
Abstract
A crucial task towards creating a sustainable chemical industry is the electrification of chemical processes that produce value-added molecules. One such molecule is 1,3-butadiene (1,3-BD), the feedstock used for manufacturing synthetic rubber. 1,3-BD is traditionally derived, as a by-product, during the energy-intensive steam cracking of naphtha to ethylene. Here we introduce an alternative approach to selectively produce 1,3-BD from the electroreduction of acetylene (e-C2H2R). By using a potassium iodide electrolyte, we created Cuδ+–Cu0 sites on a Cu2O-nanocube-derived catalyst, which are efficacious for promoting e-C2H2R to 1,3-BD. 1,3-BD was formed with a Faradaic efficiency reaching 93% at −0.85 V versus standard hydrogen electrode (SHE) and a partial current density of −75 mA cm−2 at −1.0 V versus SHE. Density functional theory calculations show that I− preserves Cuδ+–Cu0 sites, which facilitate the favourable binding of acetylene, leading to 1,3-BD formation through the coupling of *C2H3 moieties.

Similar content being viewed by others

Main
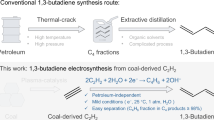
Electrifying chemical processes that produce multicarbon molecules is a pivotal step towards the much-needed decarbonization of chemical industries1. Towards this end, those chemical processes that most impact our global energy–carbon nexus must be first identified. One such process is the steam cracking of naphtha to ethylene, which also yields 1,3-butadiene (1,3-BD) as a by-product2. During naphtha steam cracking, 12–16 kg of 1,3-BD are obtained alongside every 100 kg of ethylene2,3. 1,3-BD is a vital feedstock used in the global manufacturing of synthetic rubber and engineering plastics4. In recent years, however, the production of ethylene from the dehydrogenation of ethane in shale gas has proven more cost effective5. This has resulted in a dramatic decrease in naphtha cracking. Although 1,3-BD can also be formed from the ethane feedstock, only 1–2 kg of it are now produced per 100 kg of ethylene6,7. 1,3-BD can also be synthesized by the oxidative dehydrogenation of butane or the dehydrogenation and condensation of ethanol over silicon and magnesium oxide catalysts8,9,10. All the above methods are energy intensive and require high temperatures and pressures to operate. From this perspective, a more environmentally benign strategy to synthesize 1,3-BD is highly desirable.
In recent years, acetylene has been considered as a precursor to synthesize value-added chemicals and polymers11. We describe here an approach to synthesize 1,3-BD by the electrochemical reductive coupling of acetylene (2C2H2(g) + 2H2O + 2e− → C4H6(g) + 2OH−, E0 = +0.98 V versus standard hydrogen electrode (SHE) at pH 7.0; Supplementary Note 1). C2H2 electroreduction (e-C2H2R) is potentially more sustainable because it can be conducted under relatively mild conditions, uses water as the hydrogen source for reduction, and can be powered by renewable electricity. Additionally, the acetylene feedstock11 can be synthesized from carbon dioxide or methane2,12,13, the top two greenhouse gases in the atmosphere14. Thus far, previous works have been devoted to e-C2H2R to ethylene (C2H2(g) + 2H2O + 2e− → C2H4(g) + 2OH−, E0 = +0.40 V versus SHE at pH 7.0)15,16,17,18,19,20,21,22. In the studies that showed 1,3-BD formation, typically on copper catalysts, it was formed as a by-product with current densities smaller than −4 mA cm−2, which are insufficient for industrial adoption23,24,25,26,27. Synthesizing 1,3-BD with high yields is particularly challenging because at larger overpotentials *C2H3, a key intermediate for 1,3-BD formation, hydrogenates to *C2H4 and desorbs from the catalyst surface as C2H4(g), instead of undergoing coupling to form *C4H6 (ref. 26).
In this work, we reveal a strategy to electrolyse C2H2 to 1,3-BD, forming it as a major product with high selectivity. Our approach is to introduce Cuδ+–Cu0 sites on a copper catalyst, the former having been found to promote the coupling of CO intermediates formed during the electroreduction of CO2 (CO2RR) to C2+ products28,29,30. Using in situ Raman spectroscopy and X-ray absorption spectroscopy (XAS), we show that the formation of stable Cuδ+–Cu0 active sites can be promoted using potassium iodide (KI) electrolyte31,32,33. On these sites, acetylene was reduced to 1,3-BD with a maximum Faradaic efficiency (FE1,3-BD) of 93% at −0.85 V versus SHE and a partial current density (j1,3-BD) of −75.0 mA cm−2 at −1.0 V versus SHE. These figures of merit are substantially higher than those obtained during e-C2H2R in KCl, KBr and K2SO4 electrolytes. Using density functional theory (DFT) calculations, we show that the presence of iodide helps preserve the partially oxidized copper sites. In addition, coadsorbed iodide species on the partially oxidized copper sites lead to favourable thermodynamics and surmountable coupling barriers for acetylene reduction to 1,3-BD.
Results
Characterization of Cu2O-nanocube-derived catalyst
We prepared Cu2O nanocubes (Cu2ONC) using a previously reported method34. Scanning electron microscopy (SEM) of the freshly prepared catalyst showed that it consisted of ~30 nm cubes (Fig. 1a). This catalyst was then used for e-C2H2R in 1 M KI electrolyte at a representative potential of −1.0 V versus SHE (all potentials stated hereafter are referenced to the SHE). After use for e-C2H2R, the particles’ cubic morphology was largely lost and ~60 nm agglomerates were observed instead (Fig. 1b). The morphologies of the catalyst particles after they were used for e-C2H2R in K2SO4, KCl and KBr electrolytes were also similar (Fig. 1c–e). However, double-layer capacitance measurements indicate that the roughness factors of the particles in KI were ~1.1–1.2× higher than those in K2SO4, KCl or KBr electrolytes (Fig. 1f and Supplementary Table 1). This is in line with previous works showing the roughening of copper in the presence of I− (refs. 35,36,37).

a–e, SEM images of Cu2ONC catalyst (a), before (green background) and after e-C2H2R (pink background) in KI (b), K2SO4 (c), KCl (d) and KBr (e) electrolyte. f, Double-layer capacitance (Cdl) measurements of Cu2ONC-derived catalysts after e-C2H2R. The plots denote the capacitive current density versus scan rate. The Cdl was measured in 0.5 M K2SO4. Each error bar represents the standard deviation (s.d.) of three independent measurements. g, X-ray diffractograms of Cu2ONC catalyst before and after e-C2H2R at −1.0 V versus SHE in KI and K2SO4. The X-ray diffractograms were assigned in accordance with PDF 00-003-0892 for Cu2O and PDF 00-001-1242 for copper. h, In situ Raman spectra of Cu2ONC catalyst at OCP with C2H2, after 10 min of electrolysis at −1.0 V versus SHE in KI and K2SO4, and after electrolysis. Inset: enlarged region of the Cu2O Raman peaks under OCP conditions; the Raman spectra (i) and (ii) indicate the absence and presence of oxide peaks, respectively, during e-C2H2R in KI. A graphite disc was used as a substrate for characterization.
X-ray diffraction (XRD) of Cu2ONC revealed signals corresponding to those of Cu2O (Fig. 1g). After Cu2ONC was used as an e-C2H2R catalyst in 1 M KI electrolyte at −1.0 V, two additional XRD peaks were observed at 43.5° and 50.7°, which we ascribe respectively to the (111) and (200) diffractions of metallic copper. Intense Cu2O diffractions were nonetheless still measured, which could be due to the Cu2ONC not being fully reduced at −1.0 V. This observation is consistent with a previous work, in which Cu2O XRD peaks from Cu2ONC were still present after their use for CO2RR at −1.55 V versus SHE in KHCO3 (pH 6.8)38. Cu2O and copper diffraction peaks were similarly observed on the Cu2ONC-derived catalysts after they were used for e-C2H2R in K2SO4, KCl and KBr electrolytes (Fig. 1g and Supplementary Fig. 1). We note that the surface copper atoms could have also oxidized to Cu2O during sample transfer for XRD analysis. Therefore, we resorted to in situ Raman and XAS to determine how the Cu2ONC catalyst evolves during electrolysis.
