
Abstract
Plasma p-tau217 and tau positron emission tomography (PET) are strong prognostic biomarkers in Alzheimer’s disease (AD), but their relative performance in predicting future cognitive decline among cognitively unimpaired (CU) individuals is unclear. In a head-to-head comparison study including nine cohorts and 1,474 individuals, we show that plasma p-tau217 and medial temporal lobe tau-PET signal display similar associations with cognitive decline on a global cognitive composite test (R2PET = 0.34 versus R2plasma = 0.33, Pdifference = 0.653) and with progression to mild cognitive impairment (hazard ratio (HR)PET = 1.61 (1.48–1.76) versus HRplasma = 1.57 (1.43–1.72), Pdifference = 0.322). Combined plasma and PET models were superior to the single-biomarker models (R2 = 0.35, P < 0.01). Sequential selection using plasma phosphorylated tau at threonine 217 (p-tau217) and then tau-PET reduced the number of participants required for a clinical trial by 94%, compared to a 76% reduction when using plasma p-tau217 alone. Thus, plasma p-tau217 and tau-PET showed similar performance for predicting future cognitive decline in CU individuals, and their sequential use enhances screening efficiency for preclinical AD trials.
Similar content being viewed by others


Main
In recent years, there has been a substantial increase in the availability of biomarkers that reflect core AD neuropathological hallmarks, specifically amyloid-β (Aβ) plaques and tau neurofibrillary tangles1. Tau-PET has shown excellent diagnostic accuracy and strong associations with both concurrent and longitudinal cognitive decline, outperforming established AD biomarkers like amyloid-PET and structural magnetic resonance imaging across the clinical continuum of AD1,2,3,4,5,6,7. However, tau-PET is expensive and labor intensive and has inadequate availability, and its sensitivity to detect the earliest stages of tau aggregation is limited. This is particularly troublesome in individuals with preclinical AD who harbor AD pathology but have not (yet) developed symptoms8. The recent advent of blood-based biomarkers of AD pathology potentially offers a low-cost, non-invasive and scalable alternative9. Within the swiftly evolving realm of plasma biomarkers, plasma p-tau217 has demonstrated excellent performance in detecting AD pathology, distinguishing between AD and non-AD neurodegenerative disorders and predicting future clinical progression10,11,12,13,14,15,16,17,18,19. However, in contrast with tau-PET, plasma p-tau217 provides no regional information on AD pathology, its continuous values are less representative of the full dynamic range of tau pathology and its signal represents a mix of tau and Aβ pathology and is therefore a less tau-specific biomarker20,21,22. In cognitively impaired individuals, tau-PET has shown performance superior to that of plasma p-tau217 in predicting future cognitive decline23,24. In CU individuals, however, it is yet unclear whether there is a meaningful difference in prognostic utility between tau-PET and plasma p-tau217 biomarkers25. Determining which of the two biomarkers is the strongest predictor of future cognitive deterioration in initially CU individuals is of utmost importance, as clinical trials are increasingly recruiting participants with preclinical AD to enable early intervention26. This information would become even more crucial if treatments such as lecanemab27 and donanemab28 are found to be effective in preclinical AD, as this would require large-scale screening of CU populations for AD pathology.
In previous multicenter cohort studies, we described that tau-PET29 and plasma p-tau217 (ref. 30) individually showed good prognostic performance in CU populations. To address current knowledge gaps in the literature (that is, which is the best prognostic tau biomarker in CU individuals? Is there added value in the combined use of plasma p-tau217 and tau-PET? Can the two tau biomarkers be effectively implemented in a screening approach for clinical trials?), we performed a large-scale head-to-head comparison study between tau-PET and plasma p-tau217. For tau-PET, we extracted signals from brain regions covering the medial temporal lobe (tau-PETMTL) and the temporal neocortex (tau-PETNEO)29. We assessed their associations with longitudinal cognitive decline and diagnostic progression to mild cognitive impairment (MCI). Additionally, we examined whether and how plasma p-tau217 and tau-PET can be combined to further increase their prognostic accuracy and to optimize recruitment strategies for clinical trials.
Results
Participants
We included 1,474 CU participants from nine cohorts with tau-PET and plasma p-tau217 data available at baseline, of whom 408 (27.7%) were Aβ positive on PET (see Table 1 and Supplementary Table 1 for data by cohort). The mean ± s.d. age of the participants was 69.3 ± 10.2 years, 52.8% were females, and the follow-up duration was 3.8 ± 1.8 years. The associations between tau-PET and plasma p-tau217 levels were moderate (plasma p-tau217 versus tau-PETMTL, ρ (95% confidence interval (CI) = 0.43 (0.38–0.47), P < 0.001; plasma p-tau217 versus tau-PETNEO, ρ = 0.34 (0.30–0.39), P < 0.001; Extended Data Fig. 1).
