
Abstract
Epilepsy is one of the most common symptoms in patients with gliomas; however, the mechanisms underlying its interaction are not yet clear. Moreover, epidemiological studies have not accurately identified patients with glioma-related epilepsy (GRE), and there is an urgent need to identify the molecular mechanisms and markers of its occurrence. We analyzed the demographics, transcriptome, whole-genome, and methylation sequences of 997 patients with glioma, to determine the genetic differences between glioma and GRE patients and to determine the upregulated molecular function, cellular composition, biological processes involved, signaling pathways, and immune cell infiltration. Twelve machine learning algorithms were refined into 113 combinatorial algorithms for building diagnostic recognition models. A total of 342 patients with GRE were identified with WHO grade 2 (174), grade 3 (107), and grade 4 (61). The mean age of the patients with GREs, with IDH mutations (n = 217 [63%]) and 1p19q non-codeletion (n = 169 [49%]), was 38 years old. GRE molecular functions were mainly passive transmembrane transporter protein activity, ion channel activity, and gated channel activity. Cellular components were enriched in the cation-channel and transmembrane transporter complexes. Cerebral cortical development regulates the membrane potential and synaptic organization as major biological processes. The signaling pathways mainly focused on cholinergic, GABAergic, and glutamatergic synapses. LASSO, combined with Random Forest, was the best diagnostic model and identified nine diagnostic genes. This study provides new insights and future perspectives for resolving the molecular mechanisms of GRE.
Similar content being viewed by others


Introduction
Epilepsy is the most common symptom of patients with glioma1. Glioma-related epilepsy (GRE) is influenced by the tumor ___location, histopathology, peritumoral microenvironmental features, and specific genetic alterations2,3,4,5. The probability of epilepsy varies according to the grade of the glioma6. Patients with low-grade gliomas have a 60–90% probability of epilepsy7, while those with high-grade gliomas have a 30–40% probability of epilepsy8. Compared to patients without epilepsy in the past, patients with glioma who had epilepsy before the operation had significant survival benefits9. Therefore, it is necessary to summarize and analyze the epidemiological and clinical characteristics of a large sample of patients with preoperative glioma-related epilepsy.
Abnormal cell proliferation and spread in gliomas lead to damage to neurons and neural networks, which may be an important mechanism of epilepsy10,11. Glioma cells are involved in the development of seizures through several mechanisms. They can secrete a variety of neurotransmitters or modulate neurotransmitter metabolism, such as glutamate and gamma-aminobutyric acid (GABA), thereby altering the excitatory and inhibitory balance between neurons and contributing to epilepsy12,13. Additionally, the inflammatory response around gliomas is believed to be related to the occurrence of epilepsy14,15. The release of inflammatory factors can lead to neuronal hyperactivity and destabilization of neural networks, increasing the risk of epilepsy16,17. Some signaling pathways are associated with the proliferation and survival of glioma cells and may influence epileptogenesis. For example, signaling pathways such as PI3K/Akt, MAPK, and EGFR are often activated in gliomas, and aberrant activation of these pathways may lead to cellular dysfunction and promote epilepsy18,19. Immune responses, synaptic transmission via GABA, cell cycle control, and DNA repair are involved in glioma-related epileptogenesis20,21,22. However, few studies have been conducted to comprehensively analyze the pathogenesis of glioma-related epilepsy using tumor tissue sequencing.
With the rapid development in computer technology, machine learning has become increasingly common in the medical field. Disease identification is important, and machine learning offers various linear models and decision trees for prediction, and outperforms traditional statistical methods23,24,25. A radiological prediction model was developed for the epileptic phenotype associated with GRE, using imaging features of patients with epilepsy and radiological mapping of seizure-prone regions26,27. However, few transcriptome analyses have addressed GRE versus non-epilepsy19. Additionally, the genetic background of tumor-associated epilepsy remains unknown, and few studies have investigated the genetic variability associated with GRE. Some susceptibility genes associated with epilepsy may be involved in tumor-related epilepsy28,29. One study demonstrated the potential mechanism of infiltration and provided evidence of direct physical interactions between microglia and T cells by single-cell sequencing of epileptic tissue, recruiting immune cells to create an immune microenvironment for epilepsy30. Therefore, it is particularly important to construct a model to predict the occurrence of epilepsy in patients with gliomas. Additionally, more research is needed to demonstrate that epilepsy genes affect the glioma tumor microenvironment.
The development of GRE involves various factors, including tumor ___location, tumor histopathology, peritumoral microenvironmental features, and specific genetic alterations. However, these have not yet been analyzed using large samples of transcriptomic and clinical data. In the present study, we performed a systematic investigation to reveal the genetic mechanisms of GRE genesis, and subsequently identified a GRE gene diagnostic model using transcriptome samples from 993 cases of GRE and non-epileptic tumor tissues. Probing differentially expressed genes, gene mutations, methylation, and immune cell infiltration, we hypothesized that it may be possible to individualize drugs for the treatment of patients with GREs. Finally, single-cell sequencing (scRNA-seq) was used to explore the relationship between the model genes and the tumor microenvironment.
Results
Baseline patient characteristics
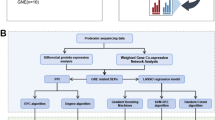
The research process was divided into six steps (Fig. 1). As shown in Supplement Table 1, we performed a comparative analysis of the clinical information between patients with GRE and those without epilepsy. Of 342 patients with GRE, 174 were WHO grade 2 (51%), 107 were WHO grade 3 (31%), and 61 were WHO grade 4 (18%). A total of 655 patients with glioma did not develop epilepsy preoperatively, 127 WHO grade 2, 188 WHO grade 3, and 340 WHO 4 grade (p < 0.001). The mean age of patients who developed glioma-related epilepsy was 38 years (32–45) and 45 years (37–54) for those who did not develop GRE (p < 0.001). Regular postoperative follow-up revealed that patients with GRE had a better prognosis: 162 (47%) patients survived and 164 (48%) died during follow-up; while 175 (27%) without epilepsy survived, and 461 (70%) died (p < 0.001). Meanwhile, 217 (63%) patients with GRE had IDH mutations, 105 (31%) had wild-type GRE, and 20 (6%) unknown. Of the patients without epilepsy, 257 (39%) had IDH mutations, 368 (56%) had wild-type IDH, and 30 (5%) unknown. Similarly, the 1p19q codeletion status in molecular pathology differed. in patients with GRE, 81 (24%) developing the deletion, 169 (49%) without codeletion, and 92 (27%) unknown. GRE did not differ according to sex, MGMT methylation, or sequencing platforms (p > 0.05).