In situ Raman spectroscopy was conducted on the Cu2ONC catalyst during C2H2 reduction in 1 M KI electrolyte (Fig. 1h and Supplementary Table 2). At open circuit potential (OCP), Cu2O was identified, as evident by its Raman peaks at 148, 218, 522 and 622 cm−1 (ref. 39). When a potential of −1.0 V was applied, these peaks were largely attenuated, although still detectable. Specifically, the 522 and 622 cm−1 peaks could be observed in spectrum (ii) in Fig. 1h. This indicates that Cu2O was not fully reduced to metallic copper. Bands at 1,134, 1,530 and 1,695 cm−1 were also measured. The 1,695 cm−1 signal could be ascribed to the ν(C≡C) stretching vibration of adsorbed C2H2 (ref. 26), while those at 1,134 and 1,530 cm−1 could be attributed to polyacetylene40. No peaks corresponding to CuI, adsorbed 1,3-BD, *C2H3 or other reduced species from e-C2H2R were observed, which could be due to their short lifetimes, small surface population or the insufficient limits of detection afforded by our spectrometer (Supplementary Fig. 2).
In situ Raman measurements during e-C2H2R in 0.5 M K2SO4 electrolyte were also conducted at −1.0 V (Fig. 1h). While we detected the ν(C≡C) stretching signal of C2H2 at 1,695 cm−1 and polyacetylene Raman peaks at 1,134 and 1,530 cm−1 (refs. 26,40), signals that could be attributed to Cu2O were not detected. Similar observations were obtained from C2H2 electrolysis in 1 M KCl and KBr electrolytes (Supplementary Fig. 3). This indicates that the use of KI electrolyte aids in preserving the Cu2O species during electrolysis. Indeed, based on predictions from the Pourbaix diagrams of Cu–H2O–X systems (X = I−, Cl−, Br− or SO42−), the reduction potential of Cu+ to Cu0 in the presence of I− is at least 0.15 V more negative compared with Cl−, Br− and SO42− (refs. 41,42,43).
When the applied negative potentials were removed, peaks at 218, 522 and 622 cm−1 were observed within 2 min on all the surfaces (Fig. 1h and Supplementary Fig. 3). This signifies that metallic copper reoxidized to Cu2O. The signals of polyacetylene at 1,020, 1,134, 1,280 and 1,530 cm−1 were also observed post electrolysis after the C2H2 gas flow was removed25,40, indicating that the formed polyacetylene remained on the catalyst surface during electrolysis.
The oxidation state of the Cu2ONC catalyst during e-C2H2R was monitored using in situ XAS in fluorescence mode (Fig. 2 and Supplementary Fig. 4). The Cu K-edge X-ray near-edge absorption spectrum (XANES) of a freshly prepared Cu2ONC catalyst corresponds to that of a Cu2O standard (Fig. 2a). During e-C2H2R in 1 M KI electrolyte at −1.0 V, the rising edge spectrum of the Cu2ONC-derived catalyst shifted towards the position observed for a metallic copper foil standard, and the absorption edge shape resembled that of metallic copper36. However, the rising edge energies still reside between those of pristine copper and Cu2O, indicating that the oxidation state of the copper catalyst lies between those of Cu0 and Cu+. Interestingly, when we conducted in situ XAS of the Cu2ONC-derived catalyst in KCl, KBr and K2SO4 electrolytes, the rising edge energies of copper were notably closer to that of copper foil. A linear combination fitting of the XANES spectra further showed that the amount of Cu+ in the catalyst is the highest in KI (31%) compared with those observed in KCl (8%), KBr (8%) and K2SO4 (6%) electrolytes (Supplementary Table 3).

a,b, In situ XANES (a) and EXAFS (b) spectra acquired on Cu2ONC catalyst at OCP during e-C2H2R at −1.0 V versus SHE in 1 M KI, KCl, KBr and 0.5 M K2SO4 electrolytes. Copper foil and Cu2O were used as standards for comparison. Inset, a: enlarged pre-edge region from 8,970 to 8,979 eV.
In situ extended X-ray absorption fine structure (EXAFS) analysis was used to determine changes in the local chemical environment of copper during e-C2H2R. Peaks corresponding to Cu–O and metallic Cu–Cu bonds were observed at 1.4 and 2.2 Å, respectively (Fig. 2b)44. Strikingly, the in situ EXAFS spectra measured in KCl, KBr and K2SO4 electrolytes showed Cu–O peak intensities to be at least 50% less intense than in KI. Instead, the Cu–Cu peak was the more dominant feature observed, indicating that metallic Cu0 is the main species present in KCl, KBr or K2SO4 electrolytes.
Overall, both operando Raman spectroscopy and XAS indicate that the presence of I− preserved a larger amount of oxidized copper species in the copper catalyst during e-C2H2R. This finding is in good agreement with a previous XANES study showing that iodide-modified copper contained a higher concentration of Cu+ sites compared with other halide-modified copper materials36.
Electrocatalytic activity of Cu2ONC for e-C2H2R
The Cu2ONC catalyst was loaded (5 mg cm−2) onto a carbon gas diffusion layer and used as an electrode for e-C2H2R in a flow cell (Supplementary Fig. 5). Unless specified, all current densities stated were normalized to the geometric surface area of the exposed electrode, which was 1 cm2. Nickel foam and an Ag/AgCl electrode were used as the counterelectrode and the reference electrode, respectively. Linear sweep voltammetry (LSV) was first conducted on the Cu2ONC electrode in a 1 M KI catholyte and a 1 M KOH anolyte (Fig. 3a). In the presence of C2H2, larger currents were measured starting from −0.85 V, as compared to that in N2 gas. This suggests that the Cu2ONC is catalytically active for e-C2H2R.

a, LSV measurements of Cu2ONC catalyst in N2 or 50% C2H2 gas feedstock. Scan rate, 50 mV s−1. b, FEs and partial current densities for 1,3-BD, C2H4, butenes and H2 from e-C2H2R as a function of the applied potential. Each electrolysis measurement lasted 50 min. Each error bar represents the s.d. of three independent measurements. c, Comparison of FEs and partial current densities for 1,3-BD with those of previously reported catalysts23,24,25,26,27. The potentials were taken with reference to the SHE. d, FEs and partial current densities from an 11 h stability experiment. A constant current of −12 mA cm−2 was applied over the course of the experiment. A 2 min oxidation pulse of +0.4 V versus SHE was introduced at the 9 h mark to remove polyacetylene from the catalyst surface. The electrolyte used in these measurements was 1 M KI.
We next investigate the effects of various C2H2 gas concentrations on the e-C2H2R performance at −1.0 V in a 1 M KI catholyte (Supplementary Table 4). The gas feedstock consisted of C2H2 balanced with N2 gas, with a total flow rate of 50 sccm into the electrolytic cell. The gaseous products were analysed using gas chromatography (Supplementary Fig. 6). When the C2H2 concentration increased from 5% to 50%, the FE1,3-BD increased from 58% to 72%, while the FEC2H4 decreased from 41% to 28%. We attribute the increased selectivity for 1,3-BD at larger C2H2 concentrations to the higher coverage of C2H2 on the catalyst surface, which promotes C–C coupling to form more 1,3-BD. However, no further increase in FE1,3-BD was observed in 100% C2H2. We also conducted e-C2H2R at different catalyst mass loadings of 1.0, 2.5 and 5.0 mg cm−2 and found that the j1,3-BD was 3–5 times higher with a 5.0 mg cm−2 catalyst loading (Supplementary Table 5). Since the optimal performance was attained at a C2H2 concentration of 50% and a catalyst loading of 5.0 mg cm−2, we utilized these conditions in all subsequent experiments.