Prediction of future decline in cognitive function
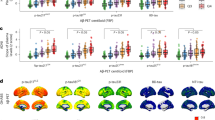
First, we examined across all participants whether the soluble phosphorylated and aggregated tau biomarkers individually and combined were associated with cognitive decline over time on the modified preclinical Alzheimer cognitive composite (mPACC5). We selected the mPACC5 because it is a sensitive measure that can reliably detect longitudinal changes over time in CU populations and is therefore often used as an outcome measure in research studies and clinical trials focusing on preclinical AD26,31. mPACC5 slopes were generated using linear mixed effects models and then used as dependent variables in linear regression models adjusting for age, sex, years of education, APOE ε4 carriership and cohort. To account for different assays and tracers across cohorts, we computed cohort-specific z scores for plasma p-tau217 and tau-PET using amyloid-PET-negative CU individuals from the same cohort as the reference group. The associations for plasma p-tau217 and tau-PET with annual mPACC5 change are shown both across (Fig. 1a) and within (Fig. 1b) cohorts. The linear regression models showed that plasma p-tau217 concentrations (R2 = 0.33, corrected Akaike information criterion (AICc) = 7,239.1), tau-PET uptake in the medial temporal lobe (tau-PETMTL, R2 = 0.34, AICc = 7,232.8) and tau-PET uptake in the temporal neocortex (tau-PETNEO, R2 = 0.33, AICc = 7,252.6) were all better predictors of longitudinal cognitive decline than basic models that included age, sex, education and cohort with APOE ε4 status (R2 = 0.24, AICc = 7,507.5) or without APOE ε4 status (R2 = 0.23, AICc = 7,524.3) (Fig. 1c and Supplementary Tables 2 and 3). There were no significant differences between single-biomarker models, that is, between plasma p-tau217 and tau-PETMTL (P = 0.653), between plasma p-tau217 and tau-PETNEO (P = 0.752) and between tau-PETMTL and tau-PETNEO (P = 0.356). Combined soluble and aggregated tau biomarker models, that is, plasma p-tau217 and tau-PETMTL (R2 = 0.35, AICc = 7,146.6) and plasma p-tau217 and tau-PETNEO (R2 = 0.35, AICc = 7,149.6), were more strongly associated with longitudinal mPACC5 decline than the single-biomarker models (all P < 0.01). The relative contribution of each biomarker in the combined models further indicated their complementary value, as, for example, in the combined plasma p-tau217 and tau-PETMTL model; of all explained mPACC5 variance, 14% was explained by plasma p-tau217 alone, 17% by tau-PETMTL alone, 22% was shared variance and the remaining 46% was explained by covariates (Fig. 1d and Supplementary Table 4). The results were overall consistent across cohorts (Fig. 1b, Supplementary Table 5 and Supplementary Fig. 1) and were largely similar when restricting the sample to CU individuals with at least 4 or 5 years of follow-up data available (Supplementary Fig. 2).

