
Abstract
Mitochondrial dysfunction plays a pivotal role in the pathogenesis of chronic obstructive pulmonary disease (COPD). This study combines bioinformatics analysis with machine learning to elucidate potential key mitochondrial-related genes associated with COPD and its immune microenvironment. We utilized the limma package and Weighted Gene Co-expression Network Analysis (WGCNA) to analyze datasets from the Gene Expression Omnibus (GEO) database (GSE57148), identifying 12 key differentially expressed mitochondrial genes (MitoDEGs). Using 12 distinct machine learning algorithms (comprising 143 predictive models), we identified the optimal diagnostic model, which includes five pivotal MitoDEGs: ERN1, FASTK, HIGD1B, NDUFA7 and NDUFB7. The diagnostic specificity and sensitivity of each gene, as well as the diagnostic model itself, were evaluated using Receiver operating characteristic (ROC) curves. This model demonstrated high specificity in the validation cohorts (GSE76925, GSE151052, GSE239897). Expression analysis revealed upregulation of ERN1 and downregulation of FASTK, HIGD1B, NDUFA7 and NDUFB7 in COPD patients. Spearman’s correlation analysis indicated a significant association between MitoDEGs and immune cell infiltration, with ERN1 expression positively correlated with neutrophil infiltration and the other genes negatively correlated. The GABA receptor modulator androstenol was identified as a potential therapeutic candidate. In vivo studies confirmed reduced mRNA expression of HIGD1B and NDUFB7 in COPD mice. These findings elucidate mitochondrial-immune interactions in COPD and highlight novel diagnostic and therapeutic targets.
Similar content being viewed by others

Introduction
Chronic obstructive pulmonary disease (COPD) is a prevalent chronic airway disease characterized by persistent airflow obstruction and progressive respiratory symptoms, with high mortality and significant socio-economic impacts1,2,3. Its pathogenesis involves gene-environment interactions, with cigarette smoke (CS) exposure being the primary risk factor4,5. Despite current therapeutic advancements, COPD management remains challenging due to disease heterogeneity and limited biomarkers for early diagnosis6,7,8. Recent studies highlight mitochondrial dysfunction as a critical player in COPD progression, linking it to inflammation, epithelial damage, and steroid resistance9.
Mitochondrial impairment in COPD manifests through multiple mechanisms: CS exposure disrupts mitochondrial dynamics (fusion/fission as well as transport) and autophagy pathways10,11, while acrolein (a major CS-derived toxin) induces bioenergetic collapse and inflammation activation12. This mitochondrial impairment is thought to contribute to heightened inflammatory responses, hindered epithelial repair processes, reduced corticosteroid sensitivity in lung tissue, and impaired macrophage phagocytosis, which are factors implicated in COPD progression13. Animal models demonstrate that mitochondria-specific antioxidant MitoQ mitigate lung injury by inhibiting mitochondrial autophagy and NLRP3 inflammasome signaling14. Clinical studies have further revealed impaired mitochondrial dysfunction in lung tissues of COPD patients, including disrupted mitochondrial fusion, increased autophagy, and accelerated cell senescence15. These changes are closely related to the aggravated airflow limitation and decreased exercise tolerance in patients16. These findings underscore the therapeutic potential of targeting mitochondrial pathways in COPD, and a number of mitochondrial-specific antioxidants have been proposed as therapeutic options17,18.
However, the molecular interplay between mitochondria-related genes and immune microenvironment regulation in COPD remains poorly understood. This study aims to identify key mitochondria-related differentially expressed genes (MitoDEGs) in COPD by using multi-omics analysis of Gene Expression Omnibus (GEO) datasets, and elucidate their regulatory roles in immune infiltration and disease progression. By integrating machine learning with network biology approaches, we expect to uncover novel biomarkers and therapeutic targets for COPD management.
Materials and methods
Microarray data collection and analysis of differentially expression genes (DEGs)
We obtained lung tissue transcriptomic datasets from the NCBI GEO database (https://www.ncbi.nlm.nih.gov/geo). The training set (GSE57148) and validation sets (GSE76925, GSE151052, GSE239897) were selected through systematic filtering by “COPD”, Homo sapiens, and lung tissue criteria. Post-quality control measures, the specifics of case inclusion across datasets are elucidated in Table 1. Data preprocessing utilized the “GEOquery” package (version 2.70.0)19, “limma” package (version 3.58.1)20, “ggplot2” (version 3.4.4) and “ComplexHeatmap” (version 2.18.0) packages in R. Differential expression analysis identified genes with adjusted P < 0.05 and log2 fold change > 0.58.
Gene set variation analysis (GSVA) and functional enrichment analysis
GSVA, an extension of the Gene Set Enrichment Analysis (GSEA) framework, allows the transformation of the gene expression matrix across samples into a gene set expression matrix to evaluate pathway enrichment in sample sets21. Utilizing the “msigdbr” package (version 7.5.1) in R, we retrieved the "c2.cp.kegg.v7.4. symbols.gmt" gene set from the Molecular Signatures Database (MSigDB) (https://www.gsea-msigdb.org/gsea/msigdb/index.jsp)22. Enrichment analysis was subsequently performed on COPD samples from GSE57148 using “GSVA” package (version 1.50.0). Functional annotation was conducted through Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses using the “ClusterProfiler” package (version 4.10.2)23. GO terms categorized molecular functions, biological processes, and cellular components, while KEGG enrichment analyses contextualized pathway information24,25. Significance thresholds were set at P-value < 0.05 or Q-value < 0.05. Results were visualized by “enrichplot” package (version 1.22.0) and "ggplot2″ package (version 3.4.4).
