
Abstract
Acute coronary syndrome (ACS) includes conditions such as unstable angina (UA), non-ST elevation myocardial infarction (NSTEMI), and ST elevation myocardial infarction (STEMI). High-sensitivity cardiac troponin I (hs-cTnI) assays have been recommended as primary biomarkers for NSTEMI diagnosis due to their high sensitivity and specificity. However, there is no consensus on the optimal diagnostic strategy for early NSTEMI detection. This study aims to evaluate the diagnostic performance of four hs-cTnI-based strategies for suspected NSTEMI: Limit of Detection (LoB), Single Cut-off, hs-cTnI 0/1 h Algorithm, and hs-cTnI 0/2 h Algorithm, in a Chinese cohort and compare the performance of assays from different manufacturers. Performance verification was conducted, including LoB, LoD, LoQ, and precision analysis. Clinical samples from 267 ACS patients were analyzed using Hybiome hs-cTnI assays. Diagnostic accuracy was assessed based on sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV), and F1-score. The LoB strategy demonstrated 100% sensitivity but low PPV (14.0%). Both the hs-cTnI 0/1 h Algorithm and hs-cTnI 0/2 h Algorithm showed high diagnostic performance, with the 0/2 h algorithm providing the best overall. 89.0% accuracy, 93.3% sensitivity, and 73.68% F1-score. The hs-cTnI 0/2 h Algorithm offers the most reliable diagnostic performance for early NSTEMI diagnosis.
Similar content being viewed by others
Background
Acute coronary syndrome(ACS) refers to unstable angina (UA), non-ST elevated myocardial infarction (NSTEMI), and ST elevated myocardial infarction (STEMI)1. Each of these conditions presents unique challenges in diagnosis and management, requiring tailored therapeutic strategies to optimize patient outcomes. STEMI is characterized by the presence of ST elevation on an ECG, UA and NSTEMI are characterized by the absence of ST elevation on an ECG. The management of NSTEMI has evolved significantly over the years. It is important and necessary to identify NSTEMI among ACS patients2. The use of high-sensitivity cardiac troponins (hs-cTn) has also enhanced the diagnostic accuracy for these conditions, allowing for more precise risk stratification and management strategies.
The European Society of Cardiology (ESC) strongly recommends the use of high-sensitivity cardiac troponin I (hs-cTnI) as the primary biomarker for the diagnosis of NSTEMI3. The absolute concentration and changes of hs-cTnI serve as highly sensitive and specific indicators for assessing myocardial injury, demonstrating significant clinical value. However, during continuous monitoring of hs-cTnI, its concentration may be influenced by factors such as assay sensitivity and coefficient of variation (CV) in analytical performance. Meanwhile, it also may be influenced by the timing of blood sampling and the progression of the disease, which can complicate its interpretation3,4.
The WHO-IFCC defines a high-sensitivity cardiac troponin I (hs-cTnI) assay as having a detection rate of more than 50% in healthy individuals and an imprecision of less than 10% at the 99 th percentile upper reference limit5,6. In addition to sensitivity and accuracy, other important assay parameters include specificity, lot-to-lot variation, and consistency comparison, all of which are required to provide consistent results in the diagnosis of NSTEMI7.
This study aims to provide a thorough analytical evaluation of a new hs-cTnI assay from Hybiome(Suzhou, China) and assess its diagnostic performance for NSTEMI in comparison with the well-established hs-cTnI Beckman Coulter(California, US) assay.
Given the complexity of absolute values, influencing factors, and the changes in hs-cTnI, the precise and rapid determination of follow-up timing for patients, alongside the formulation of early diagnostic and therapeutic strategies, remains a clinical challenge8,9. Currently, the optimal early diagnostic strategy for NSTEMI has not been established. Four exclusion strategies based on hs-cTnI recommended by many researches10,11,12,13,14,15,16: the first strategy is rule-out NSTEMI in admission by using LoD(hs-cTnI 0 h < 2ng/L); the second strategy is single cut-off value rule-out NSTEMI (hs-cTnI 0 h < the 99 th URL); the third strategy is 1 h algorithm, which indicates hs-cTnI < the 99 th URL, and 1 h absolute change value < 2ng/L; the forth strategy is 2 h algorithm, which indicates hs-cTnI < the 99 th URL, and 2 h absolute change value < 5ng/L. These four early diagnosis exclusion strategies have been evaluated in the US population but are less studied in the Chinese population11.
