
Abstract
Acinar cell carcinoma (ACC) and pancreatoblastoma (PBL) are rare pancreatic malignancies with acinar differentiation. Proteogenomic profiling of ACC and PBL revealed distinct protein expression patterns compared to pancreatic ductal adenocarcinoma (PDAC) and benign pancreas. ACC and PBL exhibited similarities, with enrichment in proteins related to RNA processing, chromosome organization, and the mitoribosome, while PDACs overexpressed proteins associated with actin-based processes, extracellular matrix, and immune-active stroma. Pathway activity differences in metabolic adaptation, epithelial-to-mesenchymal transition, and DNA repair were characterized between these diseases. PBL showed upregulation of Wnt-CTNNB1 and IGF2 pathways. Seventeen ACC-specific proteins suggested connections to metabolic diseases with mitochondrial dysfunction, while 34 PBL-specific proteins marked this pediatric cancer with an embryonic stem cell phenotype and alterations in chromosomal proteins and the cell cycle. This study provides novel insights into the proteomic landscapes of ACC and PBL, offering potential targets for diagnostic and therapeutic development.
Similar content being viewed by others


Introduction
Pancreatic acinar cell carcinoma (ACC) and pancreatoblastoma (PBL) are pancreatic neoplasms that are characterized by acinar differentiation of at least a subset of the neoplastic cells1. These diseases have distinct clinicopathological features from conventional pancreatic ductal adenocarcinomas (PDACs) and other neoplasms with ductal differentiation. ACC is a rare malignant tumor arising from the exocrine pancreas, comprises approximately 2% of all pancreatic neoplasms, and occurs mostly in adult men2. PBL is another rare malignancy that resembles ACC in acinar differentiation but occurs predominantly in infants and young children, comprising less than 1% of all pancreatic tumors3. Because ACC and PBL share many histologic and immunophenotypic features, they have been considered closely related neoplasms.
Molecular understanding of ACC and PBL is thus far confined to genomic analyses. Unlike in PDAC, KRAS mutations are rarely observed in ACC4,5,6. Mutations in SMAD4, CDKN2A, and TP53, which are frequently altered in PDAC, are observed in ACC but less frequently. The most frequent mutations in ACC are observed in the Wnt/β-catenin pathway (APC and CTNNB1), occurring in up to 25% of ACC cases7. Mutations in DNA mismatch repair genes, such as BRCA1, BRCA2, and RB1 genes, are also observed in ACC8. At the chromosomal level, BRAF rearrangement and loss of chromosome 11p have been reported in ACC4,7. Genomic analyses of PBL, in which the sample numbers have thus far been small, revealed lower somatic mutation burden compared to ACC5. Multiple studies have shown alterations in the Wnt/β-catenin signaling pathway in PBL, with APC and CTNNB1 harboring the most common gene alterations and imprinting IGF2 dysregulation as a consequence of APC alterations5,9,10. Loss of chromosome 11p has also been reported in PBL10.
As described above, genomic studies have revealed both shared and distinct genomic features between PDAC, ACC, and PBL. However, no studies at the mRNA or protein level focusing on differences between ACC and PBL have been undertaken. Given that proteins are major players that drive biological processes and the targets of therapeutic development, proteomic characterization of ACC and PBL would be beneficial for deeper biological understanding of ACC and PBL. Since current treatment options for ACC and PBL are limited to surgical resection or conventional cytotoxic chemotherapy11,12, a proteomic approach holds promise for therapeutic target development.
Studying the tumor microenvironment can lead to more effective therapeutic interventions and provide insights into disease progression. The tumor microenvironment of PDAC plays a significant role in cancer progression and treatment resistance13,14,15. No comparable studies have to date been accomplished in PBL or ACC, limiting the clinical use of immunotherapy for these diseases.
Because of the rarity of these tumors, ACC and PBL tissue samples from patients are very difficult to obtain. These tumors have barely been characterized by omics technologies other than a few genomic and transcriptomic analyses. In this study, we accumulated cohorts of tissues from ACC and PBL patients and conducted detailed global proteomic analyses accompanied by genomics to overcome current knowledge gaps in ACC and PBL. For comparison, we also profiled cohorts of conventional PDACs and benign pancreatic tissues. Our study shows, for the first time, that the two acinar neoplasms are drastically different from PDAC in their proteomic makeups. Our study also shows that ACC and PBL are similar but distinct proteomic entities with characteristic differences in protein networks and specific protein markers.
Results
Proteogenomic profiling of ACC, PBL, and PDAC
LC-MS proteome sequencing of the neoplastic and benign tissue cohorts quantified a total of 7284 proteins, with 4282 proteins being quantified in 50% or more samples of at least one histological sample type (i.e., ACC, PBL, PDAC, or benign pancreatic tissue) (Supplementary Data 1). Unsupervised UMAP of proteomic expression data showed a clear separation between disease entities (Fig. 1a). Although ACC and PBL clustered closely in the UMAP compared to PDAC and benign tissue, they were separated from each other and feature proteomic differences as represented by the heatmap (Fig. 1b).

a Unsupervised UMAP of all entities showing differences between benign pancreatic tissue, PDAC, ACC, and PBL. Note that ACC and PBL clusters are in closer proximity than the other clusters, but distinct and do not intermix. b Proteogenomic summary of the cohort (n = 41 samples). Alterations (if available) for commonly involved genes are shown. Differentially expressed proteins with log2FC > 2 or <−2 between benign tissue and PDAC, benign tissue and ACC, or benign tissue and PBL are selected, and their expression values are shown as a heatmap (n = 853 proteins). Each entity (benign, PDAC, ACC, PBL) has both shared and specific protein signatures. Gray-colored columns represent missing values (i.e., genomic information was not available for these samples). c–e Volcano plots of the proteomic profiles of PDAC, ACC, and PBL compared to those of the benign pancreatic tissue. Red and blue dots show significantly dysregulated proteins with log2FC > 1 or <−1 (vertical dashed lines) for PDAC, ACC, or PBL. Horizontal dashed lines show the significance level at 0.05 (q-value). f, g Pathway enrichment analysis results of KEGG and Hallmark gene sets (NES, normalized enrichment score). The top 10 statistically significant pathways are shown in the KEGG results. In the Hallmark results, all statistically significant pathways are shown. Red/blue NES values denote upregulation/downregulation.
Genomic sequencing revealed differences, but no clear cluster separation among these pancreatic tumors (Fig. 1b). KRAS and TP53 gene alterations were common in PDAC (10/11 patients) but less frequent in ACC (3/10 patients) or PBL (1/11 patients). Wnt/β-catenin pathway-associated APC or CTNNB1, as well as BRAF alterations, were observed in ACC and PBL but not in PDAC. In addition, SMAD2, SMAD4, BRCA1, and BRCA2 alterations were found in ACC, and SMAD4 alterations were found in PBL.
Dysregulated proteins and their associated pathways
Differential protein expression analyses were performed to identify proteins that were significantly dysregulated (q-value < 0.05) in these pancreatic neoplasms when compared to benign pancreatic tissues. From the PDAC cohort, 2635 dysregulated proteins were identified, of which 840 were upregulated and 719 were downregulated with greater than 2-fold change. From the ACC cohort, 2400 dysregulated proteins were identified, of which 1001 were upregulated and 331 were downregulated with greater than 2-fold change. From the PBL cohort, 2557 dysregulated proteins were identified, of which 1086 were upregulated and 532 were downregulated with greater than 2-fold change (Fig. 1c–e) (Supplementary Data 2a–c).
