
Abstract
Multiple next-generation molecules targeting estrogen receptor α (ERα) are being investigated in breast cancer clinical trials, encompassing thousands of women globally. Development of these molecules was partly motivated by the discovery of resistance-associated mutations in ESR1 (encodes ERα). Here, we studied the impact of ERα antagonist/degraders against Esr1 mutations expressed in mouse mammary glands. Inhibition of mutant ERα induced mixed-lineage cells, characterized by aberrant co-engagement of normally disparate master transcription factors. Lineage infidelity was also observed in Esr1-wild-type mice upon long-term estrogen deprivation. In ER+ breast cancer biopsy specimens, heavily pretreated tumors with no ESR1 mutation detected (NMD) frequently exhibited mixed-lineage features. ESR1-mutant tumors generally retained luminal features and higher ERα activity and exhibited an anti-proliferative response to the ERα antagonist giredestrant. ESR1-mutant tumors acquired mixed-lineage features following treatment. Lineage heterogeneity in advanced ER+ breast cancer may underpin the differential benefit of investigational ERα therapeutics observed in ESR1-mutant versus NMD contexts.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
27,99 € / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
118,99 € per year
only 9,92 € per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout








Similar content being viewed by others

Data availability
Raw and processed files from single-cell/bulk RNA-seq and ATAC-seq datasets have been uploaded to the NCBI Gene Expression Omnibus under the following accession IDs: GSE205573 (scRNA-seq), GSE207079 (RNA-seq), GSE205575 (RNA-seq), GSE205576 (ATAC-seq), and GSE205577 (RNA-seq). The source data for Figs. 1–6 and Extended Data figures have been provided as Source Data files. All other data supporting the findings of this study are available from the corresponding author on reasonable request; such requests will be processed within 30 days. Source data are provided with this paper.
Code availability
Analysis code is available at https://doi.org/10.5281/zenodo.13916857. For ATAC-seq, all analysis code uses publicly available R packages. For single and bulk RNA-seq, some analyses were performed using custom, in-house packages which are not publicly available, but comparable analyses can be performed using publicly available R packages.
References
Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Will, M., Liang, J., Metcalfe, C. & Chandarlapaty, S. Therapeutic resistance to anti-oestrogen therapy in breast cancer. Nat. Rev. Cancer 23, 673–685 (2023).
Mottamal, M., Kang, B., Peng, X. & Wang, G. From pure antagonists to pure degraders of the estrogen receptor: evolving strategies for the same target. ACS Omega 6, 9334–9343 (2021).
Liang, J. et al. GDC-9545 (giredestrant): a potent and orally bioavailable selective estrogen receptor antagonist and degrader with an exceptional preclinical profile for ER+ breast cancer. J. Med. Chem. 64, 11841–11856 (2021).
Liang, J. et al. Giredestrant counters a progesterone hypersensitivity program driven by estrogen receptor mutations in breast cancer. Sci. Transl. Med. 14, eabo5959 (2022).
Martín, M. et al. Giredestrant for estrogen receptor-positive, HER2-negative,previously treated advanced breast cancer: results from the randomized, phase II acelERA Breast Cancer Study. J. Clin. Oncol 42, 2149–2160 (2024).
Toy, W. et al. ESR1 ligand-binding ___domain mutations in hormone-resistant breast cancer. Nat. Genet. 45, 1439–1445 (2013).
Robinson, D. R. et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 45, 1446–1451 (2013).
Jeselsohn, R. et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin. Cancer Res. 20, 1757–1767 (2014).
Schiavon, G. et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci. Transl. Med. 7, 313ra182 (2015).
Guan, J. et al. Therapeutic ligands antagonize estrogen receptor function by impairing its mobility. Cell 178, 949–963 e918 (2019).
O’Leary, B. et al. The genetic landscape and clonal evolution of breast cancer resistance to palbociclib plus fulvestrant in the PALOMA-3 trial. Cancer Discov. 8, 1390–1403 (2018).
Turner, N. C. et al. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): a multicentre, multicohort, phase 2a, platform trial. Lancet Oncol. 21, 1296–1308 (2020).