In studies evaluating the effectiveness of existing diagnostic strategies in clinical practice, the 0/2-hour strategy has been shown to improve the early detection of NSTEMI and single cut-off value strategy has been shown to exhibit lower sensitivity17. These cohort studies have demonstrated diagnostic strategies with high sensitivity and specificity17,18,19. However, there remains a need to explore the heterogeneity of certain strategies across different populations, particularly in terms of variations in sensitivity and specificity for different diagnostic approaches.
So, this study aims to investigate the accuracy and applicability of these four strategies for ACS patients rule-out NSTEMI in early screening for emergency department triage in China. And whether diagnostic performance is consistent when using products from different reagent manufacturers.
Materials and methods
Sample processing and hs-cTnI measurement
All samples used in this study were de-identified patient specimens remaining after routine clinical testing. The Peripheral blood samples collected 5 ml of procoagulant and centrifuged at 2000 rpm for 8 min to separate the upper serum.This study was performed in compliance with the Declaration of Helsinki and was approved by the Ethics Committee of Anzhen Hospital (protocol codes KS2023034 and KS2023052).All serum samples were frozen at − 80 °C and analyzed using the AE-180 (Hybiome Suzhou Diagnostics Co., Ltd.) and UniCel DXI800 Access system(Beckman, CA, USA), respectively.
Clinical performance validation methods
The Clinical and Laboratory Standards Institute (CLSI) EP17-A2 protocol was universally used to evaluate the detection capability for clinical laboratory measurement procedures20. So, it was followed to determine the Limit of Blank (LoB), Limit of Detection (LoD), and Limit of Quantitation (LoQ). LoB Verification: Two blank samples were tested using a single reagent batch. Each sample was measured five times per day over three consecutive days, totaling 30 measurements. The mean value was calculated to determine the LoB. LoD Verification: Two samples with test values near the estimated LoD (2.5 ng/L) were analyzed using a single reagent batch. Each sample was tested five times per day over three consecutive days for a total of 30 measurements. The mean value was calculated to determine the LoD.
LoQ verification: Ten serum samples with concentrations of 2.5 ng/L, 5 ng/L, 7.5 ng/L, 10 ng/L, 12.5 ng/L, 15 ng/L, and 17.5 ng/L, representing multiples of the limit of detection (LoD = 2.5 ng/L), were analyzed. A single reagent batch was used, and each sample was tested five times per day over three consecutive days. The mean, standard deviation, and precision (expressed as the coefficient of variation, CV) were calculated for each sample. A precision curve was generated based on the raw data, and the test concentration corresponding to a CV of 20% was determined from the curve.
Three concentrations of serum samples were analyzed in precision verification: level 1 (serum with concentrations between the limit of detection (LoD) and the 99 th percentile upper reference limit (99 th URL)), level 2 (serum near the 99 th URL), and level 3 (serum exceeding five times the 99 th URL). Additionally, Bio-Rad quality control materials (low, medium, and high concentration samples) were included. Both repeatability and intermediate precision were assessed concurrently. One analytical batch was processed daily, with samples at all three levels included in each batch. Each sample was measured in triplicate over five consecutive days.
According to the CLSI EP6-A protocol21, the analytical measurement range (AMR) verification was performed by selecting patient samples at two concentration levels: one within 100–150% of the lower limit of the AMR and the other within 80–100% of the upper limit of the AMR. Select a high-value sample close to the expected upper limit and a low-value sample at the expected lower limit (or use S1 and S5 working calibration solutions), dilute the samples at the dilution ratios of 100%:0, 75%:25%, 50%:50%, 25%:75%, and 0%:100% respectively, and compare with the theoretical value, calculate y = ax + b, and verify the linear range. Repeat the measurement twice for each dilution and calculate the mean.