The activity and selectivity of e-C2H2R on the Cu2ONC-derived catalyst in KI electrolyte were quantified at constant potentials from −0.85 to −1.4 V. From −0.85 to −1.3 V, the major products formed were 1,3-BD and C2H4 (Fig. 3b and Supplementary Table 6). We found that at −0.85 V, the FE1,3-BD was as high as 93%, with a j1,3-BD of −8 mA cm−2; the remaining FE is due to C2H4. At more negative potentials, FE1,3-BD decreased while FEC2H4 increased, although the partial current densities for both compounds continued to increase. The maximum j1,3-BD attained was −75 mA cm−2 at −1.0 V. The FE1,3-BD at −0.85 V and the j1,3-BD at −1.0 V in KI electrolyte appear to be the highest reported for the formation of 1,3-BD via e-C2H2R (Fig. 3c). It is noteworthy that the j1,3-BD obtained in this work was at least 20-fold higher than those in previous works23,24,25,26,27. Minor products such as 1-butene, trans-2-butene and cis-2-butene were also detected, with the highest FE of 3.9% and j of ∼−13 mA cm−2 for these butenes occurring at −1.3 V, suggesting that applying a more negative potential further reduces 1,3-BD to these C4 olefins. We note that although n-butane and isobutane are possible products, we did not detect these even at a negative applied potential of −1.3 V (Supplementary Fig. 8). Trace amounts of ethane (FE, 0.2–0.3%) were detected starting from −1.2 V. Due to the relatively positive potentials applied, H2 gas was only produced from the parasitic hydrogen evolution reaction at −1.3 V (ref. 45). Overall, the total FEs of the products ranged from 97% to 105%, indicating that all the electrochemically formed products have been accounted for. We note that although acetylene undergoes polymerization in the presence of copper to form polyacetylene46,47, there is no associated electron transfer for that product. Thus, polyacetylene formation cannot be suitably incorporated in our FE analysis.
To ascertain that the aforementioned products were formed from e-C2H2R, we performed electrolysis at −1.0 V using a N2 feedstock. We also held the working electrode at OCP in 50% C2H2 gas (Supplementary Fig. 6). Both measurements did not yield any carbon-containing products, which show that all the products measured in the preceding e-C2H2R measurements (excluding polyacetylene and H2) were electrochemically formed from acetylene.
The catalytic stability for e-C2H2R in KI electrolyte was assessed using constant current electrolysis at −12 mA cm−2 (Fig. 3d). The FE1,3-BD was maintained at ~90 % over 8 h, while the j1,3-BD was constant at ∼−11 mA cm−2. The FE1,3-BD decreased to ~85% at the 9 h mark, which we attribute to the formation of polyacetylene and the coking of the catalyst surface. To remediate this, we introduced an oxidation pulse of +0.4 V for 2 min to remove the carbonaceous compounds from the electrode surface48. With this oxidation pulse, an FE1,3-BD of ~90% could be sustained for over 11 h. We also assessed the stability at a higher current density of −25 mA cm−2 and found that an FE1,3-BD of 72% could be maintained for 6 h (Supplementary Fig. 9).
We then determined if utilizing another anolyte can affect the selectivity for 1,3-BD. When 1 M KI was used as both the anolyte and catholyte for e-C2H2R at −1.0 V (Supplementary Table 7), the FE1,3-BD was 72% and the j1,3-BD was −61 mA cm−2, which were similar to the results obtained with a 1 M KOH anolyte. This indicates that the anolyte identity does not affect the production of 1,3-BD. We also found that the catholyte pH increased to 12.7 after electrolysis with a KI anolyte (Supplementary Table 8), which is comparable to the increase in pH observed after e-C2H2R with a KOH anolyte (catholyte pH increased from 7.0 to 12.6). This suggests that the increase in pH observed in non-buffering KI can be largely attributed to the production of OH− species during e-C2H2R. We further conducted e-C2H2R in a two-electrode flow cell to evaluate the application prospects for 1,3-BD synthesis (Supplementary Table 9 and Supplementary Fig. 10). An FE1,3-BD of 91% can be achieved at a cell voltage of −2.05 V. These two figures of merit are respectively similar to the highest FE1,3-BD obtained in a three-electrode cell at −0.85 V (Fig. 3b) and our calculated cell voltage (Supplementary Note 2).
Importance of iodide for 1,3-BD production from e-C2H2R
We then systematically investigated the influence of iodide on 1,3-BD production. A representative potential of −1.0 V was applied for all experiments, unless otherwise mentioned. We first varied the concentration of iodide in the catholyte to be 0.25, 0.5 or 1 M. K2SO4 was added to the catholyte to keep the concentration of K+ at 1 M. We found that the FE1,3-BD decreases from 72% in 1 M KI to 66% in 0.25 M KI. When only K2SO4 was used, the FE1,3-BD was 56% (Fig. 4a and Supplementary Table 10). This signifies that the quantity of I− present influences 1,3-BD production.

a, FEs for 1,3-BD and C2H4 during e-C2H2R at −1.0 V versus SHE in different concentrations of KI, b,c, Cation (b) and anion (c) electrolytes. In all experiments, the cation concentration was maintained at 1 M. In experiments without KI, 0.5 M K2SO4 was used instead. d, ECSA-normalized current densities of 1,3-BD at −1.0 V versus SHE in K2SO4, KCl, KBr and KI electrolytes. Each error bar represents the s.d. of three independent measurements.
The type of cation and anion of the electrolyte has been found to impact the selectivity for coupled products during CO2RR34,35,49. Therefore, we studied the effects of changing the electrolyte cation on 1,3-BD production during e-C2H2R (Fig. 4b and Supplementary Table 11). The FE1,3-BD and j1,3-BD attained in NaI (74%, −71 mA cm−2) and CsI (74%, −74 mA cm−2) electrolytes were similar to those observed in KI electrolyte (72%, −75 mA cm−2), which indicates that the identity of the cation does not notably affect the formation of 1,3-BD. However, varying the type of anion yielded different selectivity and activity for 1,3-BD formation (Fig. 4c and Supplementary Tables 12–14). In K2SO4, KCl and KBr electrolytes, the FE1,3-BD were remarkably similar at 56%, 54% and 55%, respectively, while the j1,3-BD ranged from −45 to −47 mA cm−2. Furthermore, the maximum j1,3-BD in K2SO4, KCl and KBr electrolytes was only –60 mA cm−2, which was attained at a more negative potential of –1.05 V and with a substantially lower FE1,3-BD of 41–45%. We also conducted e-C2H2R at −1.0 V in KF electrolyte and found that the FE1,3-BD (56%) and j1,3-BD (−44 mA cm−2) were lower than those in KI (Supplementary Table 15). Overall, these results indicate that the presence of I− species in the catholyte boosts 1,3-BD production, and that the overpotential to obtain a high FE1,3-BD and j1,3-BD is 0.05 V less negative. Interestingly, the FEs and j for butenes (1-butene, trans-2-butene and cis-2-butene) were ~2–6 times higher in K2SO4, KCl and KBr compared with in KI, indicating that the formation of these C4 olefins is more favourable in the former electrolytes.
We now assessed whether improvements in 1,3-BD production were due to differences in the surface areas of the Cu2ONC-derived catalysts immersed in the various electrolytes. To do this, we normalized the j1,3-BD with the electrochemically active surface areas (ECSAs) of the catalysts after they were used for e-C2H2R (Fig. 4d). The ECSA-normalized current density of 1,3-BD (j1,3-BD, ECSA) produced from e-C2H2R in KI electrolyte at –1.0 V was −0.080 mA cm−2, which is ~45–55% higher than the j1,3-BD, ECSA in K2SO4 (–0.055 mA cm−2), KCl (–0.055 mA cm−2) and KBr (–0.051 mA cm−2) electrolytes. The higher j1,3-BD, ECSA in KI indicates that the ECSA is not the crucial factor in improving 1,3-BD production, and that the Cu2ONC-derived catalyst is intrinsically more active for 1,3-BD production during e-C2H2R with I−. Our SEM analysis also showed that the catalysts have no clear differences in their morphologies regardless of the electrolytes used (Fig. 1a–e). Overall, we rule out differences in particle surface areas and morphology as contributing factors in 1,3-BD production.