Arnesen, S. et al. Estrogen receptor alpha mutations in breast cancer cells cause gene expression changes through constant activity and secondary effects. Cancer Res. 81, 539–551 (2021).
Nilsson, M. E. et al. Measurement of a comprehensive sex steroid profile in rodent serum by high-sensitive gas chromatography-tandem mass spectrometry. Endocrinology 156, 2492–2502 (2015).
Ku, S. Y. et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355, 78–83 (2017).
Bach, K. et al. Differentiation dynamics of mammary epithelial cells revealed by single-cell RNA sequencing. Nat. Commun. 8, 2128 (2017).
Giraddi, R. R. et al. Single-cell transcriptomes distinguish stem cell state changes and lineage specification programs in early mammary gland development. Cell Rep. 24, 1653–1666 (2018).
Soares, E. & Zhou, H. Master regulatory role of p63 in epidermal development and disease. Cell Mol. Life Sci. 75, 1179–1190 (2018).
Gatti, V. et al. p63 at the crossroads between stemness and metastasis in breast cancer.Int. J. Mol. Sci. 20, 2683 (2019).
Visvader, J. E. & Stingl, J. Mammary stem cells and the differentiation hierarchy: current status and perspectives. Genes Dev. 28, 1143–1158 (2014).
Wuidart, A. et al. Early lineage segregation of multipotent embryonic mammary gland progenitors. Nat. Cell Biol. 20, 666–676 (2018).
Keenan, A. B. et al. ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 47, W212–W224 (2019).
Li, Z. et al. ESR1 mutant breast cancers show elevated basal cytokeratins and immune activation. Nat. Commun 13, 2011 (2022).
Jhaveri, K. L. et al. Phase Ia/b study of giredestrant ±palbociclib and ±luteinizing hormone-releasing hormone agonists in estrogen receptor-positive, HER2-negative, locally advanced/metastatic breast cancer. Clin. Cancer Res. 30, 754–766 (2024).
Risom, T. et al. Transition to invasive breast cancer is associated with progressive changes in the structure and composition of tumor stroma. Cell 185, 299–310 (2022).
DeRose, Y. S. et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 17, 1514–1520 (2011).
Sikora, M. J. et al. Invasive lobular carcinoma cell lines are characterized by unique estrogen-mediated gene expression patterns and altered tamoxifen response. Cancer Res 74, 1463–1474 (2014).
Li, S. et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 4, 1116–1130 (2013).
Nguyen, Q. H. et al. Profiling human breast epithelial cells using single cell RNA sequencing identifies cell diversity. Nat. Commun. 9, 2028 (2018).
Bhat-Nakshatri, P. et al. A single-cell atlas of the healthy breast tissues reveals clinically relevant clusters of breast epithelial cells. Cell Rep. Med. 2, 100219 (2021).
Asselin-Labat, M. L. et al. Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat. Cell Biol. 9, 201–209 (2007).
Carroll, J. S. et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 122, 33–43 (2005).
Read, L. D., Greene, G. L. & Katzenellenbogen, B. S. Regulation of estrogen receptor messenger ribonucleic acid and protein levels in human breast cancer cell lines by sex steroid hormones, their antagonists, and growth factors. Mol. Endocrinol. 3, 295–304 (1989).
Saceda, M. et al. Regulation of the estrogen receptor in MCF-7 cells by estradiol. Mol. Endocrinol. 2, 1157–1162 (1988).
Overpeck, J. G., Colson, S. H., Hohmann, J. R., Applestine, M. S. & Reilly, J. F. Concentrations of circulating steroids in normal prepubertal and adult male and female humans, chimpanzees, rhesus monkeys, rats, mice, and hamsters: a literature survey. J. Toxicol. Environ. Health 4, 785–803 (1978).
Shalabi, S. F. et al. Evidence for accelerated aging in mammary epithelia of women carrying germline. Nat. Aging 1, 838–849 (2021).
Kumar, T. et al. A spatially resolved single-cell genomic atlas of the adult human breast. Nature 620, 181–191 (2023).
Bidard, F.-C. et al. Elacestrant (oral selective estrogen receptor degrader) versus standard endocrine therapy for estrogen receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: results from the randomized phase III EMERALD Trial. J. Clin. Oncol. 40, 3246–3256 (2022).