The technology roadmap consists of a total of six steps.
In the RNA-seq 693 cohort, 993 upregulated and 424 downregulated genes were identified (Supplement Fig. 1a). There were 649 upregulated and 1710 downregulated genes in cohort 325 (Supplement Fig. 1b). Moreover, 408 upregulated and 582 downregulated genes were identified in cohort 301 (Supplement Fig. 1c). GRE differential signaling pathway analysis was also performed, and we found upregulation of metabolic signaling pathways, such as steroid biosynthesis, pentose and glucuronate interconversions, glycine serine and threonine metabolism, glycosphingolipid biosynthesis, lacto and neolacto series, and sphingolipid metabolism (Supplement Fig. 1d). Meanwhile, the up-regulated signaling pathways, Notch signaling pathway, WNT signaling pathway, TGF-βsignaling pathway, and olfactory transduction were found (Supplement Fig. 1d). Downregulated metabolic signaling pathways included nicotinate and nicotinamide metabolism, tryptophan metabolism, glycan biosynthesis, glutathione metabolism, nitrogen metabolism, and propanoate metabolism (Supplement Fig. 1d). The signaling pathways that were also downregulated were the JAK-STAT signaling pathway, cytokine receptor interaction, natural killer cell-mediated cytotoxicity, and hedgehog signaling (Supplement Fig. 1d). Subsequently, we presented the analysis workflow for glioma-associated epilepsy and glioma patients (Supplement Fig. 1e). GSEA enrichment analysis also revealed that the signaling pathways activated in GRE were ribosome biosynthesis, terpenoid backbone biosynthesis, oxidative phosphorylation, and butanoate metabolism (Supplement Fig. 1f). Inhibitory signaling pathways included amino sugar and nucleotide sugar metabolism, the p53 signaling pathway, cytokine-cytokine receptor interaction, cell cycle, and the JAK-STAT signaling pathway (Supplement Fig. 1f).
Identification of glioma-related epilepsy co-expressed genes and functional enrichment
We analyzed RNA-seq 693 cohorts and found that GRE differed from glioma patients in terms of PRS type, IDH mutation, and 1p19q co-deletion (p < 0.05) (Fig. 2A). To identify genes for GRE, we performed a weighted gene co-expression network analysis (Fig. 2B, C). We found the MEmagenta module to be the most relevant module for GRE, with 852 genes. The magenta module was positively correlated with epilepsy (r = 0.33, p = 0.00015) (Fig. 2D). The MEbrown module was positively associated with non-epilepsy (r = 0.46, p < 0.001) (Fig. 2E). Separate GO and KEGG enrichment analyses were performed. Molecular function enrichment of the GRE genes revealed passive transmembrane transporters, channels, ion channels, cation channels, and gated channel activities. Cellular component enrichment revealed the presence of synaptic membranes, cation channels, transporter, and transmembrane transporter complexes. Cerebellar cortex development, membrane potential regulation, and synaptic organization were the main biological processes (Fig. 2F). The KEGG enrichment analysis revealed nicotine addiction, cholinergic synapses, GABAergic synapses, and glutamatergic synapses (Fig. 2G). Additionally, enrichment analysis of glioma patients without epilepsy revealed that MHC class II protein complex binding, integrin binding, growth factor binding, collagen binding, and extracellular matrix structural constituents were the main molecular functions. Cellular component enrichment showed cell–substrate junctions, focal adhesions, endoplasmic reticulum lumen, vesicle lumen, cytoplasmic vesicle lumen, secretory granule lumen, and collagen. Biological process enrichment analysis revealed external encapsulating structural organization, extracellular structural organization, extracellular matrix organization, collagen-containing extracellular matrix, and extracellular matrix organization (Fig. 2H). Finally, KEGG enrichment analysis showed that the main focus was on the ECM–receptor interaction, phagosome, and NF-kappa B signaling pathways (Fig. 2I).

A Differences in clinically relevant characteristics. B Hierarchical clustering trees to construct gene modules. C Different gene modules associated with glioma-related epilepsy D, E Correlation of magenta (D) and brown (E) modules with glioma-related epilepsy. F, G Glioma-related epilepsy magenta gene module Go and KEGG enrichment analysis. H, I Go and KEGG enrichment analysis of brown gene modules in gliomas without epilepsy.
Identifying glioma-related epilepsy immune landscape and expression
Immune cells are an important subgroup of cells in the tumor microenvironment. The heat map showed that most patients had low immune cell infiltration, IDH mutations, and 1p19q deletions (Fig. 3A). Thirteen immune function score analyses showed that APC costimulation, CCR, checkpoint, cytolytic activity, HLA, Inflammation-promoting, MHC class I, parainflammation, T cell costimulation, and Type I IFN response were expressed at low levels in patients with GRE (Fig. 3B). Next, an immune checkpoint analysis was performed. ADORA2A, BTNL9, C10ORF54, and SIRPA were highly expressed in GRE patients compared to glioma patients without epilepsy (p < 0.05). BTN2A1, BTN2A2, CD274, CD276, CD80, IDO1, PDCD1, PDCD1LG2. PVR, TDO2, TNFRSF14, and VTCN were lower in patients with GRE (p < 0.05) (Fig. 3C). Compared to glioma patients without epilepsy, T cell follicular helper, activated CD4 T cell, activated CD8 T cell, activated dendritic cell, central memory CD4 T cell, central memory CD8 T cell, effector memory CD8 T cell, mast cell, MDSC memory B cell, natural killer cell, natural killer T cell, neutrophil, plasmacytoid dendritic cell, regulatory T cell, T follicular helper cell, Type 1 T helper cell, Type 17 T helper cell and Type 2 T helper cell were low infiltration in GRE patients (p < 0.05) (Fig. 3D, E).