Verification of the clinical reportable range (CRR): Take a high-value sample with a known concentration of 80–120% of the upper limit of AMR, and dilute it at the highest dilution multiple (5 times) with reference to the dilution solution recommended by the manufacturer, so that the expected value of the diluted sample covers the AMR of the project as much as possible and the minimum concentration after dilution recommended by the manufacturer. Repeat the test twice for the original sample and the diluted sample, and calculate their means respectively. Compare the mean of the test value of each dilution sample with its expected value and calculate the R value (R = mean of the test value/expected value).
Study cohort in diagnostic assessment
This study included 267 patients presenting with chest pain who required confirmation of ACS classification. These patients were treated at the Emergency Center of Beijing Anzhen Hospital, affiliated with Capital Medical University, between March 2023 and February 202422.Informed consent was obtained from all subjects involved in the study. And this clinical cohort study was conducted in accordance with the Declaration of Helsinki and received approval from the Ethics Committee of Anzhen Hospital (protocol numbers KS2023034 and KS2023052).
Inclusion criteria were Chinese patients aged 18 years or older who presented with chest pain, angina, or other symptoms of myocardial ischemia (such as shortness of breath, nausea, vomiting, sweating, or fatigue) within 12 h, and who met the diagnostic criteria for NSTEMI. Exclusion criteria included patients diagnosed with STEMI (characterized by significant ST-segment elevation or T-wave inversion on electrocardiogram), those with renal failure requiring dialysis, patients with active autoimmune diseases, pregnant women, cancer patients, and individuals with severe musculoskeletal injuries.
Following detailed clinical assessments, the patients were further classified into the NSTEMI and the UA group. Samples were collected at two time points: at the time of admission and 1–2 h after admission(167 people at 1 h and 91 people at 2 h, respectively).To ensure diagnostic accuracy and comprehensiveness, cardiology experts established the final diagnosis based on a comprehensive evaluation of the patients’ medical history, physical signs, laboratory test results, and imaging findings.
According to the NSTEMI rule-in and rule-out rules, the absolute value of the 1-hour change difference is less than 2ng/L. The absolute value of the 2-hour change difference is less than 5ng/L. At the same time, it is necessary to combine the upper limit of the normal reference range. The instructions of the Beckman manufacturer illustrated that the upper limit of the healthy range for males (99 th URL) is 19.8ng/L and that for females is 11.6ng/L. The instructions of the Hybiome manufacturer defined that the upper limit of the healthy range for males is 38.7ng/L and that for females is 15.3ng/L. Early strategy comparisons will use reference ranges from different manufacturers as well as different cut-off values based on the timing of blood sampling.
Statistical analysis
Data analysis and statistical procedures analysis were performed using Python 3.10 and the matplotlib package for visualization. Use Passing-Bablok regression to assess the correlation relationship between the two manufacturers’ measurements; use Bland-Altman analysis to evaluate the differences in the measurement results23,24.
To compare different strategies for suspected early-stage NSTEMI, diagnostic performance was assessed based on accuracy, sensitivity, specificity, positive predictive value(PPV), negative predictive value(NPV), and F1 score. First, the numbers of true negatives, true positives, false negatives, and false positives were accurately counted. Sensitivity was calculated as the ratio of true positives (TP) to the sum of true positives and false negatives (FN), while specificity was the ratio of true negatives (TN) to the sum of true negatives and false positives (FP). PPV, or precision, was calculated as the ratio of TP to the sum of TP and FP, and NPV was the ratio of TN to the sum of TN and FN. These metrics provide valuable insights into the performance of diagnostic tests in identifying patients with or without NSTEMI.
Results
Distribution of clinical characteristics of patients
Through the analysis of blank samples, the LoB and LoD for Hybiome hs-cTnI assay in the clinical laboratory were determined to be 1.2 ng/L and 1.8 ng/L, respectively. A nonlinear regression curve (Fig. 1) was generated using a precision curve model based on cTnI concentrations and the corresponding coefficients of variation observed across a dilution series of 19 pools. The power function derived for LoQ determination was CV% = 30.9542 × Conc^−0.5379. The LoQ in serum was estimated to be 8.1 ng/L and 2.4 ng/L, corresponding to 10% and 20% coefficients of variation, respectively.

Determination of LoQ of hybiome hs-cTnI assay using EP17-A2 protocol.
