 (2019).
Everest, E. et al. Investigating the role of common and rare variants in multiplex multiple sclerosis families reveals an increased burden of common risk variation. Sci. Rep. 12, 16984 (2022).
Mescheriakova, J. Y. et al. Linkage analysis and whole exome sequencing identify a novel candidate gene in a Dutch multiple sclerosis family. Mult. Scler. J. 25, 909–917 (2019).
Ramagopalan, S. V. et al. Rare variants in the cyp27b1 gene are associated with multiple sclerosis. Ann. Neurol. 70, 881–886 (2011).
Dyment, D. A. et al. Exome sequencing identifies a novel multiple sclerosis susceptibility variant in the tyk2 gene. Neurology 79, 406–411 (2012).
Wang, Z. et al. Nuclear receptor nr1h3 in familial multiple sclerosis. Neuron 90, 948–954 (2016).
Maver, A. et al. Identification of rare genetic variation of nlrp1 gene in familial multiple sclerosis. Sci. Rep. 7, 3715 (2017).
Sadovnick, A. D. et al. Purinergic receptors p2rx4 and p2rx7 in familial multiple sclerosis. Hum. Mutat. 38, 736–744 (2017).
Salehi, Z. et al. Exome sequencing reveals novel rare variants in Iranian familial multiple sclerosis: The importance of pold2 in the disease pathogenesis. Genomics 113, 2645–2655 (2021).
Pytel, V. et al. Exonic variants of genes related to the vitamin d signaling pathway in the families of familial multiple sclerosis using whole-exome next generation sequencing. Brain Behav. 9, e01272 (2019).
Horjus, J. et al. Whole exome sequencing in multi-incident families identifies novel candidate genes for multiple sclerosis. Int. J. Mol. Sci. 23, 11461 (2022).
You, Y. & Gupta, V. The extracellular matrix and remyelination strategies in multiple sclerosis. Eneuro 5 (2018).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Purcell, S. et al. Plink: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Kolberg, L. et al. Update. Nucleic Acids Res. 51, W207–W212. https://doi.org/10.1093/nar/gkad347 (2023).
Halder, S. K., Sapkota, A. & Milner, R. The importance of laminin at the blood-brain barrier. Neural Regen. Res. 18, 2557–2563 (2023).
Du, F., Shusta, E. V. & Palecek, S. P. Extracellular matrix proteins in construction and function of in vitro blood-brain barrier models. Front. Chem. Eng. 5, 1130127 (2023).
Szklarczyk, D. et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638–D646 (2023).
Nielsen, N. M. et al. Familial risk of multiple sclerosis: A nationwide cohort study. Am. J. Epidemiol. 162, 774–778. https://doi.org/10.1093/aje/kwi280 (2005). https://academic.oup.com/aje/article-pdf/162/8/774/284135/kwi280.pdf.
Batzdorf, C. S. et al. Sexual dimorphism in extracellular matrix composition and viscoelasticity of the healthy and inflamed mouse brain. Biology 11, 230 (2022).
Wu, C. et al. Endothelial basement membrane laminin \(\alpha\)5 selectively inhibits t lymphocyte extravasation into the brain. Nat. Med. 15, 519–527 (2009).
Orton, S. M. et al. Expression of risk genes linked to vitamin D receptor super-enhancer regions and their association with phenotype severity in multiple sclerosis. Front. Neurol. 13, 1064008 (2022).
Zhao, K. et al. RNA sequencing-based identification of the regulatory mechanism of microRNAs, transcription factors, and corresponding target genes involved in vascular dementia. Front. Neurosci. 16, 917489 (2022).
Nii, T. et al. The bioactive peptide sl-13r expands human umbilical cord blood hematopoietic stem and progenitor cells in vitro. Molecules 26. https://doi.org/10.3390/molecules26071995 (2021).
Zhang, W. et al. Astrocytes increase exosomal secretion of oligodendrocyte precursor cells to promote their proliferation via integrin \(\beta\)4-mediated cell adhesion. Biochem. Biophys. Res. Commun. 526, 341–348 (2020).
Troletti, C. D., de Goede, P., Kamermans, A. & de Vries, H. E. Molecular alterations of the blood–brain barrier under inflammatory conditions: the role of endothelial to mesenchymal transition. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 1862, 452–460 (2016).
Jarius, S. et al. Inositol 1, 4, 5-trisphosphate receptor type 1 autoantibody (itpr1-igg/anti-sj)-associated autoimmune cerebellar ataxia, encephalitis and peripheral neuropathy: Review of the literature. J. Neuroinflamm. 19, 196 (2022).