A Differences in clinical features among patients with high and low immune cell infiltrating gliomas. B Differential expression of 13 immune cell function scores. C Differential expression of immune checkpoints in patients with glioma-related epilepsy and glioma. D, E Assessment of immune cell infiltration using CIBERSORT (D) and ssGSEA (E) algorithms. The center line represents the median, the bounds of the box indicate the interquartile range (IQR) from the 25th to 75th percentile, and the whiskers extend to 1.5 times the IQR from the 25th and 75th percentiles. Error bars represent the standard deviation (SD). (*p < 0.05, **p < 0.01, ***p < 0.001).
Metabolic signaling pathways in glioma-related epilepsy
The ssGSEA scores showed lower hypoxia and glycolytic metabolism scores in GRE patients (p < 0.001) (Supplement Fig. 2a). Tumor purity is a measure of the proportion of malignant cells in a tumor sample. Patients with GRE had higher tumor purity scores than those without epilepsy (Supplement Fig. 2b). Meanwhile, after CAMP database analysis, we found that AH.6809, TTNPB, imatinib, X5109870, and MS.275 were the top five most effective drugs for the personalized treatment of possible GRE (Supplement Fig. 2c). The stromal, immune, and estimated scores were lower in patients with GRE than in patients with non-epileptic glioma (p < 0.05) (Supplement Fig. 2d). In addition, MS.275 as a histone inhibitor, has been used as an important drug for combination immunosuppressant therapy in glioma clinical drug trials31,32,33,34. Sodium valproate is a histone inhibitor used to prevent epilepsy.
Identification of glioma-related epilepsy gene mutations and methylated genes
In our study, we found that in patients with GRE, gene mutations were identified in IDH1 (64%), TP53 (56%), ATRX (28%), CIC (21%), MUC16 (10%), EGFR (5%), TNN (3%), PTEN (3%), and NF1 (3%) (Supplement Fig. 3a). Gene mutations in non-epileptic glioma patients included TP53 (35%), IDH1 (30%), ATRX (23%), PTEN (14%), CIC (11%), NF1 (9%), EGFR (7%), PIK3CA (7%), and MUC16 (3%) (Supplement Fig. 3b). Methylated gene differential analysis was also shown in GRE patients, with cg21098323, cg13649728, cg19923810, cg26426582, and cg18992201 as downregulated genes and cg10035303 as an upregulated gene (p < 0.05) (Supplement Fig. 3c). Methylated genes were analyzed for GSEA enrichment. The activated signaling pathways were mismatch repair, RNA degradation, cell cycle, pyrimidine metabolism, and terpenoid backbone biosynthesis. The inhibited signaling pathways included linoleic acid metabolism, steroid hormone biosynthesis, the intestinal immune network for IGA production, and olfactory transduction (Supplement Fig. 3d).
Machine learning identifies patients with glioma-related epilepsy
To identify genes that may influence the occurrence of GRE by transcriptome sequencing, 113 combinations of 12 machine-learning algorithms were screened. We constructed a Wayne diagram using three cohorts of differential genes, and the epilepsy genes looked for in WGCNA by taking the intersection; a total of 33 genes were obtained (Fig. 4A). As shown in the forest plot, all genes were risk genes for glioma prognosis by one-way Cox analysis except VEPH1 (Fig. 4B). The model constructed by LASSO and Random Forest had an AUC value of 0.999 in the training set of the RNA-seq 693 cohorts. In the test cohort, the AUC of cohorts 301 and 325 were 0.685 and 0.692, respectively, with an average of 0.792 (Fig. 4C). After screening, model genes were VEPH1, ABCC3, COL5A2, VEGFA, EMP3, GOS2, GDF15, ANXA1, and TAGLN2. The LASSO screening process is shown in Fig. 4D, E). Random forests showed the weights of the model genes (Fig. 4F). Moreover, all model genes were differentially expressed in GRE and non-epilepsy glioma patients (p < 0.001) (Fig. 5A). Except for TAGLN2, all genes were highly expressed in glioma patients with poor prognosis (Supplement Fig. 4a-i). The diagnostic model genes were associated with hypoxia, glycolysis, and xenobiotic metabolism signaling pathways (p < 0.001) (Fig. 5B). Among the diagnostic model genes, the correlation coefficients between COL5A2 and VEGFA, ANXA1 and TAGLN2, GDF15, and TAGLN2 were 0.79, 0.82, and 0.79, respectively (Fig. 5C). We found that VEPH1 was negatively correlated with most immune cells, whereas the remaining model genes were positively correlated with most immune cells (Fig. 5D).

A Wenn diagram showing the intersection of differential and WGCNA co-expressed genes. B Forest plot showing a one-way Cox analysis of 33 genes. C AUC in training cohort RNA-seq 693 and validation cohorts 301 and 325 in different machine learning algorithms. D, E In the RNA-seq 693 cohort (n = 584), the optimal λ value is selected and filtered to obtain the most useful model gene. F The importance of nine model genes in the Random Forest model. The center line represents the median, the bounds of the box indicate the interquartile range (IQR) from the 25th to 75th percentile, and the whiskers extend to 1.5 times the IQR from the 25th and 75th percentiles. Error bars represent the 95% confidence intervals (CI). (*p < 0.05, **p < 0.01, ***p < 0.001).

A Beeswarm showing the difference in diagnostic model gene expression between glioma-related epilepsy and non-epilepsy. B Heatmap showing diagnostic genes correlate with metabolic signaling pathways. C Correlation analysis between model genes. D Heat map showing the correlation between model genes and immune cells. The center line represents the median, the bounds of the box indicate the interquartile range (IQR) from the 25th to 75th percentile, and whiskers extend to 1.5 times the IQR from the 25th and 75th percentiles. Error bars represent the standard deviation (SD). (*p < 0.05, **p < 0.01, ***p < 0.001).