Based on these results, we assert that enhancing the amount of Cuδ+–Cu0 species is important in promoting 1,3-BD production from e-C2H2R. This is in line with previous studies which showed an increase in C2+ products formed by C–C coupling during CO2 electrolysis in KI electrolyte32,36,50,51. In these works, the coupled products formed were found to be correlated to the higher amount of Cuδ+–Cu0 species in KI. We postulate that the adsorption of I− on Cu2O, which is the strongest among the halides52,53, prevented the Cuδ+–Cu0 species on the catalyst surface from being fully reduced, even under a negative potential. We note that it is experimentally difficult to accurately determine if Cuδ+–Cu0 ensembles are the exact active sites for 1,3-BD formation. This is because such species may only be viewed by scanning tunnelling microscopy, which further requires well-ordered substrates, and ultrahigh-vacuum and low-temperature conditions for atomic-scale imaging54. However, our e-C2H2R was conducted on powder catalysts, under ambient conditions and in an aqueous electrolyte. We further highlight, for the same aforementioned reasons, that the type of CO2RR active sites on copper catalysts has also not been experimentally proven, even after years of extensive studies55. Thus, we will now investigate, using theoretical calculations, the presence and impact of the Cuδ+–Cu0 species and I− on 1,3-BD production from e-C2H2R.
Computational modelling of 1,3-BD production
We performed DFT calculations at the level of the generalized gradient approximation to gain insight into the acetylene electroreduction pathway to 1,3-BD (Fig. 5 and Supplementary Fig. 11). Two acetylene molecules are sequentially adsorbed and hydrogenated to form a pair of *C2H3 moieties with water molecules from the environment acting as proton donors. These two moieties then couple to produce *C4H6, which desorbs as 1,3-BD (Fig. 5a). Since the pKa of water is 11 orders of magnitude smaller than that of acetylene (14 versus 25), we did not consider the latter as a proton source. We also determined the effect of halides on fully metallic, partially oxidized and fully oxidized copper active sites. Cu(100) with a square-symmetry stripe of copper atoms on top was used to represent the fully metallic phase (Fig. 5b). To simulate a partially oxidized and defective electrode, we utilized the stripe with adsorbed oxygen, denoted as stripe + *O (Fig. 5d). This choice is justified by the high activity of (100) facets for making C–C bonds56, and because oxide-derived copper active sites tend to be rough57,58,59. For the Cu2O phase, we considered a copper-terminated Cu2O(111) slab, which has copper sites in the +1 oxidation state (Fig. 5h). Such a slab was simulated because the copper-terminated Cu2O(111) surface has been previously shown to be more stable than the oxygen-terminated one at potentials under CO2RR conditions60. In all three electrodes, the presence of coadsorbed *I and/or *Cl moieties was considered (Fig. 5c,e,f,i,j; further details of the computational methodology can be found in Methods and in Supplementary Fig. 11 and Supplementary Tables 16–18). To observe clear trends made of sufficient data points, we also included calculations for stripes on Ni(100) and Ag(100) with and without *O and *I.

a, Schematic representation of the reaction pathway of acetylene reduction to 1,3-BD with water as the proton source. Carbon and hydrogen atoms appear in brown and grey, copper is shown in beige. b–j, The active sites considered in the computational models are a square-symmetry stripe on top of a Cu(100) terrace (b), stripe + *I (c), stripe + *O (d), stripe + *O + *I (e), stripe + *O + *Cl (f), stripe + *O + *HSO4 (g), Cu2O(111) (h), Cu2O(111) + *I (i) and Cu2O(111) + *Cl (j).
Using Cu2O(111), we first modelled how the removal of surface *O (*O + 2e− + H2O → * + 2OH−) is influenced by the presence of iodide and chloride. The potential to dissolve an oxygen atom from Cu2O(111) was lowered by ~100 mV when iodide is near the *O species, as compared to chloride (Supplementary Table 19). This indicates that iodide prevents the reduction of Cu2O to copper, in agreement with our experimental observations (Figs. 1h and 2).
We then determined the impact of *I and *Cl on 1,3-BD formation. The calculated activity of all active sites in Fig. 5 and their analogues on nickel and silver can be observed in Fig. 6 as a Sabatier-type volcano plot, where the activity for e-C2H2R is assessed in terms of free energy (see 'Computational methods') and plotted as a function of the free energy of adsorption of acetylene (\(\Delta {G}_{\mathrm{{C}_{2}{H}_{2}}}\)) at pH 7 and −0.85 V versus SHE, which is the least negative potential reported in our experiments. We choose \(\Delta {G}_{\mathrm{{C}_{2}{H}_{2}}}\) as a descriptor because acetylene is the main reactant and, according to the Sabatier principle, C2H2 adsorption should be neither too weak nor too strong for optimal electrocatalytic activity61. For convenience, the free energies in Fig. 6 for all materials are listed in Supplementary Tables 20–22, the acetylene binding distances are in Supplementary Table 23, and the corresponding free-energy diagrams for all the copper-based active sites are depicted in Supplementary Figs. 12 and 13.

The computed reaction free energies are plotted against the adsorption free energies of acetylene (\(\Delta {G}_{\mathrm{{C}_{2}{H}_{2}}}\)) on various electrodes. The active sites included are stripe, stripe + *O + *I, stripe + *O + *Cl, stripe + *O + *HSO4, stripe + *I and stripe + *O on copper, nickel and silver electrodes at −0.85 V versus SHE. The initial hydrogenation step is limiting on the right side of the volcano (green), and the coupling step is limiting on the left side (orange). The colour of the points indicates their limiting step, and the active sites are shown in Fig. 5. Inset: summary of the active sites of each metal. The data points represent the average activities and adsorption energies of each catalyst with different coadsorbed species, while the error bars represent the s.d. of the data in the main panel for each catalyst. Average data for Cu2O(111) are also provided for comparison (see also Supplementary Tables 20–22). The vertical line marks the energy range where acetylene is preferentially adsorbed (left-hand side) or in the gas phase (right-hand side).
Apart from delaying the reduction of the oxide, we found that the anions also modify the binding energies of acetylene differently. To evince this, consider the data points for the Cu(100) stripe (denoted as stripe) in Fig. 6, which are all located on the strong binding side of the volcano plot. With respect to the clean stripe, *O + *Cl strengthens \(\Delta {G}_{\mathrm{{C}_{2}{H}_{2}}}\) and decreases the e-C2H2R activity, while *O + *I weakens \(\Delta {G}_{\mathrm{{C}_{2}{H}_{2}}}\) and increases the activity. We also studied the effect of *O + *HSO4 on the Cu(100) stripe (Fig. 6) and found that it unsuitably strengthens \(\Delta {G}_{\mathrm{{C}_{2}{H}_{2}}}\). Similarly, the combination of *O + *I is beneficial for nickel stripes, but we note that only copper stripe sites are close to the ideal \(\Delta {G}_{\mathrm{{C}_{2}{H}_{2}}}\approx -0.34\,{\mathrm{eV}}\) at −0.85 V versus SHE because nickel binds acetylene and the reaction intermediates too strongly. Conversely, the mild adsorption energies of the copper stripes guarantee, in principle, that the surface will not be poisoned by strongly adsorbed species. In turn, silver sites bind acetylene and the other species too weakly. In fact, silver sites lie to the right of the vertical line in Fig. 6, which indicates that acetylene adsorption might be insufficient on those sites. A detailed description showing the changes in the volcano plot shape and the activities of the surface sites with three different potentials (0, −0.40 and −0.85 V versus SHE) are shown in Supplementary Figs. 14 and 15.
We further considered the nature of the active sites for 1,3-BD formation by means of the atomic partial charges on the stripe + *O + *I using Bader analysis (Supplementary Fig. 16)62. We found that the ensemble of copper stripe atoms in contact with *O and *I have in total one electron fewer than in a fully metallic surface. In addition, Supplementary Fig. 16 shows that the intermediates *C2H2 and *C2H3 adsorb on opposing bridge sites of the stripe edges where one of the copper atoms is partially positively charged and the other is neutral, suggesting that the active sites are indeed Cuδ+–Cu0.
Since the limiting step for 1,3-BD production on copper stripes at −0.85 V versus SHE is the coupling of two *C2H3 moieties, which is a non-electrochemical step, we calculated the transition state for such a coupling on the active site closest to the volcano summit, namely, the stripe + *O + *I using the nudged elastic band method63. To form the C–C bond, a diffusion barrier of 0.56 eV is needed for one *C2H3 moiety to approach the other. Subsequently, C–C coupling takes place with a barrier of 0.47 eV (Supplementary Figs. 17 and 18). As both barriers are surmountable at room temperature64, we conclude that undercoordinated stripe sites with *O and *I are both thermodynamically and kinetically poised to reduce acetylene to 1,3-BD.