AlJumah, M. et al. Familial aggregation of multiple sclerosis: Results from the national registry of the disease in Saudi Arabia. Mult. Scler. J.-Exp. Transl. Clin. 6, 2055217320960499 (2020).
Salehi, Z. et al. Consanguineous marriage among familial multiple sclerosis subjects: A national registry-based study. Heliyon (2024).
Miller, S., Dykes, D. & Polesky, H. A simple salting out procedure for extracting DNA from human nucleated cells. OUP Acad. (1988).
Kawaguchi, S., Higasa, K., Shimizu, M., Yamada, R. & Matsuda, F. Hla-hd: An accurate hla typing algorithm for next-generation sequencing data. Hum. Mutat. 38, 788–797 (2017).
Bates, D., Mächler, M., Bolker, B. & Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 67, 1–48. https://doi.org/10.18637/jss.v067.i01 (2015).
Li, H. & Durbin, R. Fast and accurate short read alignment with burrows–wheeler transform. bioinformatics 25, 1754–1760 (2009).
McKenna, A. et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Van der Auwera, G. A. et al. From FASTQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 43, 11–110 (2013).
Danecek, P. et al. Twelve years of samtools and bcftools. Gigascience 10, giab008 (2021).
Pedersen, B. S., Layer, R. M. & Quinlan, A. R. Vcfanno: Fast, flexible annotation of genetic variants. Genome Biol. 17, 1–9 (2016).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 1–14 (2016).
Wang, K., Li, M. & Hakonarson, H. Annovar: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164–e164 (2010).
Danecek, P. et al. The variant call format and vcftools. Bioinformatics 27, 2156–2158 (2011).
Pedersen, B. S. & Quinlan, A. R. Who’s who? detecting and resolving sample anomalies in human DNA sequencing studies with Peddy. Am. J. Hum. Genet. 100, 406–413 (2017).
Schaid, D. J., McDonnell, S. K., Sinnwell, J. P. & Thibodeau, S. N. Multiple genetic variant association testing by collapsing and kernel methods with pedigree or population structured data. Genet. Epidemiol. 37, 409–418 (2013).
Jassal, B. et al. The reactome pathway knowledgebase. Nucleic Acids Res. 48, D498–D503 (2020).
Kolberg, L. et al. g: Profiler—Interoperable web service for functional enrichment analysis and gene identifier mapping (2023 update). Nucleic Acids Res. 51, W207–W212 (2023).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361 (2017).
Agrawal, A. et al. Wikipathways 2024: Next generation pathway database. Nucleic Acids Res. 52, D679–D689 (2024).
Köhler, S. et al. The human phenotype ontology in 2021. Nucleic Acids Res. 49, D1207–D1217 (2021).
Shannon, P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Merico, D., Isserlin, R., Stueker, O., Emili, A. & Bader, G. D. Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PloS one 5, e13984 (2010).
Acknowledgements
Research grants for this study have been received from Istanbul University-Cerrahpasa Scientific Research Projects (No. 26360), Istanbul Technical University Scientific Research Projects (No. 41964), Turkish MS Society Award Project (No. 41), Turkish Scientific and Technological Research Council of Turkey (TUBITAK) ARDEB 1002 (No. 220S889), and ARDEB 1001 (No. 123Z072). We thank Clifon L. Dalgard and Uniformed Services University, Laboratory Core of the Collaborative Health Initiative Research Program, as well as Yale Center for Genome Analysis, for sequencing services. We would also like to thank all the MS family members for their contribution.
Author information
Authors and Affiliations
Contributions
F.B., B.G., and A.B contributed to the design of the study and did the bioinformatic analysis; U.A., Y.B., C.B., S.D.B, C.F.D., S.D., T.D., H.E., H.G., T.G., R.K., B.K, M.K., M.M., S.S(20)., M.S., S.S (22). , B.T., M.T (23)., M.T. (25), A.T., O.F.T., M.T.(20), G.U., U.U., C.U.., M.F.Y., N.Y. A.S. contributed MS families samples and clinical data; O.C., M.D.R. and U.V did the DNA isolation, M.D.R also did the demographic and clinical analysis; F. B., A. B., E.S., and U.E. contributed to discussions on the significance and functional roles of the identified genes; K.B and O.U.O contributed in the design of the bioinformatic analyses; E.E contributed the DNA isolation and collection of first 33 families; M.T (21), A.S and E.T.T contributed in the funding of the study; A.S. and E.T.T initiated and designed the study hypothesis, critically analyzed the data. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Büyükgöl, F., Gürdamar, B., Aluçlu, . et al. Exome sequencing reveals low-frequency and rare variant contributions to multiple sclerosis susceptibility in Turkish families. Sci Rep 15, 11682 (2025). https://doi.org/10.1038/s41598-025-94691-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-94691-x