Identification of glioma-related epilepsy genes and tumor immune microenvironment
Furthermore, we analyzed the expression of GRE in five cell subpopulations in the glioma microenvironment. As shown in Fig. 6A, the heatmap shows our subpopulations of marker genes, microglial cells (CSF1R, CX3CR1, P2RY12, and TMEM119), macrophages (CD163 and CD68), neutrophils (FPR2, CXCR2, and IL1R2), endothelial cells (VWF, CD34, FLT1, and CLDN5), and T cells (CD8A, CD3E, and CD3D). Additionally, we visualized each subpopulation of cells using UMAP while showing the number of cells. The 52,387 cells in the adjacent normal tissue and 60,707 cells in the tumor tissue were shown by UMAP (Fig. 6B). UMAP revealed the distribution and composition of each cell subgroup (Fig. 6C). Compared to the adjacent normal tissue, the proportion of microglia and endothelial cells decreased and the proportion of macrophages and neutrophils increased in the tumor tissue (Fig. 6D). We also analyzed the expression of diagnostic model genes in different tumor microenvironment cells. Most model genes were expressed in the tumor tissue microenvironment (Fig. 6E, F). In addition, G0S2 was expressed in all the five cell subpopulations. Compared to adjacent normal microenvironment cells, ANXA1 was more highly expressed in microglial cells, macrophages, endothelial cells, and T cells in the tumor microenvironment. EMP3 is highly expressed in microglia and endothelial cells. VEGFA was mainly expressed in macrophages and neutrophils (Fig. 6F). Moreover, we found that TGF-β had the highest activation activity in cell subpopulations, while high activation was in endothelial cells (Supplement Fig. 5a). Subsequently, we performed cell trajectory extrapolation in the tumor microenvironment to simulate cell dynamics (Supplement Fig. 5b). Endothelial cells, as the starting differentiated cells, gradually differentiated into other tumor microenvironment cells over time (Supplement Fig. 5c). After inferCNV analysis, we classified endothelial cells as malignant or benign cells (Supplement Fig. 5d). GSEA enrichment analysis of benign and malignant cells showed that the highly expressed signaling pathways were involved in focal adhesion, protein digestion and absorption, and ECM-receptor interactions (Supplement Fig. 5e). Mapping of the expression of diagnostic model genes through AUCell revealed that diagnostic model-related genes were highly expressed in malignant cells (Supplement Fig. 5f).

A Heatmap showing cell maker gene expression in different subtypes of cells. B UMAP plot showing expression of tumor microenvironment cells in adjacent normal and tumor tissues. C UMAP plot showing expression of maker genes in subpopulation cells. D Comparison of cell subpopulation ratios in adjacent normal and tumor tissues. E Diagnostic model gene expression of cell subpopulations in adjacent normal tissues. F Diagnostic model gene expression of cell subpopulations in tumor tissues. The center line represents the median, the bounds of the box indicate the interquartile range (IQR) from the 25th to 75th percentile, and whiskers extend to 1.5 times the IQR from the 25th and 75th percentiles. Outliers beyond this range are plotted individually.
Verification of model gene expression in vivo and in vitro
We analyzed the expression of model genes in the glioma cell lines U251 and T98G, and normal human astrocyte line SVGp12 using quantitative polymerase chain reaction (qPCR). Compared to normal cells, ANXA1, EMP3, GDF15, TAGLN2, VEGFA, and VEPH1 were highly expressed in tumor cell lines (p < 0.05)(Supplement Fig. 6a, b). Immunohistochemistry revealed that the expression levels of ABCC3, ANAX1, COL5A2, EMP3, GDF15, G0S2, and TAGLN2 were higher in the epilepsy group than in the non-epilepsy group, whereas the expression of VEPH1 was lower in the epilepsy group than in the non-epilepsy group (p < 0.05) (Supplement Fig. 6c, d).
Discussion
We collected transcriptomic and clinically relevant information on the largest number of patients with GRE to date and analyzed genetics, mutations, methylation, drug prediction analyses, and machine learning to identify diagnostically relevant genes, as well as cellular identifications that may influence the immune microenvironment of epilepsy. Thus, in the present study, we identified synaptic or ion channel-related signaling pathways that may contribute to the development of GRE. A predictive model capable of diagnosing glioma-associated epilepsy was constructed using various machine-learning algorithms. Moreover, we found that most model-related genes were centrally expressed in malignant endothelial cells. This may have led to an earlier onset of symptoms and resection of the lesions.
In the present study, we found that multiple signaling pathways were upregulated in patients with GRE, such as metabolism-related signaling pathways steroid biosynthesis, pentose and glucuronate interconversions, glycine serine and threonine metabolism, glycosphingolipid biosynthesis lacto and neolacto series, oxidative phosphorylation, and sphingolipid metabolism. In a metabolomic review on epilepsy published by Lai et al., alanine, aspartate, and glutamate metabolism; glycine, serine, and threonine metabolism; glycerophospholipid metabolism; glyoxylate and dicarboxylic acid metabolism; and arginine and proline metabolism were found to be involved in epilepsy. This is consistent with the findings of our study that metabolic signaling pathways may be involved in the GRE. Patients and animal models of GRE were well controlled after treatment with glutamate receptor antagonists35,36,37. Pyruvate carboxylase, pyruvate carboxylase, and glutamine synthetase have also been shown to be expressed in GRE tumor tissue38. Synapse-associated signals, such as nicotine addiction, cholinergic synapses, GABAergic synapses, and glutamatergic synapses, were enriched in our study. Thus, GRE genes may promote synapse formation. It has also been demonstrated that GRE genes promote excitatory synapse formation, leading to hyperexcitability of the cortical network surrounding the tumor, which may provide new insights into the epileptogenic mechanisms of GRE22.
Patients with GRE are usually predicted to have a lower tumor grade and better prognosis. Patients with concurrent epilepsy usually have a molecular genetic profile of IDH mutations combined with 1p19q deletions19. This finding is consistent with the results of our analysis. Low-grade gliomas are slow-growing and epileptogenic, and epilepsy is the most common initial symptom in patients39. Previous studies have found that IDH mutations lead to increased release of D-2-hydroxysteroids in the tumor cell microenvironment and that the high structural similarity of D-2-hydroxysteroids to the excitatory neurotransmitter glutamate, which increases neuronal activity and incidence of epilepsy in patients with isocitrate dehydrogenase 1 mutant gliomas40. The co-occurrence of the IDH mutation and combined 1p19q deletion is important for the diagnosis of oligodendroglioma. Patients with oligodendrogliomas containing oligodendroglial components and those with gliomas containing IDH mutations are at a high risk of developing epilepsy, although all of the above risk factors need to be clarified by pathological examination. Additionally, our study found that the average age of patients who underwent the GRE was 38 years. Therefore, in conjunction with previous literature, having the following risk factors may help physicians and patients make an early prognosis of susceptibility to preoperative GRE: age < 38 years, ___location of the glioma in the frontotemporal lobe functional area, and imaging findings suggestive of cortical involvement of the glioma41.