The inset in Fig. 6 provides a summary of the trends and shows that various active sites on the partially oxidized copper stripes are close to the top of the volcano, whereas Cu2O(111) is not as active, which suggests that a certain degree of surface reduction is necessary for copper sites to be active for 1,3-BD production. To close the computational analysis, we would like to note that our present model may be supplemented in future studies by means of constant-potential simulations65,66.
Overall, this work has illustrated the feasibility of using e-C2H2R to selectively produce 1,3-BD under mild conditions and with water as the proton source. We have used acetylene, a feedstock that can be derived from two major greenhouse gases in the atmosphere, namely, CO2 and methane. Our collective experimental and computational findings have shown that the formation of Cuδ+–Cu0 sites in the presence of I− results in a more efficient production of 1,3-BD from e-C2H2R. The 1,3-BD that is formed can be extracted from the product gas stream using pressure swing adsorption or extractive distillation3,67. We note that the state-of-the-art thermocatalytic conversion of C2H2 to 1,3-BD has a selectivity of only ~20% (ref. 68). In contrast, we have shown the synthesis of 1,3-BD from e-C2H2R with an FE of 93%. Thus, e-C2H2R is probably a more viable option for the synthesis of 1,3-BD. To further determine if e-C2H2R can be practically implemented, we estimate the energy consumption needed to synthesize 1,3-BD from e-C2H2R when starting from a methane feedstock. By first converting methane to acetylene by electric arc plasma treatment12, and then utilizing the acetylene for e-C2H2R to 1,3-BD, we estimate that the overall energy required to synthesize 1,3-BD is ∼30–85% lesser than from the conventional methods of naphtha, ethane or crude oil cracking (Supplementary Note 2). However, we note that this is only a preliminary estimate that may change in the future as the technology improves. The energy efficiency of our approach can be further improved, either by microwave plasma treatment to synthesize the acetylene feedstock69, or by utilizing the thermodynamically more favourable methanol oxidation as the anodic reaction in the electrolytic cell70.
Conclusions
We showed here that a Cu2ONC-derived electrode with KI electrolyte is an efficient catalyst for the electroreduction of acetylene to 1,3-BD. Remarkably, by employing this set-up, we were able to enhance the formation of 1,3-BD, such that it is now formed as a primary product, with an FE1,3-BD of 93% at −0.85 V versus SHE and a j1,3-BD of −75 mA cm−2 at −1.0 V versus SHE. Operando Raman and XAS spectroscopic analysis revealed that I− helped preserve the oxidized state of Cu2O, resulting in the formation of Cuδ+–Cu0 species on the catalyst under negative applied potentials, which we correlate to the larger amount of 1,3-BD produced. Conversely, e-C2H2R in Cl−-, Br−-and SO42−-based electrolytes produced a lower amount of 1,3-BD due to the substantially higher amount of metallic Cu0 present. We showed using DFT that the ensemble of undercoordinated copper sites with coadsorbed *I and *O results in a favourable adsorption of acetylene and its subsequent hydrogenation to the key intermediate *C2H3. The simulations further show that these sites display surmountable C–C coupling barriers for *C2H3 moieties, and thus enable the selective production of 1,3-BD, which corroborates our experimental findings.
In perspective, this work highlights a potentially more sustainable pathway for 1,3-BD production compared to the conventional thermocatalysis route. The diverse chemistry arising from acetylene electrolysis creates viable alternatives in the synthesis of value-added molecules, which is crucial for the decarbonization of chemical industries. However, a j1,3-BD of −75 mA cm−2 is still insufficient to replace current industrial methods. Thus, future works can focus on enhancing the j1,3-BD towards commercially relevant production rates. Further studies can also be undertaken to improve the selectivity for other C4 compounds such as 1-butene by developing tandem catalysts for e-C2H2R in an iodide electrolyte, or by co-feeding other feedstocks such as CO2 to synthesize other multicarbon compounds.
Methods
Catalyst preparation
The Cu2ONC were prepared using a previously reported method34. In brief, 4 ml of 0.1 M CuSO4·5H2O solution was first added into 360 ml of ultrapure water (18.2 MΩ) under vigorous stirring. Then, 10 ml of 1 M NaOH was added, followed by the addition of 16 ml of 0.2 M ascorbic acid. The solution was stirred for another 30 min. The Cu2ONC were extracted using centrifugation, washed with ultrapure water and ethanol, and then dried at 70 °C in an oven.
For the preparation of the working electrodes, 5 mg of Cu2ONC was mixed with 500 μl of isopropanol and 25 μl of Nafion solution, and the mixture was sonicated. The catalyst ink was then dropcast onto a gas diffusion layer (Sigracet 28BC + 20 wt% FEPD 121 wet proofing) and dried on a hot plate. The catalyst mass loading was 5 mg cm−2.
Electrochemical reduction of C2H2
A three-electrode flow cell set-up was used (Supplementary Fig. 5). Ag/AgCl (saturated KCl, Pine) and nickel foam (Xiamen Lith Machine) were used as the reference electrode and the counter electrode, respectively. Potassium iodide (Meryer, 99.99%), potassium bromide (Meryer, 99.99%), potassium chloride (Meryer, 99.99%), potassium sulfate (Meryer, 99.99%), potassium fluoride (Merck, 99.0%), sodium iodide (Sigma Aldrich, 99.5%) and caesium iodide (Sigma Aldrich, 99.99%) were used without further purification for making catholytes. The cation concentration for all electrolyte mixtures was 1 M. 1 M KOH (Meryer, 99.99%) was used as the anolyte because the oxygen evolution activity is favourable under basic conditions. The cathodic compartment (10 ml of catholyte) and anodic compartment (10 ml of anolyte) were separated by an anion-exchange membrane (Selemion AMV, AGC Asahi Glass). A mixture of C2H2 (Chem-gas, 99.8%) and N2 gas (Chem-gas, 99.99%) was used as the gas feedstock. The total gas flow rate is 50 sccm.
A potentiostat (Gamry Reference 600) was used to control and measure the electrochemical activity. The current interrupt mode was used to compensate for the iR drop during chronoamperometry. Each constant potential electrolysis was carried out for 50 min. The potentials in this work are referenced to the SHE using the average potential of the Ag/AgCl electrode (EAg/AgCl) given by equation (1):
The gaseous products (C2H4, 1,3-BD, 1-butene, trans-2-butene, cis-2-butene) were detected by an online gas chromatograph (GC, Shimadzu GC2014) with a flame ionization detector. H2 is detected with a thermal conductivity detector. The measurements were made over the course of three GC injections (with intervals of 1,140 s between consecutive injections). Details of our chromatography set-up have been previously described71. A second gas chromatograph (Wasson-ECE GCMS) with a flame ionization detector was used to further confirm the gaseous products formed during e-C2H2R.
Characterization of electrodes and electrolytes
For our SEM, ECSA, XRD and Raman spectroscopy characterization, the Cu2ONC catalyst was dropcast onto a graphite disc (Ted Pella). For our XANES characterization, Cu2ONC catalyst coated on a gas diffusion layer was used.
The surface morphologies of the catalysts were characterized by SEM (JEOL JSM-6701F, 5 keV). The pH of the electrolytes was measured using a HI-2002 Edge pH meter (Hanna Instruments).
The ECSAs of the electrodes were estimated using their double-layer capacitances (Cdl). The catalysts were first washed using ultrapure water after e-C2H2R. Their Cdl were then measured in 0.5 M K2SO4 electrolyte under N2 saturation. A three-electrode H-cell (Ag/AgCl reference electrode and a graphite counterelectrode) was used. The scan rates applied were 10, 20, 30, 40 and 50 mV s−1.
XRD analysis was used to determine the composition of the catalyst. The system used was a Bruker D8 Advance (Cu Kα 40 kV, 40 mA) with a 2D Lynxeye XE PSD counter detector, with the incoming signal fixed at 5°.