Oliveira, M. et al. Camizestrant, a next generation oral SERD vs fulvestrant in post-menopausal women with advanced ER-positive HER2-negative breast cancer: results of the randomized, multi-dose phase 2 SERENA-2 trial [abstract GS3-02]. Cancer Res. 83, GS3-02 (2023).
Quintanal-Villalonga, Á. et al. Lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nat. Rev. Clin. Oncol. 17, 360–371 (2020).
Chan, J. M. et al. Lineage plasticity in prostate cancer depends on JAK/STAT inflammatory signaling. Science 377, 1180–1191 (2022).
Metcalfe, C., Kljavin, N. M., Ybarra, R. & de Sauvage, F. J. Lgr5+ stem cells are indispensable for radiation-induced intestinal regeneration. Cell Stem Cell 14, 149–159 (2014).
Corces, M. R. et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017).
Wu, T. D. & Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26, 873–881 (2010).
Wu, T. D., Reeder, J., Lawrence, M., Becker, G. & Brauer, M. J. GMAP and GSNAP for genomic sequence alignment: enhancements to speed, accuracy, and functionality. Methods Mol. Biol. 1418, 283–334 (2016).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Liberzon, A. et al. Molecular Signatures Database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Zhang, Y. et al. Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137 (2008).
Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
McLeay, R. C. & Bailey, T. L. Motif enrichment analysis: a unified framework and an evaluation on ChIP data. BMC Bioinformatics 11, 165 (2010).
Kulakovskiy, I. V. et al. HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-seq analysis. Nucleic Acids Res. 46, D252–D259 (2018).
Acknowledgements
We thank T. Tran for contributions to the single-cell sequencing experiment; V. Dixit, L. Vermeulen, C. Siebel, F. de Sauvage and A. Chibly for helpful discussions and contributions during the preparation of the manuscript; K. L. Jhaveri, M. Bellet, N. C. Turner, S. Loi, A. Bardia, V. Boni, J. Sohn, T. G. Neilan, R. Villanueva-Vázquez, P. Kabos, L. García-Estévez, E. López-Miranda, J. A. Pérez-Fidalgo, J. M. Pérez-García and E. Lim and respective site staff for providing biopsy specimens in the clinical trial (NCT03332797); and patients and their families for participation in the clinical trial. This work was supported by Roche/Genentech. The funder was involved in the study design, data collection and data interpretation.
Author information
Authors and Affiliations
Contributions
J.L. and C.M. conceived the project, designed the experiments and wrote the manuscript. J.L. performed the animal experiments. B.-E.W. performed animal surgeries. P.A. performed the in vitro experiments. J.L. and L.C. performed tumor xenograft experiments. X.Y. created breast cancer lineage signatures from TCGA data. J.L. and M.H. performed computational analysis on NGS data from NCT03332797 and animal studies. J.G. performed pathology review of tissues/tumors. S.C. performed library preparation for ATAC-seq. Y.L. arranged specimen RNA-seq. S.D. arranged and performed single-cell RNA-seq. H.M.M., J.A., C.-W.C., M.R.G. and J.E.-W. conducted the clinical study, NCT03332797. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
All authors are or were Genentech/Roche employees and own shares of Roche. C.M. is a named co-inventor on patent 11081236 entitled “Diagnostic and therapeutic methods for the treatment of breast cancer.”
Peer review
Peer review information
Nature Cancer thanks Silvia Fre and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Characterization of ESME cells.