The power function derived for LoQ determination was CV% = 30.9542 × Conc^−0.5379. The LoQ in serum was estimated to be 8.1 ng/L and 2.4 ng/L, corresponding to 10% and 20% coefficients of variation, respectively.
The precision of the Hybiome hs-cTnI assay was determined through continuous testing over five days using serum samples at three concentration levels. Each sample was measured in triplicate within a single batch per day. Level 1: The overall mean concentration was 10.693 ng/L, with an intra-batch variance of 0.546, an intra-batch standard deviation of 0.739, and an intra-batch coefficient of variation (CV) of 6.91%.Level 2: The overall mean concentration was 20.405 ng/L, with an intra-batch variance of 1.402, an intra-batch standard deviation of 1.184, and an intra-batch CV of 5.80%.Level 3: The overall mean concentration was 84.226 ng/L, with an intra-batch variance of 4.091, an intra-batch standard deviation of 2.023, and an intra-batch CV of 2.40%.The intra-batch CV ranged from 2.40 to 6.91%, while the inter-batch CV ranged from 3.59 to 7.16%. The summarized data is presented in Table 1.
The analytical measurement range (AMR) refers to the range of analyte values that can be directly measured using the assay without any dilution, concentration, or additional pre-processing beyond the standard testing procedure. Two patient sample concentrations were selected and positioned around the lower and upper limits of the AMR (7.96 ng/L and 33,806.22 ng/L, respectively). A series of sample dilution ratios were used to calculate the linear regression equation y = ax + by = ax + by = ax + b and verify the linearity of the method. The results showed a slope (b) of 1.008 and a correlation coefficient (R) of 0.999, confirming excellent linearity within the validated range.
The purpose of Clinical Reportable Range (CRR) verification is to assess the linear relationship across various dilution concentrations and determine the maximum dilution factor, ensuring the accuracy of reported high-concentration samples. This verification is required only for assays where the CRR exceeds the AMR. The results indicated that patient sample measurements remained within the acceptable range even at a maximum dilution factor of 16, surpassing the manufacturer’s declared maximum dilution factor of 5 (corresponding to 2.5–200,000 ng/L). These findings confirm that the assay meets the requirements for clinical dynamic monitoring.
Correlation analysis of different hs-cTnI determination assays
A Passing-Bablok regression was conducted to assess the agreement between the Hybiome hs-cTnI assay and the Beckman hs-cTnI assay(Fig. 2). The regression analysis revealed a strong linear relationship.0.926 (95% CI: 0.861 ~ 0.967) The regression slope is 1.275(95% CI: 1.132 ~ 1.421), intercept is 1.4(95% CI: −0.420 ~ 2.811), and the spearman correlation coefficient R is 0.981(95% CI: 0.890 ~ 0.995) was observed. The 95% confidence interval area for the regression is shown in the plot, confirming the robustness of the correlation.
In the Bland-Altman plot, the mean difference between the two assays was 35.6%, with the limits of 1.96 standard derivation(SD) ranging from − 48.3% to + 119.4%. These findings suggest a slight bias between the methods, but the differences remain within acceptable clinical limits for most samples. However, the observed variability, while within an acceptable clinical range, may still have implications in certain clinical scenarios, such as differentiating between borderline NSTEMI and non-NSTEMI cases. The plot illustrates the dispersion of differences relative to the mean of both methods, and the limits of 1.96SD(shown as dashed lines) reflect the variability between the two assays across the tested concentration range. These results demonstrate that the Hybiome hs-cTnI assay exhibits a strong correlation with the Beckman hs-cTnI assay, with acceptable precision and minimal bias within the clinical range of concentrations tested.

Comparison of hybiome hs-cTnI and beckman hs-cTnI assays: Passing-bablok regression and bland-altman analysis. The passing-bablok regression analysis shows a strong linear relationship between the hybiome hs-cTnI and beckman hs-cTnI assays (y = 1.275x + 1.4, r = 0.981), with a mean difference of 35.6%(26.5–44.6%) and limits of 1.96SD ranging from − 48.3% to + 119.4%, confirming the accuracy of the regression model with a 95% CI.