WGCNA and key modules gene identification
We applied WGCNA (version 1.72-5) to construct a co-expression network from GSE57148 transcriptomic data26. Initial sample clustering eliminated outliers to enhance network stability. Soft thresholding power analysis optimized gene–gene correlations detection, followed by dynamic tree-cutting method to partition genes into 23 co-expression modules (Supplementary Table S1 and Supplementary Figure S1). Module eigengenes were computed to identify associations with COPD clinical features. Key modules showing significant correlations (|GS|> 0.7) were prioritized for further analysis. Hub genes within these modules were determined through intra-module connectivity analysis, highlighting candidates on the pathogenesis of COPD.
Identification of MitoDEGs
Following Fu et al.s protocol27, we identified 2030 human MitoDEGs through integrated analysis of three datasets: mitochondrial genes from MitoCarta3.0 (http://www.broadinstitute.org/mitocarta), DEGs detected by limma (v3.58.1) in GSE57148 transcriptomic data, and co-expression modules associated with COPD pathology via WGCNA (Supplementary Table S2)28,29. The intersection of these datasets was visualized via Venn diagram, ensuring comprehensive coverage of mitochondria-related genes linked to COPD.
Development and evaluation of a classification predictive model using machine learning
To identify mitochondrial gene-based biomarkers and develop a robust COPD classification model, we adapted Maimaiti et al.'s framework30, training 143 predictive models using 12 machine learning algorithms (including Lasso, Ridge, Enet, Stepglm, SVM, glmBoost, LDA, plsRglm, RF, GBM, XGBoost and NaiveBayes) to evaluate 14 MitoDEGs. Model performance assessed via receiver operating characteristic (ROC) curves. The area under the curve (AUC) values along with the corresponding 95% confidence intervals (CIs) were calculated using the “pROC” package (version 1.18.5)31. Heatmaps were used to visualize the results, clearly depicting the model performance of the various algorithms tested.
Immune infiltration analysis
We applied CIBERSORT (version 0.1.0) to quantify immune cell infiltration in COPD samples,analyzing 22 immune cell types using gene expression data32. Immune cell abundance and proportions were visualized as bar graphs (ggplot2 v3.4.4). Correlation between cell types was assessed via Pearson correlation matrices (corrplot version 0.92). Differences in cell proportions between COPD and control groups were identified by Wilcoxon test (P < 0.05), with results displayed through boxplots (ggplot2 version 3.4.4). Finally, Spearman correlation analysis linked the five hub genes to immune cells, visualized using ggplot2 (version 3.4.4).
Connectivity map (cMAP) analysis
We employed cMAP (https://clue.io), a platform analyzing differential gene expression to connect diseases, genes and small molecules33. By inputting mitochondria-associated modular gene from COPD lung samples, we identified top 10 small molecules ranked by normalized connectivity scores, highlighting their therapeutic potential. Structural data for target genes were retrieved to elucidate molecular interaction with these identified compounds.
Construction of COPD mice model and Ethical Approval
Following an established protocol34, we developed a COPD mouse model using twenty male C57BL/6 mice (6–8 weeks old) from Hunan Slake Jinda Laboratory Animal Co. Mice were housed at the Department of Laboratory Animal Science at Central South University, divided into two groups: control and COPD group. The COPD group received LPS (1 mg/kg, i.t) on day 1 and 30, plus 10 cigarettes/day (each containig 1.0 mg nicotine, 11 mg tar, Furong brand cigarettes from China) in a homemade fume box twice a day, 6 days a week, for 16 weeks. Controls were exposed to air. Lung tissues were collected after euthanasia via pentobarbital anesthesia. The protocol was approved by the Ethics Committee for Laboratory Animal Welfare at Central South University (with approval number CSU-2023-0191) and complied with ARRIVE guidelines.
Histological analysis
Left lung tissues were fixed in 4% paraformaldehyde overnight, paraffin-embedded, and sectioned into 4-μm slices. Hematoxylin–eosin staining identified histopathological changes. Leica microscopes captured images and blinded histological scoring assessed inflammatory infiltration (0–3 scale) and alveolar damage across three random fields per sample. Inflammatory infiltrates were evaluated on a four-point scale, with 0 indicating no or occasional inflammatory cells; 1 indicating a few loosely arranged inflammatory cells; 2 indicating many cells in the interstitial and intra-alveolar spaces; and 3 indicating numerous inflammatory cells in the perivascular space35.
Reverse transcription quantitative polymerase chain reaction (RT-qPCR) analysis
To extract total RNA from lung tissue, the Trizol reagent (Invitrogen) was used, followed by reverse transcription into cDNA with the PrimeScript™ II 1st Strand cDNA Synthesis Kit (Takara, Shiga, Japan). The 2 × ChamQ Universal SYBR qPCR Master Mix (Vazyme, China) was employed for amplifying each sample in a 20 μl reaction mixture. The fold changes were caculated using the 2-ΔΔCt method. Expression levels were determined by calculation and normalized to the endogenous GAPDH. Detailed primer sequences for this procedure are systematically listed in Table 2.