To further identify genetic models for diagnosing patients with GRE. Using 113 combinations of 12 machine learning algorithms, the combination of LASSO and Random Forest was identified as the optimal model, significantly reducing the dimensionality of the variables, revealing potential patterns, and contributing to the creation of a simplified and translatable model for clinical applications. As a result, the nine identified gene features accurately predicted GRE. The identified model bases were strongly correlated with metabolic signaling pathways, such as oxidative stress, glucose metabolism, and immune cell infiltration. These genes may provide a direction for further analyses of the mechanisms underlying epilepsy in gliomas. Moreover, all grades were combined as inputs to the model, making it difficult to determine whether these results were associated with the presence or absence of epilepsy. Owing to the effects of IDH mutations and different characteristics of glioblastomas, further studies are necessary to specifically investigate the association between epilepsy and glioma subtypes.
The development of GRE involves various factors, including tumor ___location, tumor histopathology, characteristics of the peritumoral microenvironment, and specific genetic alterations. Many scholars have begun to recognize that extensive and in-depth studies on glioma development in the tumor microenvironment are likely to provide new targets and therapeutic tools for tumor therapy due to the tumor microenvironment. Our results revealed differences in the expression of immune checkpoints between most patients with GRE and those without epilepsy. This may be one of the reasons why patients with GRE have a better prognosis after receiving systemic therapy. Microglia, macrophages, neutrophils, endothelial cells, and T cells are the main immune cell types in the glioma microenvironment of the brain42. Moreover, diagnostic recognition genes, such as ANXA1, EMP3, and VEGFA, are highly expressed in most of the tumor microenvironment cells. Endothelial cells are key cellular components of the blood-brain barrier. Brain endothelial cells establish a continuous complex of tight junctions and adhesion junctions along the endothelial cell-endothelial cell contact surface, providing a tight size-selective barrier43. They further express different influx and efflux transporters, several of which are capable of binding and limiting the brain penetration of a wide range of small lipophilic xenobiotic compounds and drugs43. Some patients with gliomas have difficulty controlling epilepsy after surgery and conventional antiepileptic drug therapy. Single-cell sequencing revealed that epilepsy diagnostic genes were highly expressed in malignant endothelial cells. This may provide a theoretical basis for the identification of new epilepsy drugs.
Histone modifications play important roles in epigenetic regulation. Meanwhile, after our analysis, we found that histone inhibitors may become a controlling GRE drug. Currently, sodium valproate is the most commonly used antiepileptic drug. Sodium valproate is a histone deacetylase inhibitor with anti-inflammatory, neuroprotective, and anti-convulsant properties. Despite extensive clinical drug trials, histone inhibitors are often used as adjunctive agents in combination with immunosuppressive agents for the treatment of glioblastoma but do not achieve good therapeutic results44,45. However, the results of our drug personalization analysis were based on sequencing data, and more animal and cellular experiments are needed to validate the feasibility of these drugs in the future. Thus, further studies should explore whether the use of histone inhibitors in patients with low-grade gliomas can control epilepsy while slowing tumor progression.
We collected and integrated data related to a large sample of patients with GRE, which is valuable for clinical guidance. The results of these analyses are promising; however, some limitations remain. First, all analyses were retrospective, and future validation of the diagnostic models should be performed in a prospective multicenter study. Second, some molecular, pathological, and clinical features were lacking, which may mask certain factors in the development of epilepsy in patients with gliomas. Gliomas usually occur in the white matter, and epilepsy requires the involvement of the gray matter. However, the tissue surrounding the tumor may also be an important cause of epilepsy. Additionally, many glioma-related genes exert their effects at the protein level through post-translational modifications, which may not be evident in the transcription or methylation data. Finally, the mechanistic role of the identified genes with diagnostic value in the development of epilepsy in patients with glioma remains unknown, and further in vivo and in vitro experiments are required to reveal their functions.
In conclusion, we integrated multi-omics and clinical data to identify high-risk factors that may contribute to the development of epilepsy in patients with glioma as well as diagnostic models for identification. This provides a basis for further understanding the clinical characteristics of patients with GRE and the genetic mechanisms that may influence them.
Methods
Data source and study population
GRE was defined as at least one case of preoperative epilepsy without an obvious trigger, based on the patient’s chief complaint and past medical history5. In a retrospective analysis of 997 patients with glioma who underwent craniotomy at Beijing Tiantan Hospital, 342 patients had preoperative epilepsy, and 655 patients had no preoperative epilepsy. The patients’ diagnostic criteria were as follows: (1) Diagnosis of glioma was confirmed using a combination of preoperative imaging and postoperative pathology. (2) Documented history of well-defined preoperative epilepsy and clinical signs. (3) EEG demonstrating the correlation between a patient’s tumor and epilepsy3. All patients received standard treatment, including maximum safe resection, adjuvant radiotherapy, and/or chemotherapy, depending on the tumor grade. Patient complaints, basic clinical information, and molecular pathological features were retrieved from the electronic medical record system10. Tumor grading was performed according to the World Health Organization (WHO) 2021 Classification of Tumors of the Central Nervous System46. mRNA sequencing of postoperative tumor samples was divided into RNA-seq693 (n = 507) and RNA-seq325 (n = 197) using the Illumina HiSeq platform and Array301 (n = 293) using the Agilent whole human genome (Array) platform, according to different sequencing periods. Methylation sequencing data were obtained using the Illumina Infinium HumanMethylation27 Bead-Chips platform at methyl 159 (n = 159). Data for gene mutations were obtained using the Agilent SureSelect kit v5.4 & Illumina HiSeq 4,000 platform WESeq286 (n = 286)47. All data, except for the clinical phenotype of epilepsy, were available for download from the Chinese Glioma Genome Atlas (CGGA) platform48. The scRNA-seq data of the four GBM and matched non-malignant brain samples were obtained from the Gene Expression Omnibus (GEO) database (GSE162631)49. Transcriptome data were obtained from the CGGA database (http://www.cgga.org.cn/index.jsp).
Ethics statement
This study was conducted in accordance with the Declaration of Helsinki. The data collection was approved by the Institutional Ethics Committee (Ethics Code: KY2014-002-02), and written informed consent was obtained from all patients.