(a) Flow cytometry strategy to analyze mammary epithelial lineages. (b) Comparison of (a) to another established flow cytometry panel, which resolves HS and AP cells with Sca-1. (c) Prevalence of mammary populations when sorted with Sca-1 or CD61 panels. Each point is mammary tissue analyzed from one animal; n = 3 tissues per panel. (d) Expression of known epithelial lineage marker genes for cells collected by Sca-1 or CD61 gates via bulk RNA-seq. (e) ESME frequency, grouped by epithelial lineage. For vehicle, n = 6, 7, and 7 tissues for respective genotypes and for giredestrant, n = 7 tissues across genotypes. P-values are comparisons to vehicle-treated cells of the same type. (f) ESME frequency in 3-month old animals which received ovariectomy (OVX), giredestrant or both. Ovariectomies were performed 8 days prior to tissue harvest/analysis; giredestrant was administered for 4 days prior to tissue harvest/analysis. N = 4, 4, 5, 3, 5, 6, 3, 3, 6, 4, 4, and 5 tissues by x-axis order. (g) Esr1 mutant transcript frequency for 3 and 6 month old, Gired-treated animals, inferred from bulk RNA-seq; n = 3 tissues per condition. Each point corresponds to one sample. (h-j) Whole-exome sequencing on mammary tissues; n = 7 whole-exome sequencing samples and n = 1 reference sample. (h) Experimental strategy and analysis. (i) Summary of somatic mutations detected per sample (left) and mutations detected in Esr1 gene per sample (right). (j) Analysis of detected mutations. (Left) Heatmap of gene mutations per sample, colored by mutational frequency. The majority of mutated genes were sample-specific, except for a small number present in all D542G samples across ages, but absent in the WT sample. (Right) To probe the relevance of mutations in these genes, RNA-seq reads were assessed. Heatmap represents percentage of samples harboring mutations per site by RNA reads. Approximately 50% of WT samples harbor mutations at the majority of these sites, suggesting they are not unique to D542G. (k-m) ESME cell frequencies by experimental condition: hormone supplementation without giredestrant treatment (k; n = 4, 4, 4, 4, 5, and 5 tissues by x-axis order), giredestrant treatment for 4 or 10 days, without hormone supplementation (l; n = 3, 6, 6, 3, 6, and 6 tissues by x-axis order), and Esr1-heterozygous animals treated with Gired for 4 days (m, n = 5 tissues per condition). (n) Strategy for dosing mice with giredestrant, then withdrawing drug. Each point is serum giredestrant level in one animal; n = 7 and 5 animals by x-axis order. (o) Frequency of ESME cells pre- and post-drug withdrawal; n = 5 tissues per condition. For (e-f, k-m, o), each point is the percentage of ESME cells among Lin-, EpCAM+ cells from one animal. All animals were 6 months old, unless specified in figure panel. For (c, e-g, k-o), values are mean ± S.D. All p-values were computed by two-sided Mann-Whitney U-test. AP: alveolar progenitor cells, HS: hormone-sensing luminal cells, ESME: endocrine-suppressed, Esr1 mutant-enriched cells.
Extended Data Fig. 2 Single cell RNA-sequencing of Gired-treated, Esr1-WT and mutant mammary epithelia.
(a) Analysis workflow for single-cell RNA-seq (scRNA-seq) data from the Cell Ranger pipeline; see Methods for detailed summary. (b-c) Summary statistics of UMAP clusters. (b, left) Numbers of cells per cluster and (right) proportion of cells in respective clusters across conditions. (c) Marker genes per cluster; see Methods. Unique marker genes are specific to one cluster. (d) UMAP representations, stratified by experimental condition. (e-f) Gene expression analyses; values are normalized expression scores, calculated by merging pipeline. For violin plots, endpoints depict minimum and maximum values; data points are individual cells. (e) Expression of genes encoding the FACS markers, CD49f and CD61. (f) Krt14 and Krt8 gene expression, colored by cluster. (g) Expression of marker genes; markers: Krt14 (basal), Krt8 (luminal), Krt6a and Krt16 (squamous/epidermal); Esr1, Pgr, Foxa1, and Trp63 (transcription factors). In (b, d, e-g), n = 4724, 2822, 4088, 3824, 3863, and 1616 single cells in respective clusters C1-C6.
Extended Data Fig. 3 Analyses of ESME cells by scRNA-seq.