Based on KEGG pathway enrichment analysis of these dysregulated proteins (Fig. 1f), pancreatic secretion, protein digestion and absorption, amino acid degradation and metabolism, oxidative phosphorylation, and NOD-like receptor signaling pathway alterations were shared by PDAC, ACC, and PBL. Cancer cell invasion-related pathways (e.g., focal adhesion and regulation of actin cytoskeleton) were significantly dysregulated in PDAC but not in ACC or PBL, whereas spliceosome, nucleocytoplasmic transport, and cell cycle pathways were significantly perturbed in ACC and PBL but not in PDAC. Herpes simplex virus infection pathway proteins, Th17 cell differentiation, C-type lectin receptor signaling, metabolism of xenobiotics by cytochrome P450, and glycerolipid pathways were significantly altered in ACC only. Transcriptional misregulation, chemical carcinogenesis (reactive oxygen species), fatty acid degradation, and citrate cycle (TCA cycle) pathways were significantly affected in PBL only.
Based on Hallmark pathway analysis (Fig. 1g), the dysregulated proteins were significantly associated with epithelial-to-mesenchymal transition (EMT) features in all three pancreatic neoplasms, with the highest enrichment score in PDAC. Cell cycle-related pathways (e.g., E2F targets, G2M checkpoint, and KEGG cell cycle gene sets) and DNA repair were significantly enriched in PBL and ACC. Myc-targets and PI3K-AKT-mTOR signaling were significantly altered only in PBL. Inflammatory response, apical junction, apoptosis, glycolysis, and a few other pathways were found to be enriched in PDAC but not in ACC or PBL.
As shown above, shared dysregulated pathways involved pancreatic secretion, protein digestion, and amino acid metabolism. PDAC uniquely showed dysregulation in cancer invasion pathways, while ACC showed dysregulation in viral infection and xenobiotic metabolism pathways. PBL showed dysregulation in transcriptional dysregulation and fatty acid degradation pathways. Notably, EMT features were prominent in all neoplasms, especially in PDAC.
Upregulated protein-protein interaction networks
To gain a clearer picture of the proteomic changes in these pancreatic neoplasms, we examined the protein-protein interaction networks of the proteins that were upregulated in ACC, PBL, or PDAC. The STRING network analysis showed that, for each tumor type, the upregulated proteins had significantly more interactions than expected of a random set of proteins of the same size, indicating the non-randomness of the identification of these proteins.
The upregulated protein networks of ACC and PBL were overall similar (Fig. 2). In both ACC and PBL, the networks were dominated by four functional clusters of proteins that are associated with RNA processing (Gene Ontology Biological Process, GOBP), RNA transport (GOBP), chromosome organization (GOBP), and mitochondrial translation initiation (STRING local network cluster). However, the chromosome organization network was more extensive in PBL. The most noticeable network differences involved disease-gene associations (DISEASES) database entries. Ninety-one upregulated proteins in ACC were significantly associated with “inherited metabolic diseases” (91/949, with a false discovery rate (FDR) of 7.15e-05), and 98 proteins were associated with “disease of metabolism in general” (98/1076, FDR 0.0010).

Only interactions where the proteins are part of a physical complex with a minimum interaction score of 0.9 (highest confidence setting in STRING) are included in the analysis, comprising ACC (989 upregulated proteins with 1224 interactions vs. 590 interactions expected of a random set of proteins of the same size), PBL (1073 proteins with 1549 interactions vs. 766 expected), and PDAC (818 proteins with 598 interactions vs. 159 expected), all with PPI enrichment p-values < 1.0e-16. Upregulated protein-protein interaction inferred by STRING database search shows that ACC and PBL are similar and have 4 dominant clusters (RNA processing, RNA transport, chromosome organization, and mitochondrial translation). Specifics for the ACC clusters: RNA processing (147/868 (147 proteins in the ACC network vs. 868 proteins in the entire GOBP RNA processing network), FDR 2.11e-31), RNA transport (42/159, FDR 2.24e-13), mitochondrial translation (35/112, FDR 7.92e-13), and chromosomal organization (83/968, FDR 0.00047). Specifics for PBL: RNA processing (205/868, FDR 5.27e-60), RNA transport (47/159, FDR 1.59e-15), mitochondrial translation (28/112, FDR 9.45e-08), and chromosome organization (120/968, FDR 5.67e-13). In contrast, PDAC features markedly different protein-protein interaction clusters, such as actin filament-based processes (101/609, FDR 3.88e-26), extracellular matrix organization (61/180, FDR 1.30e-28), immune response (147/1321, FDR 7.61e-23), and complement and coagulation cascades (64/196, FDR 4.57e-29).
The upregulated protein network of PDAC was drastically different from those of both ACC and PBL (Fig. 2). It was dominated by actin filament-based process (GOBP, 101/609, FDR 3.88e-26), immune response (GOBP, 147/1321, FDR 7.61e-23), and extracellular matrix organization (Reactome Pathways, 82/300, FDR 3.01e-34). Complement and coagulation cascades, including protein-lipid complex, were also highly enriched in PDAC (STRING Local Network Cluster, 64/196, FDR 4.57e-29). RNA processing, chromosome organization, or mitochondrial ribosome networks, which were highly enriched in ACC and PBL, were not enriched in PDAC. A large number of the upregulated proteins in PDAC were associated with extracellular exosomes (GO Cellular Component, 356/2096, FDR 5.86e-117) or vesicles (446/3957, FDR 8.13e-97).
The top enriched Reactome pathways, as per STRING protein-protein interaction network analysis, were processing of capped intron-containing pre-mRNA for ACC, metabolism of RNA for PBL, and immune system for PDAC (255/1979, FDR 2.82e-55). The top enriched KEGG pathways were spliceosome for ACC or PBL and complement and coagulation cascade for PDAC (40/82, FDR 1.61e-23). The top enriched WikiPathways were mRNA processing for ACC and PBL and VEGFA-VEFGR2 signaling for PDAC (69/428, FDR 7.49e-17). The top tissue expression match was glandular tissue for ACC, female reproductive system for PBL, and digestive glandular tissue for PDAC (408/2881, FDR 1.8e-114).
As shown above, protein-protein interaction networks identified significant differences between these 3 diseases. ACC and PBL had networks focused on RNA processing, transport, and chromosome organization. ACC had many proteins linked to metabolic diseases. PDAC’s network was distinct, centered on immune response and extracellular matrix organization, with many proteins associated with vesicles. Pathway analysis highlighted RNA processing for ACC and PBL, and immune response for PDAC.
EMT and metabolic pathways
Although EMT was significantly altered in all three pancreatic neoplasms, the hypoxia pathway, which is closely associated with EMT, appeared to be significantly altered only in PDAC (Fig. 1g and Supplementary Fig. 1a, b). The TGF-β signaling pathway activation, which cross-talks with the EMT pathway16, was significantly enriched in ACC, PBL, and PDAC, with PBL having the highest average score (Supplementary Fig. 1c, d). Angiogenesis, another important EMT feature, showed positive correlations with both hypoxia and EMT signatures (Supplementary Fig. 1e, f).
Several metabolic pathways were altered in the pancreatic neoplasms. Glycolysis pathway activation was significantly altered in PDAC but less in ACC and insignificantly in PBL (Supplementary Fig. 1g). Oxidative phosphorylation was significantly downregulated in PDAC and PBL but less in ACC (Fig. 1f, g and Supplementary Fig. 1h). Citrate cycle was significantly downregulated in PBL (Fig. 1g and Supplementary Fig. 1i).
Cell cycle, stemness, and other pathways
As cell cycle-related pathways were significantly enriched in ACC and PBL (Fig. 1f, g), we studied these pathways in more detail. PBL generally displayed higher CDK expression levels and cell cycle activation scores (Fig. 3a). PBL and PDAC showed significantly high stemness scores, with PBL having the highest stemness feature (Fig. 3a, b).