Raman spectroscopy was performed using a confocal Raman microscope (Horiba Jobin Yvon) in epi-illumination mode (top-down). A helium–neon laser with 633 nm wavelength (Pacific Lasertec, 05-LHP-845, 17 mW) was used as the excitation source. A water-immersion objective lens (LOMO, APO 70×; numerical aperture, 1.23) covered with a 0.013 mm thin Teflon film (American Durafilm) was used for focusing and collecting the incident and scattered laser light72. The backscattered light was filtered through a 633 nm edge filter, before being directed into a spectrometer (LabRAM HR evolution)/charge-coupled device detector (Syncerity OE). The acquisition time for each spectrum was 30 s.
XAS measurements at the Cu K-edge were performed at the XAFCA facility of the Singapore Synchrotron Light Source. The XAS spectra of the Cu2ONC catalysts were acquired under e-C2H2R reaction conditions using a home-built electrochemical cell with a platinum-wire counterelectrode and an Ag/AgCl reference (Supplementary Fig. 4). The XAS and EXAFS data analysis was performed using ATHENA and ARTEMIS, respectively, which are part of the Demeter software package73.
Computational methods
Three different copper-based electrode models were considered. We selected Cu(100) with a square-symmetry stripe on top of it to represent rough Cu0 sites. To represent oxidized copper sites, we used a copper-terminated Cu2O(111) facet. In addition, to simulate a partially oxidized and defective electrode, we utilized the stripe with adsorbed O, denoted as stripe + *O. We also considered the coadsorption of either *I or *Cl on all surface models. In total, seven different electrode models have been considered (Supplementary Fig. 11). We also considered *O + *HSO4 on the copper stripes.
Periodic slabs were constructed that had four atomic layers for the stripe and four copper atomic layers for Cu2O(111). During structural optimization, the two bottommost layers remained fixed to provide a bulk environment for the surface layers. All slab models were built from the converged Perdew–Burke-Ernzerhof (PBE) lattice constants, which were found to be a = 3.64 Å for metallic copper and a = 4.31 Å for Cu2O. The PBE lattice parameter of Cu2O was taken from the literature74. For the Cu(100) stripe, 5 × 4 supercells were used; in turn, a 2 × 2 supercell was used for Cu2O(111). Supplementary Fig. 11 provides visual representations of the slab models.
All energy values and corresponding minimum energy structures were obtained from DFT calculations carried out with the VASP code75, using the PBE exchange-correlation functional76, which provides accurate representations of transition metals in the bulk77,78 and their low Miller index surfaces79. In all model systems, the Cu(3d) shell is filled, such that no Hubbard-like (U–J) terms were necessary. The valence electron density was expanded in a plane-wave basis set with a kinetic energy cut-off of 450 eV. The projector augmented wave method80, as implemented in VASP81, was used to incorporate the effect of core electrons on the valence electron density. To facilitate convergence of the self-consistent field procedure, electronic temperature smearing at the Fermi level was used following the Methfessel–Paxton approach82 with a value of 0.2 eV for metal slabs, while a Gaussian smearing of 0.1 eV was used for the oxide. Upon convergence, all total energies were extrapolated to 0 K. The numerical integration in the reciprocal space was carried out using Monkhorst–Pack83 grids of 2 × 3 × 1 special k-points for the Cu(100) stripes and 2 × 2 × 1 for Cu2O(111). These k-point grids ensured convergence of the adsorption energies within ±0.05 eV. To prevent spurious electrostatic interactions, the periodically repeated images in the vertical direction were separated by more than 15 Å of vacuum and dipole corrections were added. Geometry optimizations utilized the conjugate-gradient optimization algorithm until the maximum residual force on all atoms was below 0.05 eV Å−1. The calculations on nickel stripes were spin unrestricted and made with a lattice constant of 3.52 Å. In turn, the calculations on silver stripes were spin restricted and made with a lattice constant of 4.16 Å. In both cases, the geometry optimizations started from analogous configurations as those found for copper stripes. Furthermore, boxes of 9 × 10 × 11 Å3 were used to calculate the energy of H2, H2O, acetylene (C2H2) and 1,3-BD (C4H6), considering the Γ-point only, using Gaussian smearing and an electronic temperature of 0.001 eV, further extrapolated to 0 K.
The reaction free energies were approximated as ∆G ≈ ∆EDFT + ∆ZPE − T∆S, where ∆EDFT is the reaction energy calculated with DFT, ∆ZPE is the zero-point energy change between reactants and products, and T∆S is the entropy correction at 298.15 K. The ZPEs of all species and vibrational S of the adsorbed species were computed from the vibrational frequencies obtained with DFT at T = 298.15 K, within the harmonic oscillator approximation. For free molecules, the values of S were calculated from data in thermodynamic tables84 at 298.15 K (Supplementary Table 16). To remove the intrinsic error of the PBE functional in the energy of gas-phase molecules, the computed total energy values of C2H2 and C4H6 were corrected by 0.12 and 0.67 eV, respectively. These corrections were obtained using the semiempirical method described by Granda-Marulanda et al.85, based on the difference between DFT-calculated and experimental formation energies. A liquid-phase correction of −0.09 eV was used for water86.
The computational hydrogen electrode was used to assess the free energy of proton–electron pairs86. The additive inverse of the activity used to make the volcano plots (−∆G) is defined as the most positive reaction free energy among the electrochemical steps at a given applied potential: ∆G = max(∆G1,∆G2,∆G3). The free energies of the electrochemical steps at pH 7 and various applied potentials were computed as: ∆Gi(U) = ∆Gi(0) + eU − 0.059pH.
We did not account for water solvation in our calculations because acetylene, butadiene and the reaction intermediates do not have polar functional groups able to make strong hydrogen bonds with water. To illustrate this, we computed the solvation stabilization for *C2H2 and *C2H2 + *C2H3 adsorbed on the copper stripe in two ways: using the VASPsol implicit solvation method implemented in VASP87,88 and using an iterative microsolvation method89,90. Supplementary Table 17 shows that water does not appreciably contribute to the stabilization of the adsorbed intermediates, as the solvation energies do not exceed 0.05 eV per adsorbate in any case. In contrast, we observe for *C2H2 and *C2H2 + *C2H3 on the stripe + *O a clear stabilization in the presence of *I and *Cl, as shown in Supplementary Table 18.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper, its Supplementary Information files and online in the Zenodo depository at https://doi.org/10.5281/zenodo.13997798. Source experimental and theoretical calculation data are provided in the Supplementary Information or are available from the corresponding authors upon reasonable request.
References
Schiffer, Z. J. & Manthiram, K. Electrification and decarbonization of the chemical industry. Joule 1, 10–14 (2017).
Weissernel, K. & Arpe, H.-J. Industrial Organic Chemistry (Wiley-VCH, 1997).
White, W. C. Butadiene production process overview. Chem. Biol. Interact. 166, 10–14 (2007).
Mori, Y., Noda, S., Shirai, T. & Kondo, A. Direct 1,3-butadiene biosynthesis in Escherichia coli via a tailored ferulic acid decarboxylase mutant. Nat. Commun. 12, 2195 (2021).
Yang, M. & You, F. Comparative techno-economic and environmental analysis of ethylene and propylene manufacturing from wet shale gas and naphtha. Ind. Eng. Chem. Res. 56, 4038–4051 (2017).
Grub, J. & Lӧser, E. in Ullmann’s Encyclopedia of Industrial Chemistry Vol. 6 (ed. Ley, C.) 381–396 (Wiley-VCH, 2012).
Sun, H. N. & Wristers, J. P. in Kirk‐Othmer Encyclopedia of Chemical Technology Vol. 4 (eds Kirk, R. E. & Othmer, D. F.) 365–392 (Wiley-VCH, 2000).
Makshina, E. V. et al. Review of old chemistry and new catalytic advances in the on-purpose synthesis of butadiene. Chem. Soc. Rev. 43, 7917–7953 (2014).
Angelici, C., Velthoen, M. E. Z., Weckhuysen, B. M. & Bruijnincx, P. C. A. Effect of preparation method and CuO promotion in the conversion of ethanol into 1,3-butadiene over SiO2–MgO catalysts. ChemSusChem 7, 2505–2515 (2014).
Shylesh, S. et al. From sugars to wheels: the conversion of ethanol to 1,3-butadiene over metal-promoted magnesia-silicate catalysts. ChemSusChem 9, 1462–1472 (2016).