(a-b) Monocle trajectory analysis of scRNA-seq dataset; the analysis identified three ‘states’ (for example trajectories). N = 4724, 2822, 4088, 3824, 3863, and 1616 single cells in respective clusters C1-C6. (a) Reduced dimension plots by states, pseudotime, and UMAP clusters. (b) Summary statistics of states and UMAP clusters. (c) Clustering was re-run for C5 and C6 cells in the ‘Y541S + E2/P4 + Gired’ sample. Analysis workflow to re-cluster Gired-treated, Y541S hormone-sensing luminal and ESME cells. See Methods for detailed summary. (d) UMAP representations of ‘Y541S + E2/P4 + Gired’, C5/C6 re-clustering. Five sub-clusters were generated, SC1-SC5; UMAPs colored by sub-clusters. (e-f) Summary statistics of sub-clusters. (g) UMAP and violin plots of cell-type gene signature scores in sub-clusters, measured as average log2 counts of genes within each signature. For violin plots, endpoints depict minimum and maximum values; data points are individual cells. In (d-e, g), n = 191, 721, 391, 391, and 667 single cells in respective clusters SC1-SC5. (h) Re-clustering of Gired-treated Y541S C5/C6 cells with previously published datasets: mammary developmental stages (GSE111113) and pregnancy/ lactation (GSE106273). GSE111113: E16 (embryonic day 16), E18 (day 18), and P4 (post-natal day 4). UMAP representations by (left to right): experimental condition/ cell type, ESME cell gene signature scores, expression of Krt14, and Krt8. N = 8,180 cells (top) and 28,276 cells (bottom). AP: alveolar progenitor cells, HS: hormone-sensing luminal cells, ESME: endocrine-suppressed, Esr1 mutant-enriched cells.
Extended Data Fig. 4 Bulk RNA-sequencing of Gired-treated, Esr1-WT and mutant mammary epithelial populations.
(a) Principal-component analysis of bulk RNA-seq samples. Principal components were calculated with the top 500 features contributing by variance. ESME cells (CD49f+/CD61low) form a distinct cluster (circle). (b) Volcano representation of differential gene expression between Gired-treated ESME cells and other lineages in Esr1-mutants. Each point represents one gene, colored by differential expression between WT basal and HS (for example lineage-defining genes); n = 15,721 (top) and 15,953 genes (bottom). (c) Gene set enrichment analysis comparing Gired-treated, Esr1 mutant ESME cells to animal-matched basal or hormone-sensing luminal cells. Analyses are stratified by GO terms and HALLMARK gene sets respectively. The top 15 significant (FDR < 0.05) ‘up’ and ‘down’ gene sets are shown per comparison, ranked by normalized enrichment score (NES). If a comparison has fewer than 15 significant ‘up’ and/or ‘down’ gene sets, all significant gene sets are shown. Data points are NES; point sizes reflect -log10 (p-values); p-values computed via the ‘fgsea’ method. Colors – green: gene sets ‘up’ in ESME cells, red: gene sets ‘down’ in ESME cells. Blue text: Keratinocyte and epidermis-related GO terms, red text: estrogen signaling. (d) Average z-score per gene across experimental conditions for gene sets. For violin plots, endpoints depict minimum and maximum values; each point represents one gene in the respective sets. N = 57 and 31 genes in respective signatures. P-values by two-sided Mann-Whitney U-test; for each group, largest p-value from respective pairwise comparisons is shown. (e) TFEA of all marker genes per cluster from single-cell RNA-seq data. Per analysis, the top 10 ‘mean-ranked’ TFs are shown. Lines: TFs are associated by ChEA3 libraries; arrows: further supported by ChIP-seq. AP: alveolar progenitor cells, HS: hormone-sensing luminal cells, ESME: endocrine-suppressed, Esr1 mutant-enriched cells.
Extended Data Fig. 5 ESME-enriched, HALLMARK estrogen response genes are associated with P63.
(a) Analysis of ESME-enriched estrogen response HALLMARK genes; ‘ESTROGEN RESPONSE EARLY’ and ‘ESTROGEN RESPONSE LATE’ genes are pooled for this analysis. Average z-score per gene across conditions for ESME-enriched estrogen response genes. For violin plots, endpoints depict minimum and maximum values. N = 54 genes; each point represents one gene. P-values by two-sided Mann-Whitney U-test. (b-c) ERα levels by immunolabeling; representative images from n = 3 experiments. (b) ERα protein in vehicle or Gired-treated Esr1-mutant tissues. (c) ERα protein in ER+ luminal cells (HS; open arrowhead) or ESME cells (closed arrowhead); dynamic range for ERα signal was adjusted to illustrate the relative similarity in ERα levels between ESME (K14+/K8+) and luminal (K8+) cells. (d) Transcription factor enrichment analysis (TFEA; see Methods) on all estrogen response genes within the HALLMARK signatures (top) or the ESME-enriched subset of genes (bottom). Per analysis, the top 10 ‘mean-ranked’ TFs are shown. Lines: TFs are associated by ChEA3 libraries; arrows: further supported by ChIP-seq. ESME: endocrine-suppressed, Esr1 mutant-enriched cells.