a Summary proteomic heatmap of cell cycle signatures. On the right side, Spearman’s rank correlation coefficients between KEGG cell cycle signature scores and the corresponding molecule/pathways are shown as bars (* indicates statistical significance). b Stemness score for each group. The stemness score is upregulated in all entities. PBL, which is a childhood cancer, shows the highest average score (Mann–Whitney U test). The box represents the 25th to 75th percentiles, with the median marked by a central line. Whiskers represent 1.5 times the interquartile range, and outliers are shown as individual dots. c Hallmark DNA repair score for each group. The DNA repair score is significantly elevated in ACC and PBL (Mann–Whitney U test). The box represents the 25th to 75th percentiles, with the median marked by a central line. Whiskers represent 1.5 times the interquartile range, and outliers are shown as individual dots. d KEGG pathway scores for four major DNA repair pathways are shown (Mann–Whitney U test). The box represents the 25th to 75th percentiles, with the median marked by a central line. Whiskers represent 1.5 times the interquartile range, and outliers are shown as individual dots. e, f Telomerase-dependent pathway scores (TelNET_TEL) and telomerase-independent pathway scores (TelNET_ALT) for each group. PBL has the highest average scores for both pathways (Mann–Whitney U test). The box represents the 25th to 75th percentiles, with the median marked by a central line. Whiskers represent 1.5 times the interquartile range, and outliers are shown as individual dots. g ALT/TEL score ratio for each group. Compared to benign tissue and PDAC, ACC and PBL show stronger dependence on the ALT pathway (Mann–Whitney U test). The box represents the 25th to 75th percentiles, with the median marked by a central line. Whiskers represent 1.5 times the interquartile range, and outliers are shown as individual dots.
The Hallmark DNA repair process was significantly upregulated in PBL and ACC but not in PDAC (Figs. 1g and 3a, c). We investigated four different DNA repair processes: mismatch repair, base excision repair, homologous recombination, and nonhomologous recombination processes. As shown in Fig. 3d, mismatch repair and homologous/nonhomologous recombination processes were significantly altered in PBL and ACC but not in PDAC.
PBL showed high activation status of the Wnt/β-catenin pathway in our proteome dataset, whereas ACC and PDAC did not show significant Wnt pathway activation (Fig. 3a).
Next, we investigated the telomere pathway because this pathway is closely connected to cell proliferation sustainability and stemness17. Both telomerase-dependent telomere lengthening (TEL) and alternative lengthening of telomeres (ALT) pathways showed elevated activation levels across all three diseases with the highest activation in PBL, consistent with PBL’s highest stemness score and cell cycle activation level (Fig. 3e, f). The ALT/TEL activation ratio was also significantly increased compared to benign tissue (Fig. 3g).
Tumor microenvironment features
To compare the tumor microenvironments of the tree pancreatic neoplasms, we deconvoluted immune cell compositions using xCell (Supplementary Fig. 2a). The overall average immune score of PDAC was much higher than those of ACC and PBL (Supplementary Fig. 2b). PDAC also had the highest enrichment in antigen processing and presentation via the MHC class I pathway (Supplementary Fig. 2a). Normal tissue and PDAC had higher levels of Treg cells, whereas ACC and PBL appeared to feature CD8+ and CD4+ T cell infiltration. Protein expression of immune proteasome components did not show large expression shifts, but proteins that transport and load peptides onto MHC class I were significantly higher in PDAC (Supplementary Fig. 2c). The IFNγ pathway, which activates the JAK/STAT pathway and IRF transcription and induces MHC class I-related proteins, was also higher in PDAC than ACC or PBL (Supplementary Fig. 2a).
Proteomic differences between ACC and PBL
To identify proteomic differences between ACC and PBL, we performed a differential expression analysis between the two and identified 261 proteins that were significantly differentially expressed (q-value < 0.05), including 59 upregulated in PBL and 116 upregulated in ACC with >2-fold change (Fig. 4a) (Supplementary Data 2d).

a Volcano plot of differential expression between PBL vs. ACC. Red and blue dots show significantly (q < 0.05) upregulated and downregulated proteins with log2FC > 1 or <−1, respectively. The horizontal dashed line shows the significance level at 0.05 (q-value). b GSE analysis of KEGG pathways. All upregulated pathways and the top 10 downregulated pathways are shown. Positive/negative NES value denotes upregulation/downregulation in PBL vs. ACC. c GSE analysis of the Hallmark gene sets. All significant results are shown. Positive/negative NES value denotes upregulation/downregulation in PBL vs. ACC. d GSE plot of SOX9, E2F, and LET based on GSEA results of MSigDB C3 (regulatory target gene sets) between ACC and PBL. A ranked gene list based on fold-change values between ACC and PBL is used as GSEA input. Target genes of these transcription factors are significantly upregulated in PBL compared to ACC at the protein level. e GSE analysis of MSigDB C8 (cell type gene signature sets). The top 10 upregulated and top 10 downregulated cell signatures are shown. NES value denotes upregulation/downregulation in PBL vs. ACC. f Venn diagrams of significantly upregulated proteins in specified pairwise comparisons (q < 0.05). For ACC: ACC vs. benign tissue, ACC vs. PDAC, ACC vs. PBL. For PBL: PBL vs. benign tissue, PBL vs. PDAC, PBL vs. ACC. Proteins with log2FC > 1 are included in each circle. 17 (ACC) and 34 (PBL) proteins are significantly upregulated in any comparison (centers of the Venn diagrams).
These differentially expressed proteins revealed that features of ECM-receptor interaction, cell cycle, and E2F targets were enriched in PBL. In contrast, ACC showed higher enrichment of exocrine features such as pancreatic secretion and oxidative phosphorylation (Fig. 4b, c). Transcription factor target gene enrichment analysis identified 3 transcription factors (E2F, SOX9, and LET) as significantly enriched regulons in PBL vs. ACC (Fig. 4d).
To test how PBL vs. ACC may differ in cell type features, we performed cell type enrichment analysis using 830 cell type signature gene sets from MSigDB. Proteins overexpressed in PBL revealed a significant resemblance to neural progenitor cells with neural crest cell features and midbrain neurotypes. In contrast, the proteins overexpressed in ACC most resembled pancreatic and fetal acinar cells (Fig. 4e).
ACC- and PBL-specific protein markers
To identify markers that are specific to ACC or PBL, we filtered for proteins that were consistently and significantly upregulated when compared across the neoplasms and benign tissue (Fig. 4f, Supplementary Data 3). Comparisons of ACC vs. PBL, ACC vs. PDAC, and ACC vs. benign pancreatic tissue identified 17 ACC-specific proteins that were consistently upregulated in ACC with >2-fold changes. Similarly, for PBL, we identified 34 consistently upregulated proteins with >2-fold changes in PBL vs. ACC, PBL vs. PDAC, and PBL vs. benign pancreatic tissue comparisons. These proteins are potential candidate biomarkers for ACC and PBL.
The 17 ACC-specific protein markers are COG7, EHHADH, NDUFAF2, BET1L, TACO1, ALDOB, BAG1, EEF1A2, SCG5, HAX1, NDUFAF3, CLPB, SPRYD4, MAOA, TOMM70A, OPLAH, and COA3 (Fig. 5a, b, Supplementary Fig. 3). These proteins were significantly associated with mitochondria, with 8 being mitochondrial proteins (e.g., TOMM70, HAX1, CLPB, COG7) (Fig. 5c). In particular, the mitochondrial respiratory chain complex assembly (GOBP) was significantly enriched (q = 0.00031), demonstrated by the protein-protein interaction network of NDUFAF3, DNUFAF2, TACO1, and COA3. At least 10 of the 17 ACC-specific proteins were associated with inherited metabolic diseases, e.g., ALDOB, OPLAH, EHHADH, and COG7.