Identify differential gene analysis and signaling pathway alterations
Moreover, we explored whether genes and signaling pathways influence the occurrence of glioma-related epilepsy. We used “edgeR” and “limma” packages from R software to analyze the different genes of GRE. The filtering criteria were p < 0.05 and | logFC | >0.5. Simultaneously, the volcano maps of RNA-seq693, RNA-seq325, and Array-301 were drawn with R package “ggpubr,” using standard | logFC| > 0.5. To further analyze the changes of signal pathways in GRE, the “GSVA” package was used to view up-regulated and down-regulated signal pathways, and the “clusterProfiler” package was used to observe activation and inhibition signal pathways50,51.
Immune infiltration and immune checkpoint evaluation
We analyzed whether immune cell infiltration affected the occurrence of epilepsy in patients with gliomas. Simultaneously, immune cell infiltration was evaluated using GREs. The immune cell infiltration was analyzed by R package “ssGSEA” and 13 kinds of immune function scores published in previous literature were scored52. To evaluate the correlation between immunocyte infiltration and the clinical and molecular pathological features of GRE, “ComplexHeatmap” was used to draw a heat map53.
Identification of co-expression network-related genes and enrichment analysis
Next, we identified the genes that influence epileptogenesis in gliomas and the signaling pathways that were affected. WGCNA was used to identify the gene modules associated with GRE and non-epilepsy (Supplement Fig. 7a). We selected MEmagenta as the set of genes related to GRE for the enrichment analysis. MEbrown module genes were selected for enrichment analysis in patients without epilepsy associated with gliomas. The results of GO functional annotation and KEGG pathway enrichment analysis were plotted using the R package “ClusterGVis.”
Building machine learning diagnostic models
We screened for genes that may affect glioma-associated epileptogenesis using multiple machine-learning algorithms. To further evaluate the identification of diagnostic genes for GRE, 12 machine learning algorithms were used: Random Forest (RF), least absolute shrinkage and selection operator (LASSO), gradient boosting machine (GBM), support vector machine (SVM), linear discriminant analysis (LDA), ridge regression (Ridge), generalized linear model boosting (glmBoost), partial least squares regression generalized linear model (plsRglm), extreme gradient boosting (XGBoost), stepwise generalized linear model (Stepglm), naive bayes classifier (NaiveBayes), and elastic network (Enet)54,55. Finally, based on the average AUC values of the three cohorts, the most appropriate machine-learning model was selected as the diagnostic model.
Identification of glioma-related epilepsy mutant genes and methylation differential genes
We analyzed whether gene mutations affected the occurrence of glioma-related epilepsy. To further identify mutations in patients with GRE, tumors and matched blood samples were analyzed using exome sequencing. For whole-exome sequencing, target capture was performed using the Agilent SureSelect kit v5.4. For targeted sequencing, a custom collection of 272 genes was captured using SureSelectXT custom kits (Agilent Technologies). Pairwise sequencing was performed using the Illumina HiSeq 4000 platform as previously described47. The R package “ComplexHeatmap” was used to visualize the different mutation types, clinical features, and the top 10 most commonly mutated genes in glioma patients.
Analysis of immune scores and The Connectivity Map in patients with glioma-related epilepsy
We analyzed multiple immune scores that influenced the occurrence of glioma-associated epilepsy and drugs that may act. To assess immunological and quantitative tumor tissue evaluation between patients with GRE and patients with gliomas without epilepsy, the R package “IOBR” was used56. The “IOBR” package is an immunological/biological study of multi-omic data on tumors, capable of revealing the relationship between the tumor microenvironment and clinical features.
To predict personalized drug therapy for GRE, an analysis was performed using the Connectivity Map (CMAP) database57. The CMAP database is based on species using similar gene expression profiles to predict therapy for an individual and is primarily used to reveal functional associations between small-molecule compounds, genes, and disease states. The CMAP score was calculated using the function “XSum.”
Epilepsy-causing genes were expressed in the tumor microenvironment
scRNA-seq data from gliomas were used to assess GRE gene expression in the tumor microenvironment. Four GBM tumor tissues and four adjacent normal tissues were analyzed. After quality control and screening, cells expressing 200–6000 genes and less than 25% of the mitochondrial genes were selected (Supplement Fig. 7b, c). Two thousand hypervariable genes were selected for subsequent analyses (Supplement Fig. 7d). The R package “harmony” was used to integrate multiple samples for de-batching (Supplement Fig. 7e). Finally, we identified the clusters of cells grouped before annotation (Supplement Fig. 7f).
The R package “InferCNV” was used to identify copy number variations in scRNA-seq analysis by pipeline parameters. According to previous literature, cells with a CNV signal greater than 0.05 and a CNV correlation coefficient greater than 0.5 were defined as malignant, and cells below these two thresholds were defined as non-malignant (Supplement Fig. 7g). Manual annotation was performed using previously published literature and the CellMaker database58. The R package “decoupleR” was also used to demonstrate the activation and inhibition of signaling pathway activity in cell subpopulations.
Cell culture, RNA extraction, cDNA synthesis, and quantitative real-time PCR
Furthermore, we validated the expression of the diagnostic model genes using the glioma cell lines U251 and T98G and the normal human astrocyte line SVGp12. The primer sequences are listed in Supplementary Table 2. The human glioma cell lines, U251, T98G, as well as the human astroglia SVGp12, were obtained from Procell (Wuhan, China) and cultured in Dulbecco’s modified Eagle medium (DMEM; Servicebio) supplemented with 10% fetal bovine serum (FBS, Excel) and 10ul/ml Penicillin–streptomycin solution, (100×, Biosharp) at a cell incubator (Hair) at 37 °C with 5% CO2.
Total RNA was extracted from tissues preserved at a temperature < −80 °C using Takara RNAiso Plus (Takara Bio. Inc., Otsu, Shiga, Japan), following the manufacturer’s protocol. cDNA was synthesized from 1 μg of RNA using the HiScriptIII Q RT SuperMix for qPCR Kit (Vazyme Medical Technology). The 2−ΔΔCt method was used to process qPCR data.