a, Scatterplots showing the association between cognitive change over time on the mPACC5 and the tau biomarkers (Q1–Q3 versus Q4) across all participants. The shaded area indicates the 95% CI around the mean derived from a linear regression model. Note that the standardized β (βSTD) coefficients and R2 statistics relate to the tau biomarker as a continuous variable and that classification into quartiles was performed for visualization purposes only. b, Standardized β coefficients and 95% CIs from linear regression models for the association between the continuous tau biomarker and annual change in the mPACC5 (adjusted for age, sex, education and APOE ε4 status) for each cohort (ordered by sample size). The size of the rhomboid resembles the sample size of each cohort. The vertical dashed line represents standardized β = 0, while the thinner vertical dashed line represents the average standardized β across all cohorts with the 95% CI indicated in gray. ADC, Amsterdam Dementia Cohort; ADRC, Alzheimer’s Disease Research Center; AIBL, Australian Imaging Biomarkers and Lifestyle Study of Ageing; MCSA, Mayo Clinic Olmsted Study of Aging; PREVENT-AD, Pre-symptomatic Evaluation of Experimental or Novel Treatments for Alzheimer’s Disease; TRIAD, Translational Biomarkers in Aging and Dementia; WRAP, Wisconsin Registry for Alzheimer’s Prevention. c, Explained variance (R2, inside the bar plot) and model fit (corrected Akaike criterion, outside the bar plot) for various models predicting longitudinal change in the mPACC5 across all participants. Error bars represent the 95% CI around the mean derived from a linear regression model. w/, with; w/o, without. d, Partial explained variance (R2) for combined biofluid and neuroimaging models predicting longitudinal change in the mPACC5 across all participants. ‘Shared’ in d refers to the explained variance shared between tau-PET and plasma p-tau217 that could not be attributed to a single tau biomarker in the model. Note that we computed cohort-specific z scores for plasma p-tau217 and tau-PET using amyloid-PET-negative CU individuals from the same cohort as the reference group. The analyses presented in this figure are based on 1,376 CU individuals. ^[18F]flortaucipir PET, ^^[18F]MK6240 PET, ^^^[18F]RO948 PET. #Lilly plasma p-tau217 immunoassay, ##Janssen plasma p-tau217+ assay. *P < 0.05, **P < 0.01, ***P < 0.001. Exact P values can be found in Supplementary Tables 2 and 3.
Next, we repeated the same set of analyses with mPACC5 as the outcome measure in amyloid-PET-positive CU individuals only (n = 396). This analysis yielded a largely similar pattern compared to analyses of the entire sample (Extended Data Fig. 2 and Supplementary Tables 6 and 7). The single-biomarker models including plasma p-tau217 (R2 = 0.33, AICc = 2,444.9), tau-PETMTL (R2 = 0.37, AICc = 2,420.0) or tau-PETNEO (R2 = 0.36, AICc = 2,405.6) outperformed the basic models with and without APOE ε4 status (R2 = 0.20, AICc = 2,535.5 and 2,536.5). Furthermore, there were no differences between the three single-biomarker models (plasma p-tau217 versus tau-PETMTL, P = 0.257; plasma p-tau217 versus tau-PETNEO, P = 0.241; tau-PETMTL versus tau-PETNEO, P = 0.725). Also, combined biomarker models, that is, plasma p-tau217 and tau-PETMTL (R2 = 0.39, AICc = 2,389.7) and plasma p-tau217 and tau-PETNEO (R2 = 0.38, AICc = 2,377.7), were more strongly associated with longitudinal mPACC5 decline than their respective single-biomarker models (both P < 0.02). Among Aβ-positive CU individuals, the relative contribution of the tau-PET measures in the combined model was substantially greater than in the entire study population, that is, tau-PETMTL 32% versus 17% and tau-PETNEO 34% versus 16%, which was not the case for plasma p-tau217 (19% versus 17%; Extended Data Fig. 2c and Supplementary Table 8).
Prediction of clinical progression to MCI
Next, we examined across all participants whether the tau biomarkers individually and combined were associated with the rate of clinical progression to MCI using Cox proportional hazard models, adjusting for age, sex, years of education, cohort and APOE ε4 carriership. The associations for plasma p-tau217 and tau-PET with clinical progression to MCI are shown both across (Fig. 2a) and within (Fig. 2b) cohorts. The analysis revealed that higher baseline plasma p-tau217 (HR = 1.57 (1.43–1.72), AICc = 1,960), tau-PETMTL (HR = 1.61 (1.48–1.76), AICc = 1,937) and tau-PETNEO (HR = 1.43 (1.30–1.52), AICc = 1,967) levels were all associated with an increased risk for future progression to MCI (all P < 0.001; Fig. 2a,b and Supplementary Tables 9 and 10). The individual fluid versus neuroimaging tau biomarker models did not differ from each other (plasma p-tau217 versus tau-PETMTL, P = 0.322; plasma p-tau217 versus tau-PETNEO, P = 0.750), while the performance of tau-PETMTL showed a trend toward better performance than tau-PETNEO (P = 0.059). The fit was improved for the plasma p-tau217 model when adding tau-PETMTL (∆AICc = −50, P ≤ 0.001) or tau-PETNEO (∆AICc = −33, P = 0.009; Supplementary Table 10). Likewise, the model fit also improved when adding plasma p-tau217 to tau-PETMTL (∆AICc = −27, P = 0.017) and to tau-PETNEO (∆AICc = −40, P = 0.004). The results were largely consistent across cohorts (Fig. 2b, Supplementary Table 11 and Supplementary Fig. 3) and were largely similar when restricting the sample to CU individuals with at least 4 or 5 years of follow-up data available (Supplementary Fig. 4).