a Heatmap of 17 ACC-specific and 34 PBL-specific protein expression abundance of all samples, including PDAC and benign pancreatic tissue. Yellow boxes highlight specifically upregulated protein expression levels for ACC and PBL, respectively. b Boxplots of expressional differences of the 17 ACC- and 34 PBL-specific proteins relative to benign tissue are shown. For each protein, log2FC values of each tumor sample relative to benign tissue median values were calculated. The plot only shows boxplots (25th, 50th, and 75th percentiles marked as bottom, center, and upper lines) and whiskers (1.5 times the interquartile range) without outlier values. c Protein-protein interaction network of 17 ACC-specific proteins based on the STRING database, showing enrichment of mitochondria-related proteins. Node colors denote mitochondrion (blue), mitochondrial respiratory chain complex assembly (pink), and inherited metabolic disease (gray). White denotes no connected biological process. d Protein-protein interaction network of 34 PBL-specific proteins based on the STRING database. Node colors denote chromosome (red), cell cycle (green), regulation of cell cycle (dark green), and embryonic structure (yellow). White denotes no connected biological process.
The 34 PBL-specific marker proteins are SMAD4, IGF2BP2, IFG2BP1, HMGA2, PLD3, CTSC, CUL1, RCC2, NASP, HIST2H3A, DPF2, FLYWCH2, CBX1, HIST1H1B, PBRM1, PGM5, RRM1, CDK2, NES, MCM3, ADAM9, GTF3C2, SRSF9, POLDIP3, EXOSC4, EXOSC6, ZCCHC3, SMARCA4, CCAR2, CIAPIN1, HNRNPLL, RBPJ, PRPF39, and CAPN6 (Fig. 5a, b, Supplementary Fig. 3). The PBL-specific marker proteins were significantly associated with chromosomes, cell cycle, and embryonic structures tissue expression (Fig. 5d). At least 14 proteins were chromosome-related proteins, including histones H3C13, H1-5, CBX1, EXOSC4, PBPJ, and NASP. DPF2, PBRM1, and SMARCA4 are part of the npBAF complex in H3K9me3-modified histone binding. At least 11 proteins were associated with the cell cycle. For example, SMAD4, MCM3, CDK2, and CUL1 are involved in cell cycle regulation, and PDF2, PBRM1, SMARCA4, and RRM1 are involved in G0/G1 and G1/S transitions. Consistent with PBL being a pediatric neoplasm, at least 12 of the 34 PBL-specific proteins were significantly associated with embryonic-type tissue expression, e.g., ADAM9, CTSC, CAPN6, POLDIP3, and NES.
To externally validate our specific proteins for ACC or PBL, we reanalyzed transcriptome data and differential expression analysis data between PBL (n = 5) and benign pancreatic tissue (n = 5)10, the only other available expressional dataset. Although comparison (only PBL vs. benign) and quantification (mRNA) were different from ours, 4 informative genes were shared with our dataset (Supplementary Data 4). ALDOB (ACC-specific) was significantly downregulated in PBL, and IGF2BP1, HMGA2, and PGM5 (PBL-specific) were significantly upregulated in PBL at the mRNA level, findings entirely consistent with our proteome results.
Connectivity map analysis revealing possible drug candidates for ACC and PBL treatment
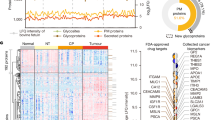
Proteins serve as the main functional components in cells and are the main targets for drugs. Leveraging our proteome data of ACC and PBL may help identify protein targets and candidate drugs for these rare diseases. Utilizing a connectivity map (CLUE webtool)18, we identified 33 and 63 small compounds with connectivity scores >70 as possible drug candidates for ACC and PBL, respectively. Small compounds with connectivity scores >80 for each disease are shown in Fig. 6. An HDAC inhibitor is a top-scoring drug for both ACC and PBL. As HDAC inhibition suppresses acinar cell fate and promotes ductal differentiation19, it may have antitumor effects on both ACC and PBL, whose morphological differentiation shows mainly acinar cell lineage. The connectivity map also identifies a PLK inhibitor as a candidate. Polo-like kinases (PLKs) are a family of serine/threonine protein kinases that play crucial roles in the regulation of cell cycle progression and other cellular processes, including DNA replication, mitosis, and cytokinesis20,21, which is compatible with the pathway results of ACC and PBL (Fig. 1, cell cycle enrichment). These findings show the potential utility of connectivity map analyses using proteomic data for generating hypotheses for drug repositioning and rational drug testing, especially for rare diseases.

Separate connectivity map analyses showing small compounds with connectivity scores >80 for ACC and PBL, respectively. Drug categories are shown on the left. Drug names are shown on the right. Gene names corresponding to proteins targeted by these drugs are shown at the bottom.
Discussion
In this study, we presented the first global proteomic characterization of tumor tissues from patients with ACC and PBL, two rare and poorly understood malignant pancreatic neoplasms. Histologically, ACC and PBL resemble each other because both show extensive acinar cell differentiation, and the cell of origin is traditionally considered the exocrine pancreas for both1. We discovered distinct protein expression profiles for ACC, PBL, and PDAC. We showed that the tissue proteomes of ACC/PBL were unambiguously different from PDAC or benign pancreatic tissue. We identified entity-specific proteins and protein networks that define ACC and PBL as different entities, rather than a single entity with differing features in children versus adults.
The proteomic data were consistent with the morphology of ACC and PBL as primarily cellular and stroma-poor neoplasms, which is in contrast to desmoplastic stroma-rich PDAC. Importantly, the proteomes revealed unique features of ACC and PBL cells with extraordinary enrichment in proteins involved in RNA processing, RNA transport, chromosome organization, and mitochondrial ribosomes. In contrast, the PDAC proteome was primarily characterized by overexpression of proteins of the actin cytoskeleton, extracellular matrix components, and an immune-active microenvironment compared to ACC/PBL. The finding of prominent RNA processing and transport machinery in ACC and PBL is new, which may open new directions for future investigation of these rare cancers. Our approach may also provide unique therapeutic targets since these processes are uniquely prominent in ACC and PBL compared to PDAC.
Solid pancreatic neoplasms can be subdivided into two categories: (1) stroma-rich, ill-defined, and desmoplastic and (2) stroma-poor, well-circumscribed, and cellular22,23. ACC and PBL have little stroma and none of the characteristic desmoplastic (cancer-associated fibrotic) stroma that is so prominent in PDAC. In our study, the contrast in the cancer microenvironment was clearly depicted by their proteomes: while the PDAC proteome was dominated by the upregulated protein-protein network of the extracellular matrix, such overexpression was not observed in the ACC or PBL proteome.
At least 56 protein components of the extracellular matrix were identified to be significantly overexpressed in PDAC, including proteoglycans (e.g., BGN, DCN), collagens, fibronectin, vitronectin, fibrillin, fibulins, and laminin. In addition, at least nine integrin proteins were found to be overexpressed in PDAC. For example, collagen COL12A1 was the most overexpressed protein (log2 fold change, 7.4) in the PDAC proteome when compared to benign tissue, which is consistent with the finding that COL12A1 acts as a prognostic marker with aberrantly high expression in pancreatic cancer24.