Study approval
All animal experiments were performed in accordance with relevant guidelines, regulations and ARRIVE guidelines. This study was approved by the ethics committee of Laboratory Animal Welfare at Central south university (grant number: CSU-2023-0191).
Statistical analyses
All statistical analyses were performed using R software (version 4.2.1, accessed on January 25, 2025, https://www.r-project.org) and GraphPad Prism (version 8.0, accessed on January 25, 2025). All results were expressed as mean ± standard error of the mean (SEM) of at least 3 independent experiments. T-tests were used for comparisons between dual groups while one-way ANOVA was used for three or more groups. Statistical significance was determined by comparing p-values, where P < 0.05 denoted significance (* P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001).
Results
Identification of DEGs between healthy controls and patients with COPD
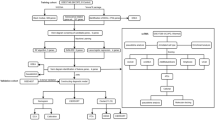
Figure 1 illustrates the study workflow. Data normalizaztion of GSE57148 identified 174 DEGs in COPD vs healthy lungs (Supplementary Table S3), with 114 upregulated and 60 downregulated genes (Fig. 2A,B). Pathway enrichment analysis via GSVA revealed significant COPD-specific alterations: upregulated pathways including the ErbB signaling pathway, prostate cancer, type 2 diabetes mellitus, phosphatidylinositol signaling system, pathways in cancer, adherens junction, neurotrophin signaling pathway, glioma, aldosterone regulated sodium reabsorption, and O-glycan biosynthesis; downregulated pathways involved proteasome, Alzheimer’s disease, the peroxisome, regulation of autophagy, the ribosome, Huntington’s disease, glutathione metabolism, base excision repair, Parkinson’s disease, and oxidative phosphorylation (Fig. 2C). Notably, mitochondrial gene sets showed marked dysregulation between the two groups (P = 5.7e−11) (Fig. 2D), underscoring mitochondrial dysfunction’s role in COPD pathogenesis alongside immune imbalance.

Flowchart of the Entire Study.

Detection of DEGs in COPD and control groups. (A) Volcano plot of DEGs; (B) Heat map of top 20 DEGs; (C) GSVA analysis based on the KEGG database; (D) Comparison of mitochondrial gene scores between two groups.
Weighted gene co-expression network construction
WGCNA identified key COPD-associated gene modules using GSE57148 dataset. After filtering low-quality samples, 91 normal and 98 COPD samples were clustered. A soft-threshold (power = 7) with R2 > 0.8 and average connectivity > 0.8 was applied, resulting in 32 modules (Fig. 3A–C). Clustering tree illustrated module evolution. Key modules showed significant correlations with COPD: yellow module (r = 0.57, P = 2e−17), pink module (r = − 0.58, P = 8e−18) and the blue module (r = − 0.48, P = 3e−12) (Fig. 3D). Strong module-gene associations were observed in all three modules: yellow module (621 genes, r = 0.73, P = 2.1e−104), pink module (351 genes, r = 0.72, P = 2.5e−57), and blue module (2634 genes, r = 0.61, P < 1e−200) (Fig. 3E–G), totaling 3606 genes for downstream analysis.

Modular genes in normal and COPD groups detected using WGCNA. (A) Determine the optimal “soft” threshold for the dataset; (B) Initial and merged modules in the clustering dendrogram; (C) Gene co-expression modules indicated in different colors under the cluster tree; (D) Identify modules that are highly correlated between the normal and COPD groups; Scatterplot of the yellow (E), pink (F) and blue (G) module.
Identification and functional analysis of MitoDEG in the context of COPD
MitoDEGs were identified through intersection of WGCNA modules, limma DEGs, and MitoCarta3.0 mitochondrial genes (Fig. 4A), yielding 14 key genes (COMTD1, ELK3, ERN1, ETFB, FASTK, HIGD1B, IFI27L2, MRPL41, MRPL55, NDUFA7, NDUFB7, NME3, PLA2G4B and ZBED3). GO enrichment analysis revealed that MitoDEGs are involved in the regulation of mitochondrial gene expression, purine ribonucleoside triphosphate biosynthesis process, mitochondrial protein-containing complexes, organellar ribosome, electron transfer activity, and structural constituent of ribosome (Fig. 4B). KEGG analysis suggested that these MitoDEGs are related to various disease pathways such as Non-alcoholic fatty liver disease, Parkinson disease, Huntington disease, Amyotrophic lateral sclerosis, Alzheimer disease, and many other and others (Fig. 4C).

Identification and functional analysis of MitoDEGs in COPD. (A) Venn diagram of key modular genes, DEGs and mitochondria-related genes; (B, C) GO and KEGG analysis of MitoDEGs.