Trotuş, I. T., Zimmermann, T. & Schüth, F. Catalytic reactions of acetylene: a feedstock for the chemical industry revisited. Chem. Rev. 114, 1761–1782 (2014).
Dinh, D. K. et al. Efficient methane-to-acetylene conversion using low-current arcs. RSC Adv. 9, 32403–32413 (2019).
McEnaney, J. M. et al. A cyclic electrochemical strategy to produce acetylene from CO2, CH4, or alternative carbon sources. Sustain Energy Fuels 4, 2752–2759 (2020).
Montzka, S. A., Dlugokencky, E. J. & Butler, J. H. Non-CO2 greenhouse gases and climate change. Nature 476, 43–50 (2011).
Davitt, H. J. & Albright, L. F. Electrochemical hydrogenation of ethylene, acetylene, and ethylene–acetylene mixtures. J. Electrochem. Soc. 118, 236–242 (1971).
Beltowska-Brzezinska, M., Luczak, T., Maczka, M., Baltruschat, H. & Müller, U. Ethyne oxidation and hydrogenation on porous Pt electrode in acidic solution. J. Electroanal. Chem. 519, 101–110 (2002).
Zhang, L. et al. Efficient electrocatalytic acetylene semihydrogenation by electron-rich metal sites in N-heterocyclic carbene metal complexes. Nat. Commun. 12, 6574 (2021).
Gao, Y., Tsuji, H., Hattori, H. & Kita, H. New on-line mass spectrometer system designed for platinum-single crystal electrode and electroreduction of acetylene. J. Electroanal. Chem. 372, 195–200 (1994).
Chen, Z., Cai, C. & Wang, T. Identification of copper as an ideal catalyst for electrochemical alkyne semi-hydrogenation. J. Phys. Chem. C 126, 3037–3042 (2022).
Jia, S. et al. Preparation of trimetallic electrocatalysts by one-step co-electrodeposition and efficient CO2 reduction to ethylene. Chem. Sci. 13, 7509–7515 (2022).
Zhao, B. H. et al. Economically viable electrocatalytic ethylene production with high yield and selectivity. Nat. Sustain 6, 827–837 (2023).
Xue, W. et al. Electrosynthesis of polymer-grade ethylene via acetylene semihydrogenation over undercoordinated Cu nanodots. Nat. Commun. 14, 1–9 (2023).
Huang, B., Durante, C., Isse, A. A. & Gennaro, A. Highly selective electrochemical hydrogenation of acetylene to ethylene at Ag and Cu cathodes. Electrochem. Commun. 34, 90–93 (2013).
Shi, R. et al. Room-temperature electrochemical acetylene reduction to ethylene with high conversion and selectivity. Nat. Catal. 4, 565–574 (2021).
Bu, J. et al. Selective electrocatalytic semihydrogenation of acetylene impurities for the production of polymer-grade ethylene. Nat. Catal. 4, 557–564 (2021).
Wang, S. et al. Highly efficient ethylene production via electrocatalytic hydrogenation of acetylene under mild conditions. Nat. Commun. 12, 7072 (2021).
An, S. et al. Functional aqueous zinc–acetylene batteries for electricity generation and electrochemical acetylene reduction to ethylene. Angew. Chem. Int. Ed. 61, e202116370 (2022).
Arán-Ais, R. M., Scholten, F., Kunze, S., Rizo, R. & Roldan Cuenya, B. The role of in situ generated morphological motifs and Cu(I) species in C2+ product selectivity during CO2 pulsed electroreduction. Nat. Energy 5, 317–325 (2020).
Xiao, H., Goddard, W. A., Cheng, T. & Liu, Y. Cu metal embedded in oxidized matrix catalyst to promote CO2 activation and CO dimerization for electrochemical reduction of CO2. Proc. Natl Acad. Sci. USA 114, 6685–6688 (2017).
Lv, X. et al. Grain refining enables mixed Cu+/Cu0 states for CO2 electroreduction to C2+ products at high current density. Appl Catal. B 324, 122272 (2023).
Arán-Ais, R. M., Gao, D. & Roldan Cuenya, B. Structure- and electrolyte-sensitivity in CO2 electroreduction. Acc. Chem. Res. 51, 2906–2917 (2018).
Gao, D., Scholten, F. & Roldan Cuenya, B. Improved CO2 electroreduction performance on plasma-activated Cu catalysts via electrolyte design: halide effect. ACS Catal. 7, 5112–5120 (2017).
Ma, W. et al. Electrocatalytic reduction of CO2 to ethylene and ethanol through hydrogen-assisted C–C coupling over fluorine-modified copper. Nat. Catal. 3, 478–487 (2020).
Ke, W. H., Hsia, C. F., Chen, Y. J. & Huang, M. H. Synthesis of ultrasmall Cu2O nanocubes and octahedra with tunable sizes for facet-dependent optical property examination. Small 12, 3530–3534 (2016).
Huang, Y., Ong, C. W. & Yeo, B. S. Effects of electrolyte anions on the reduction of carbon dioxide to ethylene and ethanol on copper (100) and (111) surfaces. ChemSusChem 11, 3299–3306 (2018).
Gao, D. et al. Selective CO2 electroreduction to ethylene and multicarbon alcohols via electrolyte-driven nanostructuring. Angew. Chem. Int. Ed. 58, 17047–17053 (2019).
Varela, A. S., Ju, W., Reier, T. & Strasser, P. Tuning the catalytic activity and selectivity of Cu for CO2 electroreduction in the presence of halides. ACS Catal. 6, 2136–2144 (2016).
Möller, T. et al. Electrocatalytic CO2 reduction on CuOx nanocubes: tracking the evolution of chemical state, geometric structure, and catalytic selectivity using operando spectroscopy. Angew. Chem., Int. Ed. 59, 17974–17983 (2020).
Ren, D. et al. Selective electrochemical reduction of carbon dioxide to ethylene and ethanol on copper(I) oxide catalysts. ACS Catal. 5, 2814–2821 (2015).
Shirakawa, H., Ito, T. & Ikeda, S. Raman scattering and electronic spectra of poly(acetylene). Polym. J. 4, 460–462 (1973).
Tromans, D. & Silva, J. C. Anodic behavior of copper in iodide solutions: comparison with chloride and effect of benzotriazole‐type inhibitors. J. Electrochem. Soc. 143, 458–465 (1996).
Aben, T. & Tromans, D. Anodic polarization behavior of copper in aqueous bromide and bromide/benzotriazole solutions. J. Electrochem. Soc. 142, 398–404 (1995).
Tromans, D. & Silva, J. C. Behavior of copper in acidic sulfate solution: comparison with acidic chloride. Corrosion 53, 171–178 (1997).
Keil, P., Frahm, R. & Lützenkirchen-Hecht, D. Native oxidation of sputter deposited polycrystalline copper thin films during short and long exposure times: comparative investigation by specular and non-specular grazing incidence X-ray absorption spectroscopy. Corros. Sci. 52, 1305–1316 (2009).
Cave, E. R. et al. Trends in the catalytic activity of hydrogen evolution during CO2 electroreduction on transition metals. ACS Catal. 8, 3035–3040 (2018).
Sahin, Y., Pekmez, K. & Yıldız, A. Electrochemical polymerization of acetylene with copper catalyst on platinum and copper electrodes. Synth. Met. 129, 117–121 (2002).
Pässler, P. et al. in Ullmann’s Encyclopedia of Industrial Chemistry Vol. 1 (ed. Ley, C.) 277–326 (Wiley-VCH, 2011).
Shiratsuchi, R., Aikoh, Y. & Nogami, G. Pulsed electroreduction of CO2 on copper electrodes. J. Electrochem. Soc. 140, 3479–3482 (1993).
Singh, M. R., Kwon, Y., Lum, Y., Ager, J. W. & Bell, A. T. Hydrolysis of electrolyte cations enhances the electrochemical reduction of CO2 over Ag and Cu. J. Am. Chem. Soc. 138, 13006–13012 (2016).
Li, H. et al. High-rate CO2 electroreduction to C2+ products over a copper–copper iodide catalyst. Angew. Chem. Int. Ed. 60, 14329–14333 (2021).
Gao, D., Arán-Ais, R. M., Jeon, H. S. & Roldan Cuenya, B. Rational catalyst and electrolyte design for CO2 electroreduction towards multicarbon products. Nat. Catal. 2, 198–210 (2019).