2.3 Four strategies of hs-cTnI in the early diagnosis of suspected NSTEMI cohorts.
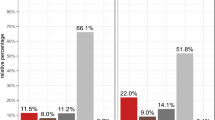
The diagnostic performance of various algorithms for suspected NSTEMI was evaluated using four methods: Limit of Detection (LoB), Single Cut-off, hs-cTnI 0/1 h Algorithm, and hs-cTnI 0/2 h Algorithm. The LoB strategy for rule-out NSTEMI has a relatively high sensitivity but was limited by a low Positive Predictive Value(PPV). For NSTEMI cases (N = 267), the LoB model showed an accuracy of 17.2%, with a sensitivity of 100% and a negative predictive value (NPV) of 100%. However, its positive predictive value (PPV) was low at 14.0%, indicating a high number of false positives. The F1-score was 24.56%.
The Single Cut-off (Gender-specific cut-off values)method provided improved accuracy and a better balance between sensitivity and specificity18. For NSTEMI (N = 267), the accuracy improved to 85.4%, with a sensitivity of 91.7%, specificity of 84.4%, and an NPV of 98.48%. The PPV was 47.8%, and the F1-score was 62.85%, reflecting better balance between sensitivity and specificity compared to the LoB method.
The hs-cTnI 0/1 h Algorithm and hs-cTnI 0/2 h Algorithm demonstrated high accuracy and sensitivity, with the 0/2 h algorithm outperforming others in terms of overall accuracy and F1-score. When hs-cTnI 0/1 h Algorithm was applied in suspected NSTEMI cohort(N = 167), this method showed the accuracy of 88.6%, with the sensitivity of 90.9% and the specificity of 88.3%. The PPV was 54.1%, and the NPV was 98.5%. The F1-score was 68.54%, indicating strong performance in distinguishing between NSTEMI and non-NSTEMI cases.
Hs-cTnI 0/2 h Algorithm provides the most reliable performance for diagnosing NSTEMI, particularly in terms of sensitivity and specificity. For NSTEMI (N = 91), the accuracy was 89.0%, with a sensitivity of 93.3% and specificity of 88.2%. The PPV was 60.9%, and the NPV was 98.5%, with an F1-score of 73.68%, reflecting the best overall performance among the methods evaluated.
The diagnostic performance of these four strategies is further illustrated in Fig. 3, which compares four diagnostic strategy algorithm in terms of accuracy, sensitivity, specificity, and F1-score. Figure 3 provides a visual representation of the comparative strengths and limitations of each diagnostic approach, confirming the reliability of the hs-cTnI 0/2 h Algorithm in clinical applications for early NSTEMI detection.

Diagnostic performance of various algorithms for suspected NSTEMI: comparison of limit of detection (LoB), single cut-off, hs-cTnI 0/1 h algorithm, and hs-cTnI 0/2 h algorithm. It summarizes accuracy, sensitivity, specificity, and F1-score for each method across different NSTEMI patient cohorts.
Discussion
In this study, we evaluated the diagnostic performance of four strategies for early diagnosis of suspected NSTEMI using the Hybiome hs-cTnI assay. These strategies included Limit of Detection (LoB), Single Cut-off, hs-cTnI 0/1 h Algorithm, and hs-cTnI 0/2 h Algorithm. Additionally, we performed rigorous analytical performance validation to ensure the accuracy and reliability of the hs-cTnI assay, which played a crucial role in interpreting the diagnostic performance.
The diagnostic performance of the four strategies was assessed using accuracy, sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV). The LoB strategy demonstrated 100% sensitivity for ruling out NSTEMI but had a low PPV of 14.0%, indicating a higher likelihood of false positives. This is because LoB level is a very low level of cTnI detection. The possibility of myocardial injury cannot be determined simply by detecting the presence of free troponin I in serum. While its high sensitivity ensures that true NSTEMI cases are not missed, it also means that clinicians must consider other factors, such as clinical symptoms and additional diagnostic tests, to avoid overdiagnosis and optimize patient management. False positives could lead to unnecessary testing and treatments, causing patient distress and increased healthcare costs.