Selection of candidate hub genes using machine learning algorithms
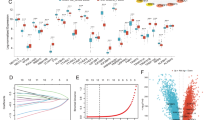
We trained 143 machine learning models using 12 machine learning algorithms to identify MitoDEGs among 14 candidates (Fig. 5A). Evaluated on GSE57148 training and three external validation cohorts (GSE57148, GSE76925, and GSE151052), the glmBoost and RF algorithm achieved optimal performance (AUC = 0.72, accuracy = 0.662, recall = 0.667, precision = 0.738, and F1 score = 0.696) (Supplementary Figure S2), identifying five key genes: ERN1, FASTK, HIGD1B, NDUFA7, and NDUFB7. Compared to controls, COPD samples showed decreased expression in FASTK, HIGD1B, NDUFA7, and NDUFB7 and increased ERN1 (Fig. 5B). The diagnostic model built from these genes demonstrated strong accuracy: AUC = 0.881 (95% CI: 0.832–0.930) in training (Fig. 5C), and 0.613–0.881 in validation (Fig. 5D–F). Individual gene AUCs ranged from 0.774 (HIGD1B) to 0.855 (ERN1) in training (Fig. 5G–K), confirming their diagnostic value for COPD.

Construction and testing of mitochondria-related genes score. (A) AUC values of 143 machine learning algorithms combinations in 4 sets; (B) Expression of the 5 candidate genes (ERN1, FASTK, HIGD1B, NDUFA7, and NDUFB7) in the training sets; (C–F) ROC curves for the diagnostic model in the training set GSE57148, and validation sets (GSE76925, GSE151052, GSE239897); (G–K) ROC curves for the 5 candidate genes (ERN1, FASTK, HIGD1B, NDUFA7 and NDUFB7).
Immune cell infiltration in COPD
CIBERSORT analysis of GSE57148 dataset revealed distinct immune cell infiltration patterns between COPD and control groups (Fig. 6A). Monocyte and neutrophil infiltration exhibited strong positive correlation (r = 0.379, P = 0.0008), while activated and resting mast cells showed significant negative correlation (r = − 0.668, P = 2.72e−07) (Fig. 6B). Differential analysis highlighted enrichment of activated dendritic cells (DCs), macrophages M0, activated mast cells, and neutrophils in COPD patients contrasted with dominance of macrophages M2, resting natural killer (NK) cells and T follicular helper (Tfh) cells in controls (P < 0.05) (Fig. 6C). Five key MitoDEGs displayed unique immune associations: Upregulated ERN1 correlated with activated DCs, eosinophils, activated mast cells, monocytes, neutrophils, and resting CD4 memory T cells (P < 0.01), while downregulated FASTK, HIGD1B, NDUFA7, and NDUFB7 preferentially linked to memory B cells, macrophages M2, resting NK cells, and Tfh cells (P < 0.01) (Fig. 6D). Notably, neutrophils infiltration was significantly higher in COPD (Fig. 6C), with ERN1 showing the strongest positive correlation (r = 0.52, P = 2.04e−14). Conversely, FASTK (r = − 0.55, P = 5.87e−16), HIGD1B (r = − 0.42, P = 3.43e−09), NDUFA7 (r = − 0.46, P = 5.19e−11), and NDUFB7 (r = − 0.51, P = 5.94e−14) exhibited marked negative correlations with neutrophil levels. These findings underscore immune dysregulation and mitochondrial-gene interactions in COPD pathogenesis, particularly highlighting neutrophil-driven inflammation modulated by key MitoDEGs like ERK1 and NDUFA7.

Analysis of immune cell infiltration in COPD and control groups. (A) Stacked bar chart of immune cell proportions; (B) Heatmap of the correlation between 22 immune cell; (C) Boxplots showing the difference in immune cells between the COPD and healthy control; (D) Correlation between 5 hub genes and 22 immune cells.
Prediction of candidate drugs
We identified 10 mitochondria-related compounds with potential therapeutic effects on COPD via cMAP analysis (Fig. 7A). Key candidates included mitomycin-c (a DNA alkylating drug), RITA (a thioredoxin reductase inhibitor), SN-38 (a topoisomerase inhibitor), celastrol (an anti-inflammatory agent), spectinomycin (a 30S ribosomal subunit inhibitor), SB-216763 (a glycogen synthase kinase inhibitor), PTB1 (an AMPK activator/tyrosine phosphatase inhibitor), androstenol (a GABA receptor modulator), PKC beta-inhibitor, and 2-aminopurine (a serine/threonine protein kinase inhibitor) (Fig. 7A). Among these, androstenol emerged as the most promising candidate due to its high score and association with GABA-mediated airway mucus regulation36. Pathways linked to glycogen synthase kinase and thioredoxin reductase were prioritized for their roles in COPD-related inflammation and oxidative stress37,38 (Fig. 7B). Figure 7C–L described the chemical structures of these 10 small molecule compounds.

Screening of potential small molecule compounds for mitochondria-related COPD genes by cMAP analysis. (A) The top 10 compounds with the highest negative enrichment scores based on cMAP analysis; (B) Pathway descriptions associated with the 10 compounds. (C–L) Chemical structures of the 10 compounds.
Validation of core gene expression in lung tissue of COPD mice
To validate bioinformatics findings, we established a COPD mouse model using CS/LPS induction. Lung pathological scores and mean linear intercept (MLI) were significantly elevated in COPD mice vs. controls (Fig. 8), confirming successful emphysema modeling. qPCR analysis of ERN1, FASTK, HIGD1B, NDUFA7, and NDUFB7 in lung tissues revealed: HIGD1B and NDUFB7 were significantly downregulated (P < 0.05), while FASTK, NDUFA7, and ERN1 showed non-significant expression changes (P > 0.05) (Fig. 9). These results partially contradicted bioinformatics predictions but highlighted HIGD1B and NDUFB7 as key players in COPD inflammation, underscored by their robust diagnostic relevance despite limited statistical significance for other genes.