Our study discovered that mitochondrial ribosomes (mitoribosomes) were uniquely enriched in ACC and PBL, but not in PDAC. Ultrastructurally, electron microscopy did reveal rich mitochondria in several cases of ACC/PBL, e.g., an ACC case highlighting the tumor cells with numerous mitochondria and distinctive electron-dense intramitochondrial inclusions25, and two cases of pancreatic solid pseudopapillary tumors with possible differential diagnoses of ACC, PBL, and endocrine tumors showing the tumor cells rich in mitochondria, containing abundant and often disintegrated granules of variable sizes26. Mitoribosomes translate mtDNA that encodes membrane integral core subunits of the oxidative phosphorylation system, which we also found upregulated in our data. Abnormal expression of mitoribsomal proteins may lead to mitochondrial metabolic disorders and cell dysfunction.
In this study, we identified 17 ACC-specific protein markers. Because ACC has rarely been studied by proteomic means, dysfunction in none of these proteins or their coding genes has been reported for ACC. Several ACC marker proteins that were highly overexpressed are crucial players in metabolic processes, suggesting ACC may have a distinct metabolic profile compared to other entities. The ACC-specific markers, together with the overexpression of mitoribosomal components, reveal ACC as a pancreatic neoplasm with unique features of mitochondrial metabolic disturbance.
For example, ACC-specific marker protein NDUFAF3 is a part of the assembly of complex I of the mitochondrial respiratory chain and plays a crucial role in the electron transport chain. Mutations in NDUFAF3 cause mitochondrial complex I deficiency, a fatal neonatal disorder of the oxidative phosphorylation system27,28. MAOA is a mitochondrial enzyme that catalyzes the oxidative deamination of amines29. OPLAH uses ATP hydrolysis to catalyze the conversion of 5-oxo-L-proline to L-glutamine30. EHHADH is a bifunctional enzyme with enoyl-CoA hydratase and 3-hydroxyl-CoA dehydrogenase activities31.
Intriguingly, by gene-disease association, several ACC-specific protein markers are associated with inherited metabolic diseases. COG7 is associated with congenital disorder of glycosylation32. ALDOB is associated with hereditary fructose intolerance33. CLPB is associated with type VIIb 3-methylglutaconic aciduria, an autosomal recessive inborn error of metabolism34. COA3 is associated with Cox deficiency, a rare mitochondrial disorder that affects various body parts35.
We identified 34 PBL-specific protein markers. Of these, at least 14 proteins are chromosomal-related proteins, 12 are associated with embryonic/progenitor-like tissue phenotype, and 11 are linked to the cell cycle. These PBL-specific markers suggest that changes in chromosomal structure and cell cycle define this neoplasm which affects primarily young children.
Gene mutations in SMAD4 have been identified in a subset of pancreatic neoplasms with acinar differentiation5. Our study, for the first time, identified the overexpression of SMAD4 protein as a PBL-specific marker. In contrast, protein overexpression of SMAD4 was detected in neither ACC nor PDAC when compared to benign pancreatic tissues.
Genomically, PBL is characterized by a high frequency of aberrant activation of the Wnt signaling pathway, either via (1) somatic CTNNB1 mutations or (2) copy-neutral loss of heterozygosity of APC with imprinting dysregulation of IGF2 gene as a universal consequence5,9,10. Our study identified the protein overexpression of CDK2 as a PBL-specific marker. CDK2 is the catalytic subunit of the cyclin-dependent protein kinase that regulates cell cycle progression36. CDK2 phosphorylates CTNNB1 protein and modulates its degradation, thus fine-tuning a key player in the Wnt/β-catenin pathway37. In line with the IGF2 gene dysregulation, our study found that the IGF2 protein was overexpressed in PBL, whereas its receptor IGF2R was overexpressed in PBL, ACC, and PDAC. In addition, while the IGF2 binding protein IGFBP3 was overexpressed in both PBL and PDAC, IGF2BP1 and IGF2BP2 were specific to PBL. Therefore, our findings illustrate that the Wnt signaling pathway may be altered in PBL by either ß-catenin or IGF2 via their binding partners at the protein level.
HMGA2 and NES are recognized stem cell markers. HMGA2, a transcription factor, is highly expressed in embryonic stem cells during embryogenesis and mediates motility and self-renewal in normal and cancer stem cells. The oncofetal HMGA2-IGF2BP2-RAS axis was reported as a critical oncogenic driver in embryonic rhabdomyosarcoma38. Interestingly, our study found both IGF2BP2 and HMGA2 proteins as PBL-specific protein markers. NES-positive cells isolated from fetal pancreas possess the characteristics of pancreatic progenitor cells and also share many phenotypic markers with mesenchymal stem cells derived from bone marrow39. Thus far, NES expression was reported in one other case of PBL by immunohistochemistry40. The expression of these embryonic/stem cell markers is concordant with PBL as a cancer of pediatric predilection.
Comparison with the only available prior analysis of PBL and benign tissue10, whose cohort (n = 5) was smaller than ours and limited to transcriptome only, partially validated our disease-specific proteins (ALDOB, PGM5, HMGA2, and IGF2BP1, Supplementary Data 4), supporting the notion that our proteome analysis is robust and reliable.
Since squamous morules are a key morphological feature of PLB and distinguish PBL from ACC, we attempted to analyze whether our proteomic analysis would capture this histological feature. Although there is no specific protein marker reported, squamoid nests are positive for epithelial membrane antigens (also known as MUC-1) or CK541. MUC-1 was quantified in our proteomes, but showed no significant differences between ACC and PBL (p = 0.92, Mann–Whitney U test). This finding may be due to the fact that squamous nests only occupy a very small tissue volume in PBL. Currently, there are no diagnostic IHC markers for PBL. Therefore, the disease-specific proteins for PBL and ACC described in our study are expected to be promising candidates for useful differential diagnostic IHC markers and might serve as a good starting point for future test development.
Drug repurposing is an important opportunity, especially for rare diseases, as available treatment options are generally very limited and dedicated drug development opportunities and clinical trials are difficult relative to those for more common cancers. To identify possible treatment targets for ACC and PBL, we searched 2429 small compounds in silico using the CLUE webtool with 1 million perturbation experiments and identified over 30 drug candidates for each entity. This result generates new hypotheses for drug repurposing in patients with ACC or PBL.
There are some limitations to our study. Our study offers insights into two extremely rare pancreatic tumor types. The rarity of the tumors constrained our cohort sizes. Nevertheless, we gathered and analyzed a meaningful sample set, providing significant first-of-kind experimental data and laying a foundation for future research. Due to the tumors’ low incidence rates, we had to use formalin-fixed paraffin-embedded (FFPE) tissue samples, as no frozen tissue was available. Global proteome correlation coefficients between FFPE and frozen tissue samples have been shown to exceed 0.9, a method we used in our study42. This allowed us to effectively utilize existing samples and derive valuable insights. Due to very limited sample material, all available tissue samples were used for proteome analyses, limiting additional validation via Western blotting or IHC testing. However, comprehensive proteome analysis by mass spectrometry provides robust quantitative data on a tumor’s protein expression. Our previous studies have confirmed a positive correlation between mass spectrometry-based proteome data and IHC assessment43,44,45,46,47. Studies by CPTAC and other groups have also confirmed concordance between proteome data and IHC assessment48,49,50,51,52,53,54. The lack of well-characterized disease models, such as cell lines or patient-derived xenografts for PBL and ACC, limited our ability to conduct functional experiments. Nonetheless, our findings offer a starting point for developing these models, which will further elucidate disease mechanisms and potential therapeutic targets. Despite these limitations, our study provides new data and insights, paving the way for future research on both ACC and PBL.