Immunohistochemical verification of model gene expression
We used tumor tissues from three patients with GRE and three without epilepsy to validate the model gene expression. Every pathological section was evaluated by two experienced pathologists, with diagnostic conclusions aligned with both the imaging results and clinical assessments. All the samples were fixed in 10% formalin. Subsequently, the tumor tissues were dehydrated, clarified, and embedded in paraffin. Tissues were consistently sectioned at 4–5 μm and stained with hematoxylin–eosin (HE). After selecting the representative lesion areas, the sections were baked and deparaffinized, followed by three 5-minute washes in PBS. Subsequently, the samples were immersed in citrate buffer (pH 6.0) for repair. After the nonspecific sites were saturated with 5% normal goat serum, the sections were incubated overnight at 4 °C with primary antibodies (ABCC3, #DF3874, EMP3, DF14661, Transgelin 2 #DF12053, 1:200, Affinity; G0S2, #A9970, GDF15, #A0185, #ANXA1, #A0767, VEGFA, #A0280, 1:200, Abclonal; COL5A2, #CSB-PA072697, 1:100, Cusabio). They were then washed thrice with PBS for 5 min each. The sections then received a secondary antibody treatment at 37 °C for 10 min, followed by another PBS wash and DAB staining. Staining was halted using PBS. After dehydration with ethanol, sections were sealed using xylene. The criteria for determining positive staining were based on the color range from yellow to brown-yellow. The gradation for positive staining was as follows: (3, ≥50%; 2, ≥5% and <50%; 1, ≥1% and <5%; and 0, <1%).
Data availability
The data presented in this study are deposited in the Sequence Read Archive (SRA) repository with the accession numbers PRJCA001747, PRJCA001746, and PRJCA001636. All data were sourced from the CGGA database (http://www.cgga.org.cn/) (mRNAseq_693, mRNAseq_325, mRNA-array_301, methyl_159, WESeq_286) and the TCGA database (http://www.cgga.org.cn/). Additional clinical information can be obtained from the corresponding author.
Code availability
All code for this study is available on GitHub.
References
Pallud, J. et al. Epileptic seizures in diffuse low-grade gliomas in adults. Brain 137, 449–462 (2014).
van Breemen, M. S., Wilms, E. B. & Vecht, C. J. Epilepsy in patients with brain tumours: epidemiology, mechanisms, and management. Lancet Neurol. 6, 421–430 (2007).
Liang, S. et al. Clinical practice guidelines for the diagnosis and treatment of adult diffuse glioma-related epilepsy. Cancer Med. 8, 4527–4535 (2019).
Wang, Y. et al. Localizing seizure-susceptible brain regions associated with low-grade gliomas using voxel-based lesion-symptom mapping. Neuro Oncol. 17, 282–288 (2015).
Zhou, X. W. et al. Biomarkers related with seizure risk in glioma patients: a systematic review. Clin. Neurol. Neurosurg. 151, 113–119 (2016).
Di Bonaventura, C. et al. Epileptic seizures heralding a relapse in high grade gliomas. Seizure 51, 157–162 (2017).
Kerkhof, M. & Vecht, C. J. Seizure characteristics and prognostic factors of gliomas. Epilepsia 54, 12–17 (2013).
Kerkhof, M. et al. Effect of valproic acid on seizure control and on survival in patients with glioblastoma multiforme. Neuro Oncol. 15, 961–967 (2013).
Marku, M. et al. Prediagnosis epilepsy and survival in patients with glioma: a nationwide population-based cohort study from 2009 to 2018. J. Neurol. 269, 861–872 (2022).
Fang, S. et al. Altering patterns of sensorimotor network in patients with different pathological diagnoses and glioma-related epilepsy under the latest glioma classification of the central nervous system. CNS Neurosci. Ther. 29, 1368–1378 (2023).
Venkatesh, H. S. et al. Electrical and synaptic integration of glioma into neural circuits. Nature 573, 539–545 (2019).
Gillespie, S. & Monje, M. An active role for neurons in glioma progression: making sense of Scherer’s structures. Neuro Oncol. 20, 1292–1299 (2018).
Zorec, R. et al. Astroglial excitability and gliotransmission: an appraisal of Ca2+ as a signalling route. ASN Neuro 4, e00080 (2012).
Tewari, B. P. et al. Perineuronal nets decrease membrane capacitance of peritumoral fast spiking interneurons in a model of epilepsy. Nat. Commun. 9, 4724 (2018).
John Lin, C. C. et al. Identification of diverse astrocyte populations and their malignant analogs. Nat. Neurosci. 20, 396–405 (2017).
Vezzani, A., Aronica, E., Mazarati, A. & Pittman, Q. J. Epilepsy and brain inflammation. Exp. Neurol. 244, 11–21 (2013).
Vezzani, A. et al. Infections, inflammation and epilepsy. Acta Neuropathol. 131, 211–234 (2016).
Mangani, D. et al. Limited role for transforming growth factor-β pathway activation-mediated escape from VEGF inhibition in murine glioma models. Neuro Oncol. 18, 1610–1621 (2016).
Li, L. et al. Expression changes in ion channel and immunity genes are associated with glioma-related epilepsy in patients with diffuse gliomas. J. Cancer Res. Clin. Oncol. 148, 2793–2802 (2022).
Feyissa, A. M. et al. Analysis of intraoperative human brain tissue transcriptome reveals putative risk genes and altered molecular pathways in glioma-related seizures. Epilepsy Res. 173, 106618 (2021).
Berntsson, S. G., Malmer, B., Bondy, M. L., Qu, M. & Smits, A. Tumor-associated epilepsy and glioma: are there common genetic pathways? Acta Oncol. 48, 955–963 (2009).
Wang, Y. H. et al. Glioma-derived TSP2 promotes excitatory synapse formation and results in hyperexcitability in the peritumoral cortex of glioma. J. Neuropathol. Exp. Neurol. 80, 137–149 (2021).
Van Calster, B. & Wynants, L. Machine learning in medicine. N. Engl. J. Med. 380, 2588 (2019).
van Niftrik, C. H. B. et al. Machine learning algorithm identifies patients at high risk for early complications after intracranial tumor surgery: registry-based cohort study. Neurosurgery 85, E756–e764 (2019).
Swinburne, N. C. et al. Machine learning for semi-automated classification of glioblastoma, brain metastasis and central nervous system lymphoma using magnetic resonance advanced imaging. Ann. Transl. Med. 7, 232 (2019).
Yunhe, M., Yuan, Y., Xiang, W., Yanhui, L. & Qing, M. Mapping seizure foci and tumor genetic factors in glioma associated seizure patients. J. Neurosurg. Sci. 64, 456–463 (2020).