Histological changes in the lungs of control (CTL) and COPD (CS + LPS) mouse. (A) Hematoxylin–eosin staining of lung tissues. Lung pathological scores (B) and MLI analysis (C) of mice in the control and COPD groups. n = 3 mice per group, data are expressed as mean ± SEM, Student’s t-test. Compared with the control group, * P < 0.05; ** P < 0.01.

Expression verification of 5 hub genes in lung tissues of COPD mouse. (A) ERN1 (B) FASTK (C) HIGD1B (D) NDUFA7 (E) NDUFB7. n = 5 mice per group, data are expressed as mean ± SEM, Student’s t-test. Compared with the control group, * P < 0.05; ** P < 0.01; ns: not significant.
Discussion
COPD is a common chronic inflammatory airway disease linked to mitochondrial dysfunction39, involves dysregulation of mitochondrial protein expression, structure, and function triggered by external stimuli like inflammation, infection, and CS40. This dysfunction perpetuates chronic airway inflammation and immune injury41. Using MitoCarta 3.0, we identified mitochondrial-related genes and explored their correlation with COPD pathogenesis and immune microenvironment via bioinformatics methods. Significant differences in mitochondrial gene scores were observed between COPD patients and healthy controls, and a diagnostic model based on these genes demonstrated high accuracy for COPD.
Machine learning identified 5 hub MitoDEGs (ERN1, FASTK, HIGD1B, NDUFA7, and NDUFB7) associated with COPD. Animal experiments confirmed downregulation of HIGD1B and NDUFB7 in COPD, aligning with bioinformatics predictions. The hypoxia-inducible ___domain (HIGD) gene family primarily consists of five genes, including HIGD-1A, -1B, -1C, -2A, and -2B, are crucial for mitochondrial integrity and respiratory chain complex IV42,43. Limited studies on HIGD1B suggest its involvement in tumorigenesis and pituitary adenomas progression44. Pang et al.45 demonstrated that HIGD1B maintains mitochondrial integrity under hypoxia, promoting cell survival via caspase-3/9 inhibition, while its knockdown induces mitochondrial fragmentation. NDUF (NADH-ubiquinone oxidoreductase), the first enzyme of the mitochondrial electron transport chain, plays a critical role in oxidative phosphorylation46. Its subunits NDUFA7 and NDUFB7 are linked to inflammatory diseases: elevated levels were observed in COVID-19, Acute lung injury (ALI) and psoriatic arthritis patients47,48, reflecting tissue damage. These findings highlight connections between HIGD and NDUF families and inflammation. Combined with our data, we propose that HIGD1B and NDUFB7 downregulation in COPD disrupts mitochondrial function, exacerbating airway inflammation and disease progression, though further validation is required.
Although ERN1, FASTK, and NDUFA7 showed non-significant expression differences between groups, their trends suggest biological relevance given COPD’s complexity and sample limitations49. Despite constrained validation sample size, we integrated multiple datasets to strengthen findings. Existing studies link these molecules to inflammation. The endoplasmic reticulum to nucleus signaling 1 (ERN1) gene encodes the transmembrane protein kinase inositol-requiring enzyme 1 (IRE1), a key mediator of endoplasmic reticulum (ER) stress that regulates neutrophils activation and contributes to inflammatory diseases. In ALI mice model, C5a receptor-mediated ER stress induces neutrophils granule release and lung injury via the IRE1α-TRAF2-NF-κB pathway50,51. Similarly, IRE1α overactivation in non-alcoholic fatty liver disease promotes liver inflammation by recruiting macrophages through extracellular vesicle release52. While in asthma models, it exacerbates neutrophilic airway inflammation via Th17 cell activation53. IRE1α inhibitors reduce apoptosis and fibrosis in alveolar epithelial cells54. The Fas-activated serine/threonine kinase (FASTK) gene, a serine/threonine protein kinase and key post-transcriptional regulator of mitochondrial gene expression55,56, is linked to liver and cardiac diseases. Genetic deletion of FASTK attenuated hepatic steatosis and inflammation in chronic alcoholic liver diseases57, while its downregulation in alcoholic cardiomyopathy correlates with ethanol-induced suppression via reactive oxygen species, destabilizing FASTK mRNA58. These studies suggest a potential role for FASTK in COPD-associated inflammation, though further large-scale studies are needed to clarify its mechanistic involvement.
Mitochondrial-related genes hold significant potential for COPD prevention and treatment. Our studies demonstrate that their expression levels can serve as biomarkers for early COPD risk prediction, while specific genes correlate with immune cell infiltration patterns, guiding immunotherapy strategies59. Therapeutically, targeting mitochondrial dysfunction has already made some progress in COPD: SUL-151, a novel compound with mitochondria protective properties, reduced neutrophil infiltration and lung inflammation in COPD mice60; Dysregulation of mitochondrial iron homeostasis is closely related to mitochondrial dysfunction; iron chelators significantly alleviated CS-induced mucociliary clearance impairment and pulmonary inflammation in COPD mice61; and taurine/3-methyladenine restored mitochondrial gene expression in emphysema models62. We have also identified 10 small molecule compounds with potential therapeutic effects in COPD. Notably, androstanol- a volatile steroid found in male sweat- emerges as a candidate compound63. Although direct COPD efficacy data are lacking, its possible biological activity as a volatile steroid and its influence on physiological responses warrant further investigation. Exogenous supplementation of androgens has been reported to alleviate pulmonary artery hypertension, increase serum insulin-like growth factor (IGF)-1 and IGF-binding protein-1, reverse the loss in diaphragm force-generating capacity, improve mitochondrial and muscle function, increase myosin expression, and attenuate pulmonary epithelial inflammation in COPD64,65. Future studies should explore the potential of androstenol in COPD, particularly in its ability to modulate inflammatory pathways, enhance muscle function, and interact with the endocrine system. Investigating its mechanism and therapeutic efficacy could provide novel insights into COPD management and expand the scope of steroid-based interventions.