In summary, we present the first proteomic characterization of two rare pancreatic neoplasms (ACC and PBL) and discovered proteomic features that distinguish these neoplasms from conventional pancreatic adenocarcinoma and from each other. In addition, we identified entity-specific protein markers for each disease. This result opens new hypotheses for drug repurposing in patients with ACC or PBL.
Methods
Clinical specimens and pathological data
We collected 10 ACC, 11 PBL, 11 PDAC, and 9 benign pancreatic tissues (mainly comprised of normal pancreatic acini) for this study. The study was approved by the MSKCC Institutional Review Board (16-1683) and the BIDMC Institutional Review Board (2023P000933). Formalin-fixed paraffin-embedded (FFPE) blocks were selected from the sample repository after pathological review. Data were acquired retrospectively and in an anonymized manner. Histological type and other clinicopathological parameters were extracted from diagnostic pathology reports (Supplementary Data 5), and tumor content for all samples was checked by gastrointestinal subspecialty pathologists.
MSK-IMPACT sequencing
Targeted gene sequencing results of 11 PDAC, 1 ACC, and 4 PBL were retrieved from the medical records. Sequencing results for 6 ACC cases were obtained from a previous report4. For the cases with missing genomic information, we checked FFPE tissue availability and found that 1 PBL and 3 ACC cases had sufficient remaining tissue available for sequencing. Six PBL cases did not have sufficient remaining material available for sequencing analysis. MSK-IMPACT sequencing was performed as previously described55. In brief, genomic DNA from FFPE tumor tissues and patient-matched normal tissues were extracted using the Qiagen DNeasy Tissue kit and EZ1 Advanced XL system (Qiagen, Valencia, CA). The extracted DNA was sheared using a Covaris E200 instrument (Covaris, Woburn, MA, USA). Custom DNA probes were designed for targeted sequencing of all exons and selected introns. Probes were synthesized using the NimbleGen SeqCap EZ library custom oligo system and biotinylated to allow for sequence enrichment by capture using streptavidin-conjugated beads. Sequencing libraries were prepared using the KAPA HTP protocol (Kapa Biosystems, Wilmington, MA, USA) and Biomek FX system (Beckman Coulter, Brea, CA). Pooled libraries containing the captured DNA fragments were sequenced using an Illumina HiSeq 2500 to obtain high, uniform coverage (>500× median coverage). All classes of genomic alterations, including substitutions, indels, copy number alterations, and rearrangements, were determined and called against the patient’s matched normal sample.
Tissue proteome extraction from FFPE tissue
Proteome extraction was performed according to a previous proteome study of FFPE tissue with modifications42. In brief, 10 unstained sections of 10-μm thickness were cut from FFPE tissue blocks. Adjacent 4-μm-thick sections were stained with H&E to confirm tumor content and tumor areas were manually dissected from the corresponding 10 sections. After dewaxing with xylene, a lysis buffer containing 100 mM Tris and 5% SDS was added to the tissue. The tissue mixture was sonicated and incubated at 98 °C for 20 min at 1000 rpm, and subsequently incubated at 80 °C for 2 h at 1000 rpm. After centrifugation at 14,000 × g for 30 min at 4 °C, the supernatant containing all the soluble proteins was collected. Protein concentration was determined using a BCA assay (Pierce).
Protein digestion and peptide preparation
Protein samples of 100 µg were double-digested with Trypsin/Lys-C Mix (Mass Spec Grade, Promega) and processed using the S-Trap system (ProtiFi) according to the manufacturer’s protocol. The peptides were then desalted on a lab-made C18 StageTip. Desalted peptides were dried in a SpeedVac vacuum concentrator, re-dissolved in 3% acetonitrile/0.1% formic acid, and stored at −80 °C until mass spectrometry analysis.
Quality control sample preparation for mass spectrometry analysis
As a quality control measure, we prepared pooled protein samples by mixing the same amount of protein extracts from each sample within this cohort. All individual patient samples were processed side-by-side with pooled quality control samples in batches. In each mass spectrometry run, we ran one pooled quality control sample per sample batch and calculated the pairwise correlation coefficients between each batch to ensure instrument inter-run stability. All Spearman’s correlation coefficients were greater than 0.96, indicating high reproducibility.
Proteome sequencing by LC-MS
Peptide samples were injected into a C18 capillary column (Peptide BEH 1.7 μm × 75 μm × 250 mm) on a nano ACQUITY UPLC system (Water), which was coupled to a Q Exactive Plus mass spectrometer (Thermo Scientific) via a Proxeon 2 nano electrospray source. The peptides were eluted with a non-linear 200 min gradient of 2–35% with buffer B (0.1% (v/v) formic acid, 100% acetonitrile) and buffer A (0.1% formic acid and 100% HPLC-grade water) at a flow rate of 300 nl/min. After each gradient, the column was washed with 90% buffer B for 5 min and re-equilibrated with 98% buffer A. MS data were acquired with an automatic switch between a full scan and 10 data-dependent MS/MS scans (TopN method). The target value for the full scan MS spectra was 1 × 106 ions in the 380–1600 m/z range with a maximum injection time of 50 ms and a resolution of 70,000 at 200 m/z, with data collected in profile mode. Precursors were selected using a 1.5 m/z isolation width. Precursors were fragmented by high-energy C-trap dissociation (HCD) with a normalized collision energy of 27 eV. MS/MS scans were acquired at a resolution of 17,500 at 200 m/z with an ion target value of 5 × 104, maximum injection time of 50 ms, and dynamic exclusion for 15 s, with data collected in centroid mode.
Protein identification and quantification
Label-free protein quantification was carried out with MaxQuant56,57 with human UniProt FASTA (canonical version downloaded 09/2018) and “Match between runs” option. The search assumed trypsin and Lys-C digestions with up to 2 missed cleavages. Allowed fixed variable modification included carbamidomethylation of cysteine. Allowed variable modifications included oxidation of methionine and acetylation of the protein N-terminus. Precursor and fragment ion mass tolerance was set to 20 ppm. Protein and peptide level FDR was set as 0.01. A minimum of 1 peptide was required for protein identification, but 2 uniquely mapped peptides were required to calculate a protein level ratio. Using the MaxLFQ algorithm58 as implemented in MaxQuant, we normalized protein quantification values (LFQ values). After quantification and data normalization in MaxQuant, protein expression data were processed by Perseus software59,60. In this step, only proteins whose expression values were valid in over 50% of samples from at least one group (i.e., benign pancreatic tissue, PDAC, ACC, or PBL) were kept, and missing values were imputed with shifted Gaussian distribution, followed by differential expression analyses (t-test) using Perseus software59,60. Q-values were calculated from nominal p-values (t-test) by using the qvalue (v2.34.0) R package61. Statistical analyses were considered significant with q < 0.05.
Gene set enrichment analysis
ClusterProfiler (version 3.16.1) with a ranked gene list ordered by fold-change values was used to identify enriched pathways/biological processes. Enrichment tests on the GOBP, Hallmark, KEGG, and Reactome gene sets were carried out with the Molecular Signatures Database (MSigDB version 7.4). Functional characterization of each sample was performed with single sample Gene Set Enrichment Analysis (ssGSEA) (https://github.com/broadinstitute/ssGSEA2.0). Normalized enrichment scores (NES) of cancer-relevant gene sets were computed by projecting the matrix of normalized protein expression onto GO, Hallmark, KEGG and Reactome pathway gene sets with the following parameters: sample.norm.type = “log”, Weight=0.75, statistic = “area.under.RES”, output.score.type = “NES”, nperm=1000, min.overlap=3, correl.type = “z.score”. To elucidate gene set enrichment in each sample, ssGSEA was applied to normalized proteome data62, and NES was calculated for MSigDB terms.