Extended Data Fig. 6 Bulk ATAC-sequencing of Gired-treated, Esr1-WT and mutant mammary glands.
(a) Principal-component analysis of top 500 features contributing by variance for bulk ATAC-seq. Circle: ESME cells. (b) Representative ATAC tracks for Krt6a and Trp63 loci; each track is representative of n = 3 biological replicates, each replicate is sorted cells from one tissue. (c-d) Top 10 enriched motifs per comparison via AME. Percentage of motif enrichment is calculated as ‘% true positive (TP) - % false positive (FP)’ from AME output. Adjusted p-values by one-sided Fisher’s exact test. (c) Comparison of WT basal cell- or HS cell-enriched peaks to each other. (d) Comparison of ESME-unique peaks to those shared between ESME-basal cells or ESME-HS cells. (e) Genes nearest to each ESME-unique peak were identified. Of these, 30% were enriched in ESME cells, shown by z-score heatmap; n = 855 genes. Remaining genes were not captured (10% of genes) or lacked differential expression (60%). All animals are 6 months old. AP: alveolar progenitor cells, HS: hormone-sensing luminal cells, ESME: endocrine-suppressed, Esr1 mutant-enriched cells.
Extended Data Fig. 7 Esr1-mutant associated lineage plasticity and effects of giredestrant.
(a) Average z-score per gene across WT and Esr1-mutant basal cells for lineage-specific gene signatures. Each point represents one gene from a gene signature; n = 45, 31, and 20 genes in respective signatures (top to bottom). (b) Differentially-accessible ATAC peaks between giredestrant vs. vehicle in hormone-sensing luminal cells per genotype. (c) Volcano plot of differential gene expression between WT hormone-sensing luminal cells, with vehicle or giredestrant. Each point is one gene (non-significant genes omitted); n = 2,483 genes shown. (d) GSEA scores between WT HS cells treated with vehicle or giredestrant; p-values computed via the ‘fgsea’ method. Blue text denote estrogen/hormone-associated gene sets. (e) Average z-score per gene across conditions for either giredestrant-repressed genes (n = 1147 genes) or induced genes (n = 1309 genes); each point is one gene defined in (c). (f) Differentially-accessible ATAC peaks between D542G/Y541S vs. WT in HS cells per treatment. (g) Volcano plot of differential ATAC peaks between WT hormone-sensing luminal cells, with vehicle or giredestrant. Each point is one ATAC peak; n = 276,927 peaks total. Peaks are colored by differential accessibility in either (left) WT basal vs. WT luminal cells or (right) Esr1-mutant luminal vs. WT luminal cells. (h) Motif enrichment analysis of these peaks via AME. For each analysis, the opposing peak set was used as a reference. P-values by one-sided Fisher’s exact test. For (a, e), p-values by two-sided Mann-Whitney U-test. For (b-c, f-g), statistical cutoffs: log2-fold change > 1 or < -1, FDR < 0.05. For all violin plots, endpoints depict minimum and maximum values. All animals are 6 months old. AP: alveolar progenitor cells, HS: hormone-sensing luminal cells, ESME: endocrine-suppressed, Esr1 mutant-enriched cells.
Extended Data Fig. 8 Analysis of WT ESME cells by bulk RNA-seq.