Choon, H. B. N. & Wai, Y. F. Shape evolution of Cu2O nanostructures via kinetic and thermodynamic controlled growth. J. Phys. Chem. B 110, 20801–20807 (2006).
Lefevre, G., Walcarius, A., Ehrhardt, J. J. & Bessiere, J. Sorption of iodide on cuprite (Cu2O). Langmuir 16, 4519–4527 (2000).
Shields, S. S. & Gupta, J. A. STM study of surface restructuring of oxidized Cu(100). Surf. Sci. 740, 122403 (2024).
Stephens, I. E. L. et al. 2022 roadmap on low temperature electrochemical CO2 reduction. J. Phys. Energy 4, 042003 (2022).
Li, H., Li, Y., Koper, M. T. M. & Calle-Vallejo, F. Bond-making and breaking between carbon, nitrogen, and oxygen in electrocatalysis. J. Am. Chem. Soc. 136, 15694–15701 (2014).
Verdaguer-Casadevall, A. et al. Probing the active surface sites for CO reduction on oxide-derived copper electrocatalysts. J. Am. Chem. Soc. 137, 9808–9811 (2015).
Pique, O., Vines, F., Illas, F. & Calle-Vallejo, F. Elucidating the structure of ethanol-producing active sites at oxide-derived Cu electrocatalysts. ACS Catal. 10, 10488–10494 (2020).
Cheng, D. et al. The nature of active sites for carbon dioxide electroreduction over oxide-derived copper catalysts. Nat. Commun. 12, 395 (2021).
Nie, X., Griffin, G. L., Janik, M. J. & Asthagiri, A. Surface phases of Cu2O(111) under CO2 electrochemical reduction conditions. Catal. Commun. 52, 88–91 (2014).
Medford, A. J. et al. From the Sabatier principle to a predictive theory of transition-metal heterogeneous catalysis. J. Catal. 328, 36–42 (2015).
Henkelman, G., Arnaldsson, A. & Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 36, 354–360 (2006).
Sheppard, D., Xiao, P., Chemelewski, W., Johnson, D. D. & Henkelman, G. A generalized solid-state nudged elastic band method. J. Chem. Phys. 136, 074103 (2012).
Nitopi, S. et al. Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte. Chem. Rev. 119, 7610–7672 (2019).
Goodpaster, J. D., Bell, A. T. & Head-Gordon, M. Identification of possible pathways for C–C bond formation during electrochemical reduction of CO2: new theoretical insights from an improved electrochemical model. J. Phys. Chem. Lett. 7, 1471–1477 (2016).
Sundararaman, R., Goddard, W. A. & Arias, T. A. Grand canonical electronic density-functional theory: algorithms and applications to electrochemistry. J. Chem. Phys. 146, 114104 (2017).
Kishida, K. et al. Recognition of 1,3‐butadiene by a porous coordination polymer. Angew. Chem. Int. Ed. 55, 13784–13788 (2016).
Agbaba, Ö., Trotuş, I. T., Schmidt, W. & Schüth, F. Light olefins from acetylene under pressurized conditions. Ind. Eng. Chem. Res. 62, 1819–1825 (2023).
Wnukowski, M., van de Steeg, A. W., Hrycak, B., Jasiński, M. & van Rooij, G. J. Influence of hydrogen addition on methane coupling in a moderate pressure microwave plasma. Fuel 288, 119674 (2021).
Badreldin, A., Youssef, E., Djire, A., Abdala, A. & Abdel-Wahab, A. A critical look at alternative oxidation reactions for hydrogen production from water electrolysis. Cell Rep. Phys. Sci. 4, 101427 (2023).
Ting, L. R. L., Peng, Y. & Yeo, B. S. Mechanistic insights into the selective electroreduction of crotonaldehyde to crotyl alcohol and 1-butanol. ChemSusChem 14, 2963–2971 (2021).
Deng, Y. & Yeo, B. S. Characterization of electrocatalytic water splitting and CO2 reduction reactions using in situ/operando Raman spectroscopy. ACS Catal. 7, 7873–7889 (2017).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537–541 (2005).
Islam, M. M., Diawara, B., Maurice, V. & Marcus, P. Bulk and surface properties of Cu2O: a first-principles investigation. J. Mol. Struct. THEOCHEM 903, 41–48 (2009).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Janthon, P., Kozlov, S. M., Viñes, F., Limtrakul, J. & Illas, F. Establishing the accuracy of broadly used density functionals in describing bulk properties of transition metals. J. Chem. Theory Comput. 9, 1631–1640 (2013).
Janthon, P. et al. Bulk properties of transition metals: a challenge for the design of universal density functionals. J. Chem. Theory Comput. 10, 3832–3839 (2014).
Vega, L., Ruvireta, J., Viñes, F. & Illas, F. Jacob’s Ladder as sketched by Escher: assessing the performance of broadly used density functionals on transition metal surface properties. J. Chem. Theory Comput. 14, 395–403 (2018).
Blochl, P. E. Projector augmented wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Methfessel, M. & Paxton, A. T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 40, 3616–3621 (1989).
Monkhorst, H. J. & Pack, J. D. Special points for Brillonin-zone integrations. Phys. Rev. B 13, 5188–5192 (2019).
Lide, D. R. CRC Handbook of Chemistry and Physics (CRC Press, 2005).
Granda-Marulanda, L. P. et al. A semiempirical method to detect and correct DFT-based gas-phase errors and its application in electrocatalysis. ACS Catal. 10, 6900–6907 (2020).
Nørskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886–17892 (2004).
Mathew, K., Kolluru, V. S. C., Mula, S., Steinmann, S. N. & Hennig, R. G. Implicit self-consistent electrolyte model in plane-wave density-functional theory. J. Chem. Phys. 151, 234101 (2019).
Mathew, K., Sundararaman, R., Letchworth-Weaver, K., Arias, T. A. & Hennig, R. G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 140, 084106 (2014).
Rendón-Calle, A., Builes, S. & Calle-Vallejo, F. Substantial improvement of electrocatalytic predictions by systematic assessment of solvent effects on adsorption energies. Appl. Catal. B 276, 119147 (2020).
Romeo, E., Illas, F. & Calle-Vallejo, F. Evaluating adsorbate–solvent interactions: are dispersion corrections necessary? J. Phys. Chem. C 127, 10134–10139 (2023).
Acknowledgements
The authors thank Shell Global Solutions International BV (A-8001111-00-00), the Ministry of Education Singapore (A-8001571-00-00), the National Research Foundation of Singapore (Urban Solutions and Sustainability, Industry Alignment Fund (Pre-Positioning) Programme (A-0004543-00-00), the projects PID2021-127957NB-I00, TED2021-132550B-C21, PID2021-126076NB-I00, and the María de Maeztu Excellence Unit CEX2021-001202-M funded by the Spanish MCIN/AEI/10.13039/501100011033 and the European Union for the financial support of this research. We thank the Red Española de Supercomputación for computational resources (grants QHS-2023-2-0007 and QHS-2023-3-0006). E.R. thanks the Spanish MCIN for an FPI PhD grant (MDM-2017-0767-20-1). The authors thank V. Urade and S. van Bavel for their valuable advice and feedback throughout this project.
Author information
Authors and Affiliations
Contributions
B.S.Y. and F.C.-V. coordinated the project. W.J.T. designed the experiments and analysed the results. E.R. and F.C.-V. carried out the computational simulations and modelling. S.X. assisted with the XAFS measurements. B.R. and F.I. provided feedback and advice on the experimental/computational procedures and results. All authors read and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Catalysis thanks Christian Durante, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Notes 1 and 2, Figs. 1–18 and Tables 1–23.
Supplementary Data 1
Direct coordinates for DFT calculations
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Teh, W.J., Romeo, E., Xi, S. et al. Selective electroreduction of acetylene to 1,3-butadiene on iodide-induced Cuδ+–Cu0 sites. Nat Catal 7, 1382–1393 (2024). https://doi.org/10.1038/s41929-024-01250-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41929-024-01250-0
This article is cited by
-
Rekindling the use of acetylene as a chemical building block
Nature Chemical Engineering (2025)