STRING analysis
We performed STRING network analysis with stringent settings63. Only protein-protein interactions from known physical complexes were included, whereas those with only functional associations were excluded. The highest confidence level (0.9 on a scale of 0–1.0) was set as a cut-off for the minimum required interaction score. Enriched networks and pathways were searched with STRING 2023 database version 12.0.
Telomerase pathway and alternative lengthening pathway assessment
Because direct quantification of telomerase is difficult because of its very low expression level, we inferred the activation level of the telomerase-dependent telomere lengthening pathway (TEL) and alternative lengthening pathway (ALT) using the ssGSEA algorithm and TelNet database, which contains gene lists of both TEL-related and ALT-related genes. To infer telomerase (TEL) and ALT pathway activation status, we created custom gene sets based on the TelNet database64, which provides functional connectivity for telomere lengthening mechanisms. Based on the TelNet database, we created a TEL pathway gene set (185 genes named TelNET_TEL gene set in this study) and an ALT pathway gene set (152 genes named TelNET_ALT gene set in this study) and then calculated the ssGSEA score and added 10 to each score for each sample to make scores positive while keeping data distribution invariant.
Immune score calculation
Using the ESTIMATE package (v. 1.0.13) in R (https://bioinformatics.mdanderson.org/public-software/estimate/), we calculated immune scores from the normalized protein expression matrix of all samples.
Immune cell type enrichment deconvolution
To infer the immune cell type composition of each sample, we used the xCell tool65, which performs cell type enrichment analysis based on gene expression. We input the protein abundance matrix of this study into xCell using the expression level ranking.
Cell type enrichment analysis
Using the differential expression analysis between ACC and PBL together with an MSigDB cell type signature gene set (C8 gene set containing 830 gene sets), we performed GSEA and calculated the NES of cell type signatures.
Connectivity map analyses for drug repurposing
The top 150 upregulated and downregulated valid gene names between ACC and benign pancreatic tissue or PBL and benign pancreatic tissue were uploaded to the CLUE webtool, which executes a connectivity map analysis using 1 million curated expression profiles18. We then ran a query against the gene expression (L1000) dataset (version 1.0) and obtained the CMap connectivity score, ranging from −100 to 100, for 2429 small compounds.
General statistical analysis
The Mann–Whitney U test was used for continuous variables unless otherwise specified. All statistical analyses were performed using R (v4.3.2). Boxplots show 25th, 50th (bar), and 75th percentiles with whiskers indicating ±1.5 × IQR. All statistical analyses were considered significant with p < 0.05.
Data availability
The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD036998.
References
Thompson, E. D. & Wood, L. D. Pancreatic neoplasms with acinar differentiation: a review of pathologic and molecular features. Arch. Pathol. Lab. Med. 144, 808–815 (2020).
Klimstra, D. S., Heffess, C. S., Oertel, J. E. & Rosai, J. Acinar cell carcinoma of the pancreas. A clinicopathologic study of 28 cases. Am. J. Surg. Pathol. 16, 815–837 (1992).
Gu, W. Z., Zou, C. C., Zhao, Z. Y., Liang, L. & Tang, H. F. Childhood pancreatoblastoma: clinical features and immunohistochemistry analysis. Cancer Lett. 264, 119–126 (2008).
Chmielecki, J. et al. Comprehensive genomic profiling of pancreatic acinar cell carcinomas identifies recurrent RAF fusions and frequent inactivation of DNA repair genes. Cancer Discov. 4, 1398–1405 (2014).
Jiao, Y. et al. Whole-exome sequencing of pancreatic neoplasms with acinar differentiation. J. Pathol. 232, 428–435 (2014).
Furukawa, T. et al. Whole exome sequencing reveals recurrent mutations in BRCA2 and FAT genes in acinar cell carcinomas of the pancreas. Sci. Rep. 5, 8829 (2015).
Abraham, S. C. et al. Genetic and immunohistochemical analysis of pancreatic acinar cell carcinoma: frequent allelic loss on chromosome 11p and alterations in the APC/beta-catenin pathway. Am. J. Pathol. 160, 953–962 (2002).
Liu, W. et al. DNA mismatch repair abnormalities in acinar cell carcinoma of the pancreas: frequency and clinical significance. Pancreas 43, 1264–1270 (2014).
Abraham, S. C. et al. Distinctive molecular genetic alterations in sporadic and familial adenomatous polyposis-associated pancreatoblastomas: frequent alterations in the APC/beta-catenin pathway and chromosome 11p. Am. J. Pathol. 159, 1619–1627 (2001).
Isobe, T. et al. Integrated molecular characterization of the lethal pediatric cancer pancreatoblastoma. Cancer Res. 78, 865–876 (2018).
Liu, T., Zhao, T., Shi, C. & Chen, L. Pancreatoblastoma in children: clinical management and literature review. Transl. Oncol. 18, 101359 (2022).
Ikezawa, K. et al. Comprehensive review of pancreatic acinar cell carcinoma: epidemiology, diagnosis, molecular features and treatment. Jpn. J. Clin. Oncol. 54, 271–281 (2024).
Ho, W. J., Jaffee, E. M. & Zheng, L. The tumour microenvironment in pancreatic cancer—clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 17, 527–540 (2020).
Orth, M. et al. Pancreatic ductal adenocarcinoma: biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 14, 141 (2019).
Luo, W., Wen, T. & Qu, X. Tumor immune microenvironment-based therapies in pancreatic ductal adenocarcinoma: time to update the concept. J. Exp. Clin. Cancer Res. 43, 8 (2024).
Xu, J., Lamouille, S. & Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 19, 156–172 (2009).
Noureen, N. et al. Integrated analysis of telomerase enzymatic activity unravels an association with cancer stemness and proliferation. Nat. Commun. 12, 139 (2021).
Subramanian, A. et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 171, 1437–1452.e1417 (2017).
Haumaitre, C., Lenoir, O. & Scharfmann, R. Histone deacetylase inhibitors modify pancreatic cell fate determination and amplify endocrine progenitors. Mol. Cell Biol. 28, 6373–6383 (2008).
Takai, N., Hamanaka, R., Yoshimatsu, J. & Miyakawa, I. Polo-like kinases (Plks) and cancer. Oncogene 24, 287–291 (2005).
Liu, Z., Sun, Q. & Wang, X. PLK1, a potential target for cancer therapy. Transl. Oncol. 10, 22–32 (2017).
Wang, Q. & Reid, M. D. Cytopathology of solid pancreatic neoplasms: an algorithmic approach to diagnosis. Cancer Cytopathol. 130, 491–510 (2022).
Masugi, Y. The desmoplastic stroma of pancreatic cancer: multilayered levels of heterogeneity, clinical significance, and therapeutic opportunities. Cancers 14, 3293 (2022).
Song, Y., Wang, L., Wang, K., Lu, Y. & Zhou, P. COL12A1 acts as a novel prognosis biomarker and activates cancer-associated fibroblasts in pancreatic cancer through bioinformatics and experimental validation. Cancers 15, 1480 (2023).
Romanish, M. G. & Seethala, R. R. True oncocytic acinic cell carcinoma: a case image. Head. Neck Pathol. 17, 883–885 (2023).
Lompo, O. et al. [Solid and pseudopapillary tumor of the pancreas: immunohistochemical and ultrastructural study of 2 pediatric cases]. Ann. Pathol. 20, 221–224 (2000).
Fassone, E. & Rahman, S. Complex I deficiency: clinical features, biochemistry and molecular genetics. J. Med. Genet. 49, 578–590 (2012).