(a) Example of a rare P63+/HR+ cell (arrowhead) in WT tissues; representative image from n = 3 experiments. (b) Experimental strategy for performing low-input RNA-sequencing from rare ESME cells (CD49f+/CD61low); n = 3 biological replicates per condition; each biological replicate is sorted cells from one animal. Per biological replicate, a minimum of 1,000 cells were sequenced, pooled from multiple WT tissues. (c) Principal-component analysis of samples from (b). Principal components were calculated with the top 500 features contributing by variance. WT CD49f+/CD61low cells clustered with mutant ESME cells (circled). HS: hormone-sensing luminal cells. (d) Venn diagram representation of differential gene expression between giredestrant-treated ESME cells vs. hormone-sensing luminal cells for WT and Esr1-mutants respectively. Each circle represents genes up (left) or down (right) in ESME cells compared to hormone-sensing luminal cells. Statistical cutoffs: log2-fold change > 1 or < -1, FDR < 0.05. (e-f) GSEA scores between WT ESME and hormone-sensing luminal cells. (e) Top 10 enriched GO terms for each cell type. (f) GO terms associated with skin/epidermis or keratinocytes; normalized enrichment scores shown for respective contrasts between WT and Esr1-mutants, ESME vs. hormone-sensing luminal. P-values computed via the ‘fgsea’ method. (g) Transcription factor enrichment analysis for genes enriched in WT ESME cells compared to hormone-sensing luminal cells. See Methods. (h) Heatmap of z-scores for key ESME cell-associated transcription factors for WT/mutant HS and ESME cells. All samples were treated with giredestrant. Animals were 6 months old. AP: alveolar progenitor cells, HS: hormone-sensing luminal cells, ESME: endocrine-suppressed, Esr1 mutant-enriched cells.
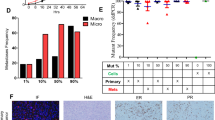
Extended Data Fig. 9 Analysis of lineage markers in PDX models of ER+ breast cancer.
(a) ERα immunolabeling for PDX tumors; markers: K8: luminal, K14: basal. (b) Percentage of PDX tumor cells with KI67-positivity; each point is one tumor. P-values by two-sided Mann-Whitney U-test. (c) Proliferating K14+ tumor cells for each model. (d) Categorization of proliferating cells in each model by K8 and/or K14 positivity. In (a, c), images are from n = 1 experiment and representative of respective conditions. In (b, d), n = 5, 5, 4, 4, 4, and 4 tumors by x-axis order; values are means ± S.D. All animals were approximately 2 months old at tumor injection.
Extended Data Fig. 10 Analyses of lineage-associated transcription factor-programs in breast cancer.
(a) Workflow for analyzing the transcription factor (TF) landscape of n = 1,096 TCGA breast cancer and normal cases by voom-normalized RNA-seq data. The top 500 most variably expressed transcription factors across the cohort were analyzed. (b) K-means clustering of cases; optimal k = 4. (c) UMAP representation and numbers of cases, colored by PAM50 subtype (top) or histological receptor subtype (bottom). N = 173, 304, 486, and 133 tumors by x-axis order. ‘Normal’ refers to normal breast tissue. ‘Unknown’ refers to samples for which data was not unavailable. (d) One-way Welch test to identify most variable transcription factors. F is calculated as the variation of a TF between cluster means / variation within clusters; a larger F-test score represents TFs that are more specific to individual clusters. Each point is one TF; n = 1,271 TFs total. (e) Correlation matrix on top 100 ranked TFs to identify co-occuring transcription factors (for example ‘programs’); four programs identified. (f) Running score and pre-ranked list plots of GSEA results. Plots are shown for ESR1-mutant cases; NMD cases were omitted since all comparisons were non-significant.
Supplementary information
Supplementary Table 1
Supplementary Table 1: Mutations per sample via whole-exome sequencing.
Supplementary Table 2
Supplementary Table 2: Gene signatures used in this study.
Supplementary Table 3
Supplementary Table 3: Marker genes for clusters C1-C6 via single-cell RNA-seq.
Supplementary Table 4
Supplementary Table 4: Summary of GO39932 patient biopsies and K14 immunolabeling.
Source data
Source Data All Figs
Source data for plots in Figs. 1, 2, 5 and 6 and Extended Data Figs. 1–7, 9 and 10.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liang, J., Yao, X., Aouad, P. et al. ERα dysfunction caused by ESR1 mutations and therapeutic pressure promotes lineage plasticity in ER+ breast cancer. Nat Cancer 6, 357–371 (2025). https://doi.org/10.1038/s43018-024-00898-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43018-024-00898-8