False-positive and false-negative results in hs-cTnI detection can arise from several factors. False positives may occur in conditions like renal failure, chronic inflammation, or the presence of macrotroponins, which can elevate hs-cTnI levels without actual myocardial injury. Cross-reactivity with proteins such as skeletal muscle troponin or other cardiac markers can also contribute to elevated hs-cTnI levels in non-cardiac conditions. Conversely, false negatives may happen when troponin levels do not rise sufficiently above the detection threshold due to sample timing or early-stage myocardial injury, potentially leading to missed diagnoses and delayed treatment. Clinicians should interpret hs-cTnI results alongside other clinical findings and consider the timing of sample collection to minimize the risk of false negatives.
The Single Cut-off strategy improved the accuracy to 85.4%, with a better balance of sensitivity (91.7%) and specificity (84.4%) compared to LoB. This method demonstrated an NPV of 98.48% and PPV of 47.8%, which is clinically useful for ruling out NSTEMI while minimizing false positives.
Both the hs-cTnI 0/1 h Algorithm and hs-cTnI 0/2 h Algorithm provided high diagnostic accuracy. The hs-cTnI 0/1 h Algorithm achieved an accuracy of 88.6%, with sensitivity (90.9%) and specificity (88.3%) reflecting strong diagnostic performance. The F1-score of 68.54% demonstrated the ability of this algorithm to distinguish NSTEMI from non-NSTEMI cases effectively. However, the hs-cTnI 0/2 h Algorithm outperformed the others in terms of overall accuracy (89.0%), sensitivity (93.3%), and F1-score (73.68%). This algorithm’s ability to detect NSTEMI with high specificity and sensitivity makes it particularly useful for clinical decision-making, especially in emergency settings where quick, reliable results are necessary.
The LoB and LoD for the Hybiome hs-cTnI assay were determined to be 1.2 ng/L and 1.8 ng/L, respectively, based on the EP17-A2 protocol. These values confirm the assay’s ability to detect low levels of cardiac injury, which is essential for early NSTEMI detection. The LoQ determined using the precision curve model was calculated to be 8.1 ng/L at a 10% coefficient of variation. These findings indicate that the assay maintains acceptable precision across a wide range of concentrations, reinforcing its reliability for routine clinical use.
The precision verification of the hs-cTnI assay showed excellent performance across different concentration levels. These values demonstrate the assay’s robustness and precision, with acceptable variation even at the extremes of the AMR. Furthermore, the inter-batch CV ranged from 3.59 to 7.16%, showing consistent results across different batches. These findings demonstrate that the assay provides accurate and consistent measurements across the expected clinical range, ensuring reliable results in routine testing.
The performance validation results of the Hybiome hs-cTnI assay support its clinical applicability in the early detection of NSTEMI. With strong linearity, high sensitivity, and minimal bias, the assay meets the analytical requirements for clinical use, and its performance is consistent with the established standards of Beckman hs-cTnI.
The assay’s LoB, LoD, and LoQ validation data provide a solid foundation for its use in real-world clinical scenarios, ensuring accurate and reliable results. These findings highlight the assay’s potential in clinical applications, improving the accuracy of early NSTEMI diagnosis and enhancing patient care.
In comparison to previous studies, our findings also support the use of hs-cTnI-based algorithms for early NSTEMI diagnosis25,26,27. However, this study highlights the need for further validation of these algorithms in diverse patient populations, particularly in Chinese cohorts, where the applicability of these strategies has been less extensively studied. Moreover, while the 0/2 h algorithm showed superior performance, its practical implementation in real-world clinical settings needs further exploration, particularly regarding the optimal cut-off values for different populations and the potential need for adjustments based on local clinical practices28.
Compared to the multi-center Chinese cohort validating the ESC 0/1 h algorithm26, our optimized 0/2 h protocol achieved comparable rule-out safety (NPV 99.1% vs. 99.7%) with fewer required blood draws, while maintaining high clinical safety (30-day MACE 0.6% vs. 0.4%). However, our specificity (89.3%) was slightly lower than their optimized 0/1 h approach (92.1%), likely due to differences in cohort risk profiles and sampling protocols.
While this study demonstrates the effectiveness of the Hybiome hs-cTnI assay, some limitations must be considered. First, the study cohort was primarily from a single center, our findings may be influenced by selection bias (e.g., preferential referral of higher-risk patients to our center) and institutional protocols (e.g., uniform blood sampling intervals, standardized ESC guideline adherence) which may limit the generalizability of the results.