This study uncovered mitochondria-related genes and COPD immune microenvironment through bioinformatics analysis. However, there are some limitations. Firstly, although rigorous bioinformatics analysis was conducted, the data were sourced from public databases with limited sample sizes, and the variations in data composition, geographical distribution, and collection methods can introduce bias. Secondly, due to the specific characteristics of the datasets used, the lack of relevant clinical data limited our ability to adjust or correct for common covariates such as smoking status, ethnicity, and comorbidities, which can lead to confounding bias. Additionally, while direct validation in human tissue would be ideal, limitations in sample access made us rely on computational methods and indirect validation using available COPD animal tissues. Finally, we identified five key genes, but their exact functional roles in the pathogenesis of COPD remain speculative. Further cellular and animal experiments are needed to explore the specific roles of these genes in COPD. We acknowledge these constraints, highlight the preliminary nature of our findings, and suggest that future studies should consider incorporating larger and more diverse datasets as well as additional in vivo and in vitro experiments to more effectively address these potential confounders and enhance the robustness of the research findings.
Conclusions
This study revealed distinct mitochondrial gene expression and immune infiltration patterns between COPD and healthy controls, uncovering the interplay between mitochondrial metabolism and immune responses. Machine learning identified five key MitoDEGs (ERN1, FASTK, HIGD1B, NDUFA7, and NDUFB7) and developed a classification diagnostic model for the early COPD diagnosis. ERN1 expression positively correlated with neutrophil infiltration, while FASTK, HIGD1B, NDUFA7 and NDUFB7 showed negative correlations. Animal validation confirmed differential expression of HIGD1B and NDUFB7 in COPD, underscoring their co-regulatory roles in mitochondrial metabolism and immune crosstalk.
Data availability
The transcriptome datasets analyzed during the current study are available in the public NCBI GEO database (https://www.ncbi.nlm.nih.gov/geo/). Accession numbers and link URL are GSE57148 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE57148), GSE76925 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE76925), GSE151052 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE151052) and GSE239897 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE239897). To ensure the reproducibility of our results, the complete ML code and all R code used in the research are available and provided as a separate document in the Supplementary Material.
Abbreviations
- ALI:
-
Acute lung injury
- AUC:
-
The area under the curve
- CIs:
-
Confidence intervals
- cMAP:
-
Connectivity map
- COPD:
-
Chronic obstructive pulmonary disease
- CS:
-
Cigarette smoke
- DCs:
-
Dendritic cells
- DEGs:
-
Differentially expressed genes
- GEO:
-
Gene expression omnibus
- GO:
-
Gene ontology
- GSEA:
-
Gene set enrichment analysis
- GSVA:
-
Gene set variation analysis
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- LPS:
-
Lipopolysaccharide
- MitoDEGs:
-
Mitochondria-related differentially expressed genes
- MLI:
-
Mean linear intercept
- MSigDB:
-
Molecular signatures database
- NK:
-
Natural killer
- ROC:
-
Receiver operating characteristic
- ROS:
-
Reactive oxygen species
- RT-qPCR:
-
Reverse transcription quantitative polymerase chain reaction
- SEM:
-
Standard error of the mean
- Tfh:
-
T Follicular helper cells
- WGCNA:
-
Weighted gene co-expression network analysis
References
Mei, F. et al. Chronic obstructive pulmonary disease (COPD) mortality trends worldwide: An update to 2019. Respirology 27(11), 941–950 (2022).
Whittaker, H., Rothnie, K. J. & Quint, J. K. Cause-specific mortality in COPD subpopulations: A cohort study of 339 647 people in England. Thorax 79(3), 202–208 (2024).
Christenson, S. A. et al. Chronic obstructive pulmonary disease. Lancet 399(10342), 2227–2242 (2022).
Kaur, M. et al. Particulate matter in COPD pathogenesis: An overview. Inflamm. Res. 71(7–8), 797–815 (2022).
Agustí, A. et al. Pathogenesis of chronic obstructive pulmonary disease: Understanding the contributions of gene-environment interactions across the lifespan. Lancet Respir. Med. 10(5), 512–524 (2022).
Bolger, G. B. Therapeutic targets and precision medicine in COPD: Inflammation, ion channels, both, or neither?. Int. J. Mol. Sci. 24(24), 17363 (2023).
Tiew, P. Y., Meldrum, O. W. & Chotirmall, S. H. Applying next-generation sequencing and multi-omics in chronic obstructive pulmonary disease. Int. J. Mol. Sci. 24(3), 2955 (2023).