Saada, A. et al. Mutations in NDUFAF3 (C3ORF60), encoding an NDUFAF4 (C6ORF66)-interacting complex I assembly protein, cause fatal neonatal mitochondrial disease. Am. J. Hum. Genet. 84, 718–727 (2009).
Bach, A. W. et al. cDNA cloning of human liver monoamine oxidase A and B: molecular basis of differences in enzymatic properties. Proc. Natl Acad. Sci. USA 85, 4934–4938 (1988).
Van der Werf, P., Orlowski, M. & Meister, A. Enzymatic conversion of 5-oxo-L-proline (L-pyrrolidone carboxylate) to L-glutamate coupled with cleavage of adenosine triphosphate to adenosine diphosphate, a reaction in the -glutamyl cycle. Proc. Natl Acad. Sci. USA 68, 2982–2985 (1971).
Hoefler, G. et al. cDNA cloning of the human peroxisomal enoyl-CoA hydratase: 3-hydroxyacyl-CoA dehydrogenase bifunctional enzyme and localization to chromosome 3q26.3-3q28: a free left Alu Arm is inserted in the 3’ noncoding region. Genomics 19, 60–67 (1994).
Jaeken, J. Congenital disorders of glycosylation. Handb. Clin. Neurol. 113, 1737–1743 (2013).
Kim, A. Y. et al. Pitfalls in the diagnosis of hereditary fructose intolerance. Pediatrics 146, e20193324 (2020).
Wortmann, S. B. et al. CLPB mutations cause 3-methylglutaconic aciduria, progressive brain atrophy, intellectual disability, congenital neutropenia, cataracts, movement disorder. Am. J. Hum. Genet. 96, 245–257 (2015).
Ostergaard, E. et al. Mutations in COA3 cause isolated complex IV deficiency associated with neuropathy, exercise intolerance, obesity, and short stature. J. Med. Genet. 52, 203–207 (2015).
Ghafouri-Fard, S. et al. A review on the role of cyclin dependent kinases in cancers. Cancer Cell Int. 22, 325 (2022).
Park, C. S. et al. Modulation of beta-catenin phosphorylation/degradation by cyclin-dependent kinase 2. J. Biol. Chem. 279, 19592–19599 (2004).
Li, Z. et al. Oncogenic NRAS, required for pathogenesis of embryonic rhabdomyosarcoma, relies upon the HMGA2-IGF2BP2 pathway. Cancer Res. 73, 3041–3050 (2013).
Zhang, L. et al. Nestin-positive progenitor cells isolated from human fetal pancreas have phenotypic markers identical to mesenchymal stem cells. World J. Gastroenterol. 11, 2906–2911 (2005).
Ohike, N. et al. Immunohistochemical analysis of nestin and c-kit and their significance in pancreatic tumors. Pathol. Int. 57, 589–593 (2007).
Bien, E. et al. Pancreatoblastoma in children: EXPeRT/PARTNER diagnostic and therapeutic recommendations. Pediatr. Blood Cancer 68, e29112 (2021).
Marchione, D. M. et al. HYPERsol: high-quality data from archival FFPE tissue for clinical proteomics. J. Proteome Res. 19, 973–983 (2020).
Tanaka, A. et al. DEAD-box RNA helicase protein DDX21 as a prognosis marker for early stage colorectal cancer with microsatellite instability. Sci. Rep. 10, 22085 (2020).
Tanaka, A. et al. Maspin as a prognostic marker for early stage colorectal cancer with microsatellite instability. Front. Oncol. 10, 945 (2020).
Tanaka, A. et al. STAT1 as a potential prognosis marker for poor outcomes of early stage colorectal cancer with microsatellite instability. PLoS ONE 15, e0229252 (2020).
Tanaka, A. et al. Prolyl 4-hydroxylase alpha 1 protein expression risk-stratifies early stage colorectal cancer. Oncotarget 11, 813–824 (2020).
Tanaka, A. et al. Proteogenomic characterization of primary colorectal cancer and metastatic progression identifies proteome-based subtypes and signatures. Cell Rep. 43, 113810 (2024).
Clark, D. J. et al. Integrated proteogenomic characterization of clear cell renal cell carcinoma. Cell 179, 964–983.e931 (2019).
Gao, Q. et al. Integrated proteogenomic characterization of HBV-related hepatocellular carcinoma. Cell 179, 561–577.e522 (2019).
Vasaikar, S. et al. Proteogenomic analysis of human colon cancer reveals new therapeutic opportunities. Cell 177, 1035–1049.e1019 (2019).
Chen, Y. J. et al. Proteogenomics of non-smoking lung cancer in East Asia delineates molecular signatures of pathogenesis and progression. Cell 182, 226–244.e217 (2020).
Gillette, M. A. et al. Proteogenomic characterization reveals therapeutic vulnerabilities in lung adenocarcinoma. Cell 182, 200–225.e235 (2020).
Krug, K. et al. Proteogenomic landscape of breast cancer tumorigenesis and targeted therapy. Cell 183, 1436–1456.e1431 (2020).
Satpathy, S. et al. A proteogenomic portrait of lung squamous cell carcinoma. Cell 184, 4348–4371.e4340 (2021).
Cheng, D. T. et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagn. 17, 251–264 (2015).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Cox, J. et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 (2011).
Cox, J. et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell Proteom. 13, 2513–2526 (2014).
Tusher, V. G., Tibshirani, R. & Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl Acad. Sci. USA 98, 5116–5121 (2001).
Tyanova, S. et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016).
Storey, J. D., Bass, A. J., Dabney, A. & Robinson, D. qvalue: Q-value estimation for false discovery rate control. GitHub http://github.com/jdstorey/qvalue (2024).
Barbie, D. A. et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112 (2009).
Szklarczyk, D. et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638–D646 (2023).
Braun, D. M., Chung, I., Kepper, N., Deeg, K. I. & Rippe, K. TelNet—a database for human and yeast genes involved in telomere maintenance. BMC Genet. 19, 32 (2018).
Aran, D., Hu, Z. & Butte, A. J. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 18, 220 (2017).
Acknowledgements
M.H.R. acknowledges grants from the NIH/NCI (R21 CA251992, R21 CA263262, and U01 CA263986), a Cycle for Survival Equinox Innovation Grant, an Investigator Grant from the Neuroendocrine Tumor Research Foundation (NETRF), and support from the Farmer Family Foundation. Parts of the study were supported by the MSKCC NCI Cancer Center Support Grant (P30 CA008748). The funding bodies were not involved in the design of the study and the collection, analysis, and interpretation of data.
Author information
Authors and Affiliations
Contributions
A.T. performed the experiments, data analyses, and manuscript drafting. M.O. and Y.Z. helped with the experiments and data analysis. R.C.H., M.M.M., and Z.L. supported mass spectrometric experiments and data analyses. D.S.K. consulted on the study and provided partial funding. J.Y.W. consulted on the study, analyzed the data, and co-wrote the manuscript. M.H.R. designed and supervised the study, analyzed the data, wrote and edited the manuscript, and provided funding.
Corresponding authors
Ethics declarations
Competing interests
A.T., J.Y.W., Y.Z., M.O., and R.C.H. declare no conflicts of interest related to this study. D.S.K. is a consultant for and equity holder in Paige.AI and a consultant for Merck. M.H.R. is a member of the Scientific Advisory Board of Universal Diagnostics (UDX). None of these companies had any role in the support, design, execution, data analysis, or any other aspect of this study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tanaka, A., Ogawa, M., Zhou, Y. et al. Proteomic basis for pancreatic acinar cell carcinoma and pancreatoblastoma as similar yet distinct entities. npj Precis. Onc. 8, 221 (2024). https://doi.org/10.1038/s41698-024-00708-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-024-00708-5