Second, the limited sample size and imbalanced cohort composition (e.g., a higher proportion of NSTEMI vs. non-NSTEMI cases) may influence the generalizability and validity of the findings. Future studies with larger, multi-center cohorts are necessary to confirm these findings.
Additionally, while the assay showed strong linearity and high precision in the validation process, real-world clinical conditions, including patient heterogeneity and comorbidities, might impact its performance. Further studies should explore these variables to assess the assay’s robustness across diverse clinical settings. Finally, The time window of symptom-to-blood-draw interval was not an exclusion criterion, which may influence the sensitivity of early hs-cTnI measurements.
Conclusion
In conclusion, the Hybiome hs-cTnI assay demonstrated high precision, strong analytical performance, and excellent diagnostic accuracy for the early detection of NSTEMI (sensitivity: 96.8%; specificity: 89.3%). The hs-cTnI 0/2 h Algorithm outperformed other strategies in terms of accuracy (89.0%), sensitivity (93.3%), and F1-score (73.68%), making it a promising tool for NSTEMI detection in clinical practice. These findings underscore the potential of hs-cTnI-based strategies in improving early diagnosis and patient management in suspected NSTEMI cases. To translate these findings into practice, we recommend: Multi-center validation to confirm generalizability across diverse populations and clinical workflows; Integration with machine learning models to refine risk stratification in borderline cases.These advancements could redefine early diagnostic pathways for suspected NSTEMI, prioritizing both accuracy and resource efficiency.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Thygesen, K. et al. Fourth universal definition of myocardial infarction (2018). Circulation 138, 1 (2018).
Collet, J. P. et al. 2020 ESC guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Rev. Esp. Cardiol. (Engl Ed). 74, 544 (2021).
Byrne, R. A. et al. 2023 ESC guidelines for the management of acute coronary syndromes. Eur. Heart J. Acute Cardiovasc. Care. 13, 55–161 (2024).
Apple, F. S., Sandoval, Y. & Jaffe, A. S. Cardiac Troponinassays: guide to Understanding analytical characteristicsand their impact on clinical care. J] Clin. Chem..
Apple, F. S. et al. IFCC educational materials on selected analytical and clinical applications of high sensitivity cardiac troponin assays. Clin. Biochem. 48, 201–203 (2015).
Wu, A. H. B. et al. Clinical laboratory practice recommendations for the use of cardiac troponin in acute coronary syndrome: expert opinion from the academy of the American association for clinical chemistry and the task force on clinical applications of cardiac Bio-markers of the international federation of clinical chemistry and laboratory medicine. Clin. Chem. 64, 645–655 (2018).
Landin, B., Sisowath, C. & Strålfors, A. Factors affecting the evaluation and use of a hemoglobin A method - Lot-to-lot variation, long-term deviation and carry-over. Clin. Chim. Acta. 544, 117332 (2023).
Krintus, M. & Panteghini, M. Laboratory-related issues in the measurement of cardiac troponins with highly sensitive assays. Clin. Chem. Lab. Med. 58, 1773–1783 (2020).
Warner, J. V. & Marshall, G. A. High incidence of Macrotroponin I with a high-sensitivity troponin I assay. Clin. Chem. Lab. Med. 54, 1821–1829 (2016).
Boeddinghaus, J. et al. Clinical validation of a novel high-sensitivity cardiac troponin I assay for early diagnosis of acute myocardial infarction. Clin. Chem. 64, 1347–1360 (2018).
Boeddinghaus, J. et al. Direct comparison of 4 very early Rule-Out strategies for acute myocardial infarction using High-Sensitivity cardiac troponin I. Circulation 135, 1597–1611 (2017).
Arslan, M., Dedic, A., Boersma, E. & Dubois, E. A. Serial high-sensitivity cardiac troponin T measurements to rule out acute myocardial infarction and a single high baseline measurement for swift rule-in: A systematic review and meta-analysis. Eur. Heart J. Acute Cardiovasc. Care. 9, 14–22 (2020).