Moll, M. & Silverman, E. K. Precision approaches to chronic obstructive pulmonary disease management. Annu. Rev. Med. 75, 247–262 (2024).
Harrington, J. S. et al. Mitochondria in health, disease, and aging. Physiol. Rev. 103(4), 2349–2422 (2023).
Wang, Q., Unwalla, H. & Rahman, I. Dysregulation of mitochondrial complexes and dynamics by chronic cigarette smoke exposure Utilizing MitoQC reporter mice. Mitochondrion 63, 43–50 (2022).
Tulen, C. B. M. et al. Dysregulated mitochondrial metabolism upon cigarette smoke exposure in various human bronchial epithelial cell models. Dis. Model Mech. 15(3), dmm049247 (2022).
Tulen, C. B. M. et al. Acrolein inhalation acutely affects the regulation of mitochondrial metabolism in rat lung. Toxicology 469, 153129 (2022).
Zong, Y. et al. Mitochondrial dysfunction: Mechanisms and advances in therapy. Signal Transduct. Target Ther. 9(1), 124 (2024).
Sang, W. et al. Antioxidant mitoquinone ameliorates EtOH-LPS induced lung injury by inhibiting mitophagy and NLRP3 inflammasome activation. Front. Immunol. 13, 973108 (2022).
Li, C. et al. Role of mitochondrial fusion proteins MFN2 and OPA1 on lung cellular senescence in chronic obstructive pulmonary disease. Respir. Res. 24(1), 319 (2023).
Haji, G. et al. Mitochondrial dysfunction in airways and quadriceps muscle of patients with chronic obstructive pulmonary disease. Respir. Res. 21(1), 262 (2020).
Liu, Y. B. et al. The role of mitochondrial quality control in chronic obstructive pulmonary disease. Lab. Invest. 104(2), 100307 (2024).
Fairley, L. H. et al. Mitochondria-targeted antioxidants as a therapeutic strategy for chronic obstructive pulmonary disease. Antioxidants (Basel) 12(4), 973 (2023).
Davis, S. & Meltzer, P. S. GEOquery: A bridge between the gene expression omnibus (GEO) and BioConductor. Bioinformatics 23(14), 1846–1847 (2007).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43(7), e47 (2015).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7 (2013).
Liberzon, A. et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 1(6), 417–425 (2015).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb.) 2(3), 100141 (2021).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28(1), 27–30 (2000).
Kanehisa, M. et al. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 49(D1), D545–D551 (2021).
Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Chang, J. et al. Constructing a novel mitochondrial-related gene signature for evaluating the tumor immune microenvironment and predicting survival in stomach adenocarcinoma. J. Transl. Med. 21(1), 191 (2023).
Rath, S. et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 49(D1), D1541–D1547 (2021).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102(43), 15545–15550 (2005).
Maimaiti, A. et al. DNA methylation regulator-mediated modification patterns and risk of intracranial aneurysm: A multi-omics and epigenome-wide association study integrating machine learning, Mendelian randomization, eQTL and mQTL data. J. Transl. Med. 21(1), 660 (2023).
Robin, X. et al. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 12, 77 (2011).
Chen, B. et al. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol. Biol. 1711, 243–259 (2018).
Subramanian, A. et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 171(6), 1437–52.e17 (2017).
Jiang, C. et al. Screening of lipid metabolism-related genes as diagnostic indicators in chronic obstructive pulmonary disease. Int. J. Chron. Obstruct. Pulmon. Dis. 18, 2739–2754 (2023).
Geng, Y. et al. Effect of PD-1 inhibitor combined with X-Ray irradiation on the inflammatory microenvironment and lung tissue injury in mice. J. Inflamm. Res. 15, 545–556 (2022).
Patel, B. S. et al. Class C GPCRs in the airway. Curr. Opin. Pharmacol. 51, 19–28 (2020).
Qin, Y. et al. Effective-component compatibility of Bufei Yishen formula alleviates chronic obstructive pulmonary disease inflammation by regulating GSK3β-mediated NLRP3 inflammasome activation. Biomed. Pharmacother. 168, 115614 (2023).
Huang, J. et al. A thioredoxin reductase and/or thioredoxin system-based mechanism for antioxidant effects of ambroxol. Biochimie 97, 92–103 (2014).
Plichta, J., Kuna, P. & Panek, M. Biologic drugs in the treatment of chronic inflammatory pulmonary diseases: Recent developments and future perspectives. Front. Immunol. 14, 1207641 (2023).
Xu, M. et al. Mitochondrial quality control: A pathophysiological mechanism and potential therapeutic target for chronic obstructive pulmonary disease. Front Pharmacol. 15, 1474310 (2024).
Pokharel, M. D. et al. Mitochondrial network dynamics in pulmonary disease: Bridging the gap between inflammation, oxidative stress, and bioenergetics. Redox Biol. 70, 103049 (2024).
Timón-Gómez, A. et al. HIGD-driven regulation of cytochrome c oxidase biogenesis and function. Cells 9(12), 2620 (2020).
Timón-Gómez, A. et al. Distinct roles of mitochondrial HIGD1A and HIGD2A in respiratory complex and supercomplex biogenesis. Cell Rep. 31(5), 107607 (2020).
Zhou, Y. et al. Identification of key genes with differential correlations in lung adenocarcinoma. Front. Cell Dev. Biol. 9, 675438 (2021).