Wang, Y. et al. Predicting the type of tumor-related epilepsy in patients with low-grade gliomas: a radiomics study. Front. Oncol. 10, 235 (2020).
Xu, J., Sun, M., Wang, Y., Xie, A. & Gao, J. Identification of hub genes of mesio temporal lobe epilepsy and prognostic biomarkers of brain low-grade gliomas based on bioinformatics analysis. Cell Transpl. 29, 963689720978722 (2020).
Kong, B. et al. Protein-protein interaction network analysis and gene set enrichment analysis in epilepsy patients with brain cancer. J. Clin. Neurosci. 21, 316–319 (2014).
Kumar, P. et al. Single-cell transcriptomics and surface epitope detection in human brain epileptic lesions identifies pro-inflammatory signaling. Nat. Neurosci. 25, 956–966 (2022).
Krauze, A. V. et al. A phase 2 study of concurrent radiation therapy, temozolomide, and the histone deacetylase inhibitor valproic acid for patients with glioblastoma. Int. J. Radiat. Oncol. Biol. Phys. 92, 986–992 (2015).
Su, J. M. et al. Phase I/II trial of vorinostat and radiation and maintenance vorinostat in children with diffuse intrinsic pontine glioma: a children’s oncology group report. Neuro Oncol. 24, 655–664 (2022).
Iwamoto, F. M. et al. A phase I/II trial of the histone deacetylase inhibitor romidepsin for adults with recurrent malignant glioma: North American Brain Tumor Consortium Study 03-03. Neuro Oncol. 13, 509–516 (2011).
Ghiaseddin, A. et al. Phase II study of bevacizumab and vorinostat for patients with recurrent world health organization grade 4 malignant glioma. Oncologist 23, 157–e121 (2018).
Dunn-Pirio, A. M. et al. Adjunctive perampanel for glioma-associated epilepsy. Epilepsy Behav. Case Rep. 10, 114–117 (2018).
Mayer, J. et al. Perampanel attenuates epileptiform phenotype in C6 glioma. Neurosci. Lett. 715, 134629 (2020).
Lange, F. et al. Perampanel add-on to standard radiochemotherapy in vivo promotes neuroprotection in a rodent F98 glioma model. Front. Neurosci. 14, 598266 (2020).
Buccoliero, A. M. et al. Angiocentric glioma-associated seizures: the possible role of EATT2, pyruvate carboxylase and glutamine synthetase. Seizure 86, 152–154 (2021).
Ge, H. et al. Does epilepsy always indicate worse outcomes? A longitudinal follow-up analysis of 485 glioma patients. World J. Surg. Oncol. 20, 297 (2022).
Chen, H. et al. Mutant IDH1 and seizures in patients with glioma. Neurology 88, 1805–1813 (2017).
Li, L. et al. Glioma-related epilepsy in patients with diffuse high-grade glioma after the 2016 WHO update: seizure characteristics, risk factors, and clinical outcomes. J. Neurosurg. 136, 67–75 (2022).
Klemm, F. et al. Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell 181, 1643–1660.e1617 (2020).
Daneman, R. & Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 7, a020412 (2015).
Puduvalli, V. K. et al. A Bayesian adaptive randomized phase II multicenter trial of bevacizumab with or without vorinostat in adults with recurrent glioblastoma. Neuro Oncol. 22, 1505–1515 (2020).
Bukowinski, A. et al. A phase 1 study of entinostat in children and adolescents with recurrent or refractory solid tumors, including CNS tumors: Trial ADVL1513, Pediatric Early Phase-Clinical Trial Network (PEP-CTN). Pediatr. Blood Cancer 68, e28892 (2021).
Louis, D. N. et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neurooncology 23, 1231–1251 (2021).
Zhao, Z. et al. WES data from 286 diffuse gliomas under the 2021 WHO Classification of Tumors of the Central Nervous System. Sci. Data 9, 692 (2022).
Zhao, Z. et al. Chinese Glioma Genome Atlas (CGGA): a comprehensive resource with functional genomic data from chinese glioma patients. Genomics Proteom. Bioinforma. 19, 1–12 (2021).
Xie, Y. et al. Key molecular alterations in endothelial cells in human glioblastoma uncovered through single-cell RNA sequencing. JCI Insight 6, e150861 (2021).
Wu, T. et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation 2, 100141 (2021).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinforma. 14, 7 (2013).
He, Y., Jiang, Z., Chen, C. & Wang, X. Classification of triple-negative breast cancers based on Immunogenomic profiling. J. Exp. Clin. Cancer Res. 37, 327 (2018).
Gu, Z. & Hübschmann, D. Make Interactive Complex Heatmaps in R. Bioinformatics 38, 1460–1462 (2022).
Liu, Z. et al. Machine learning-based integration develops an immune-derived lncRNA signature for improving outcomes in colorectal cancer. Nat. Commun. 13, 816 (2022).
Wang, L. et al. Comprehensive machine-learning survival framework develops a consensus model in large-scale multicenter cohorts for pancreatic cancer. Elife 11, e80150 (2022).
Zeng, D. et al. IOBR: multi-omics immuno-oncology biological research to decode tumor microenvironment and signatures. Front. Immunol. 12, 687975 (2021).
Musa, A. et al. A review of connectivity map and computational approaches in pharmacogenomics. Brief. Bioinform. 18, 903 (2017).
Zhang, X. et al. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 47, D721–d728 (2019).
Acknowledgements
Thanks to the main data sources provided by the Chinese Glioma Genome Atlas Database and the participation of all patients. This study was funded by the Major Scientific and Technological Innovation Project (2021ZD0201605) and the National Natural Science Foundation of China (82072786).
Author information
Authors and Affiliations
Contributions
Jinwei Li organized the data, analyzed and wrote the paper. Shengrong Long and Yang Zhang performed the experimental validation and cell and tissue experiments. Wei Wei and Shuangqi Yu proofread the article and conducted some of the experiments. Quan Liu, Xuhui Hui, Xiang Li, and Yinyan Wang constructed the experimental idea and reviewed the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, J., Long, S., Zhang, Y. et al. Molecular mechanisms and diagnostic model of glioma-related epilepsy. npj Precis. Onc. 8, 223 (2024). https://doi.org/10.1038/s41698-024-00721-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-024-00721-8