Greenslade, J. H. et al. Diagnostic accuracy of a new High-Sensitivity troponin I assay and five accelerated diagnostic pathways for ruling out acute myocardial infarction and acute coronary syndrome. Ann. Emerg. Med. 71, 439–451e3 (2018).
Johannessen, T. R. et al. Rapid rule-out of acute myocardial infarction using the 0/1-hour algorithm for cardiac troponins in emergency primary care: the OUT-ACS implementation study. BMC Prim. Care. 26, 34 (2025).
Clerico, A., Zaninotto, M. & Plebani, M. Rapid rule-in and rule-out protocols of acute myocardial infarction using hs-cTnI and hs-cTnT methods. Clin. Chem. Lab. Med. 62, 213–217 (2024).
Twerenbold, R. et al. Prospective validation of the 0/1-h algorithm for early diagnosis of myocardial infarction. J. Am. Coll. Cardiol. 72, 620–632 (2018).
Shah, A. S. V. et al. High-sensitivity cardiac troponin I at presentation in patients with suspected acute coronary syndrome: a cohort study. Lancet 386, 2481–2488 (2015).
Mueller, C. et al. Rapid rule out of acute myocardial infarction: novel biomarker-based strategies. Eur. Heart J. Acute Cardiovasc. Care. 6, 218–222 (2017).
Ambavane, A. et al. Correction: economic evaluation of the one-hour rule-out and rule-in algorithm for acute myocardial infarction using the high-sensitivity cardiac troponin T assay in the emergency department. PLoS One. 13, e0191348 (2018).
Fitzgerald, R. L. et al. Analytical performance evaluation of the Elecsys® troponin T gen 5 STAT assay. Clin. Chim. Acta. 495, 522–528 (2019).
He, M. & Zhang, J. Evaluation of four commercial biuret reagent kits of serum total protein by the American association for clinical chemistry reference measurement procedure. Clin. Chem. Lab. Med. 49, 989–992 (2011).
Wu, J. et al. Machine Learning Diagnostic Model for Early Stage NSTEMI: Using hs-cTnI 1/2 h Changes and Multiple Cardiovascular Biomarkers. Diagnostics (Basel) 14, (2024).
Ayan, D. & Soylemez, S. Measuring plasma ferritin levels with two different methods: A comparison of Roche Cobas e601 versus Roche Cobas c501 (integrated modular system Roche Cobas 6000). J. Med. Biochem. 39, 13–18 (2020).
Solak, Y. Comparison of serum sodium levels measured by blood gas analyzer and biochemistry autoanalyzer in patients with hyponatremia, eunatremia, and hypernatremia. Am. J. Emerg. Med. 34, 1473–1479 (2016).
Sörensen, N. A. et al. Prognostic Value of a Novel and Established High-Sensitivity Troponin I Assay in Patients Presenting with Suspected Myocardial Infarction. Biomolecules 9, (2019).
Lin, Y. et al. A multicenter prospective study on the ESC algorithm for the early diagnosis of non-ST-elevation myocardial infarction. iScience 27, 110643 (2024).
Bohula May, E. A. et al. Prognostic performance of a high-sensitivity cardiac troponin I assay in patients with non-ST-elevation acute coronary syndrome. Clin. Chem. 60, 158–164 (2014).
Diaz-Garzon, J. et al. Biological variation of cardiac troponins in health and disease: A systematic review and Meta-analysis. Clin. Chem. 67, 256–264 (2021).
Acknowledgements
We would like to thank Suzhou Hybiome Biomedical Engineering Co., Ltd. for providing reagent support in cardiac biomarker testing.
Funding
Capital Medical University Undergraduate Scientific Research Innovation Training Project (XSKY2024304).
Author information
Authors and Affiliations
Contributions
Conceptualization, methodology, software, J.W.; validation, formal analysis, data curation, Y.H. and Y.G.; writing -original draft preparation, J.W; Sample and reagent resource management K. C., S.C. and J. Y. Supervision, H.Y.; All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, J., Hua, Y., Ge, Y. et al. Clinical performance validation and four diagnostic strategy assessments of high-sensitivity troponin I assays. Sci Rep 15, 14442 (2025). https://doi.org/10.1038/s41598-025-99273-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-99273-5