Pang, Y. et al. HIGD-1B inhibits hypoxia-induced mitochondrial fragmentation by regulating OPA1 cleavage in cardiomyocytes. Mol. Med. Rep. 24(2), 549 (2021).
Okoye, C. N., Koren, S. A. & Wojtovich, A. P. Mitochondrial complex I ROS production and redox signaling in hypoxia. Redox Biol. 67, 102926 (2023).
Chen, Z. Z. et al. Mitochondria and cytochrome components released into the plasma of severe COVID-19 and ICU acute respiratory distress syndrome patients. Clin. Proteomics 20(1), 17 (2023).
Zhu, J. et al. Identification of proteins associated with development of psoriatic arthritis in peripheral blood mononuclear cells: A quantitative iTRAQ-based proteomics study. J. Transl. Med. 19(1), 331 (2021).
Shrine, N. et al. Multi-ancestry genome-wide association analyses improve resolution of genes and pathways influencing lung function and chronic obstructive pulmonary disease risk. Nat. Genet. 55(3), 410–422 (2023).
Hu, R. et al. Endoplasmic reticulum stress of neutrophils is required for ischemia/reperfusion-induced acute lung injury. J. Immunol. 195(10), 4802–4809 (2015).
Ye, L. et al. Endoplasmic reticulum stress is involved in ventilator-induced lung injury in mice via the IRE1α-TRAF2-NF-κB pathway. Int. Immunopharmacol. 78, 106069 (2020).
Dasgupta, D. et al. IRE1A stimulates hepatocyte-derived extracellular vesicles that promote inflammation in mice with steatohepatitis. Gastroenterology 159(4), 1487–503.e17 (2020).
Wu, D. et al. Unconventional activation of IRE1 enhances TH17 responses and promotes airway neutrophilia. Am. J. Respir. Cell Mol. Biol. 71(2), 169–181 (2024).
Thamsen, M. et al. Small molecule inhibition of IRE1α kinase/RNase has anti-fibrotic effects in the lung. PLoS ONE 14(1), e0209824 (2019).
Yang, X. et al. The Vsr-like protein FASTKD4 regulates the stability and polyadenylation of the MT-ND3 mRNA. Nucleic Acids Res. 53, gkae1261 (2024).
Ohkubo, A. et al. The FASTK family proteins fine-tune mitochondrial RNA processing. PLoS Genet. 17(11), e1009873 (2021).
Zhang, F. et al. Genetic ablation of fas-activated serine/threonine kinase ameliorates alcoholic liver disease through modulating HuR-SIRT1 mRNA complex stability. Free Radic. Biol. Med. 166, 201–211 (2021).
Zhang, F. et al. Accelerated FASTK mRNA degradation induced by oxidative stress is responsible for the destroyed myocardial mitochondrial gene expression and respiratory function in alcoholic cardiomyopathy. Redox Biol. 38, 101778 (2021).
Chellappan, D. K. et al. Targeting the mitochondria in chronic respiratory diseases. Mitochondrion 67, 15–37 (2022).
Wang, L. et al. SUL-151 decreases airway neutrophilia as a prophylactic and therapeutic treatment in mice after cigarette smoke exposure. Int. J. Mol. Sci. 22(9), 4991 (2021).
Cloonan, S. M. et al. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat. Med. 22(2), 163–174 (2016).
Li, X. et al. Taurine ameliorates particulate matter-induced emphysema by switching on mitochondrial NADH dehydrogenase genes. Proc. Natl. Acad. Sci. USA 114(45), E9655–E9664 (2017).
Cowley, J. J. & Brooksbank, B. W. Human exposure to putative pheromones and changes in aspects of social behaviour. J. Steroid. Biochem. Mol. Biol. 39(4b), 647–659 (1991).
Bonnet, S. et al. Dehydroepiandrosterone (DHEA) prevents and reverses chronic hypoxic pulmonary hypertension. Proc. Natl. Acad. Sci. USA 100(16), 9488–9493 (2003).
Wang, X. et al. Testosterone attenuates pulmonary epithelial inflammation in male rats of COPD model through preventing NRF1-derived NF-κB signaling. J. Mol. Cell Biol. 13(2), 128–140 (2021).
Funding
This study was supported by the National Natural Science Foundation of China (82370055) and the Fundamental Research Funds for the Central Universities of Central South University (2024ZZTS0171).
Author information
Authors and Affiliations
Contributions
Chen, Jiang was responsible for the conception of the study and drafting of the article. JN, Lin and MJ, Peng played a significant role in data collection, analysis, interpretation, and visualization. Bin, Xie conducted a thorough review of the article. Qiong, Chen and ZY, Dai provided financial support. The final manuscript received unanimous approval from all authors. All authors actively contributed to the articles content and endorsed its submission in its current form.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics declaration
This study was reviewed and approved by the Ethics Committee for Laboratory Animal Welfare at Central South University, with the approval number CSU-2023-0191. The human participant data utilized in our study were downloaded from public database, and this research did not require ethical review or approval according to local legislation and institutional requirements.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Peng, M., Jiang, C., Dai, Z. et al. Identification and verification of mitochondria-related genes biomarkers associated with immune infiltration for COPD using WGCNA and machine learning algorithms. Sci Rep 15, 14347 (2025). https://doi.org/10.1038/s41598-025-99002-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-99002-y