a, Survival curves for progression to MCI (Q1–Q3 versus Q4) across all participants, including a table of the total number of participants available at each time point. The shaded area indicates the 95% CI around the mean derived from Cox proportional hazard models. Note that the HR and Akaike information criterion (AIC) statistics relate to the tau biomarker as a continuous variable and that classification into quartiles was only done for visualization purposes. b, HRs and 95% CIs from Cox proportional hazard models for the association between the tau continuous biomarker and progression to MCI (adjusted for age, sex, education and APOE ε4 status) for each cohort (ordered by sample size). The size of the rhomboid relates to the sample size of each cohort. The vertical dashed line represents standardized HR = 1, while the thinner vertical dashed line represents the average HR across all cohorts with the 95% CI indicated in gray. c, Model fit (corrected Akaike criterion) for various models predicting future clinical progression to MCI across all participants. Error bars represent the 95% CI around the mean derived from Cox proportional hazard models. d, HRs and 95% CIs around the mean from Cox proportional hazard models in simple models (that is, modeling one tau biomarker at a time, top three HRs) and combined models (that is, modeling plasma and PET tau biomarkers simultaneously, bottom four HRs) when assessing all cohorts together. Vertical dashed line shows the HR = 1 (no effect). Note that we computed cohort-specific z scores for plasma p-tau217 and tau-PET using amyloid-PET-negative CU individuals from the same cohort as the reference group. The analyses presented in this figure are based on 1,426 CU individuals. ^[18F]flortaucipir PET, ^^[18F]MK6240 PET, ^^^[18F]RO948 PET. #Lilly plasma p-tau217 immunoassay, ##Janssen plasma p-tau217+ assay. *P < 0.05, **P < 0.01, ***P < 0.001. Exact P values can be found in Supplementary Tables 9 and 10.
When repeating the same set of analyses in Aβ-positive CU individuals only (n = 396), we found a largely similar pattern (Extended Data Fig. 3 and Supplementary Tables 10 and 11). Baseline plasma p-tau217 (HR = 1.58 (1.38–1.80), AICc = 1,094), tau-PETMTL (HR = 1.53 (1.39–1.70), AICc = 1,072) and tau-PETNEO (HR = 1.34 (1.25–1.44), AICc = 1,088) levels were all associated with an increased risk for future progression to MCI (all P < 0.001). The individual tau biomarker models did not differ from each other (plasma p-tau217 versus tau-PETMTL, P = 0.200; plasma p-tau217 versus tau-PETNEO, P = 0.711; tau-PETMTL versus tau-PETNEO, P = 0.209). Adding tau-PET to plasma p-tau217 models improved the model fit when adding tau-PETMTL (∆AICc = −39, P = 0.007) and tau-PETNEO (∆AICc = −24, P = 0.031; Supplementary Tables 12 and 13). The model fit also slightly improved when adding plasma p-tau217 to either tau-PETMTL (∆AICc = −17, P = 0.042) and at trend level for tau-PETNEO (∆AICc = −18, P = 0.061).
A two-step approach to reduce the sample size in clinical trials
Next, we tested whether and how a two-step sequential approach (that is, plasma p-tau217 followed by tau-PET) could reduce the number of participants needed for a preclinical AD trial using longitudinal changes in cognitive function as the primary outcome. Substantial sample size reductions can already be achieved in the first step, that is, using only plasma p-tau217. When using the mPACC5 as the outcome measure, assuming 80% power and α = 0.05 in a 4-year clinical trial with annual repeated testing, selecting participants with plasma p-tau217 levels in quartiles 2–4 (that is, Q2–Q4, excluding the lowest 25% of plasma p-tau217) would result in a 32% (14–41%) reduction in the number of required participants compared to including the entire study population (Fig. 3 and Supplementary Table 14). Selecting participants in plasma p-tau217 Q3–Q4 and Q4 further reduced the required sample size by 64% (51–72%) and 82% (73–86%), respectively. Using clinical progression to MCI as an outcome measure yielded similar results, that is, plasma p-tau217 Q2–Q4, 28% (21–36%); Q3 and Q4, 55% (48–64%); and Q4, 82% (77–87%) (Fig. 4 and Supplementary Table 14). In the second step, tau-PET measures further reduced the required sample size. For example, in the population with plasma p-tau217 concentrations in Q3 and Q4, selecting participants with tau-PET in Q4 would further reduce the sample size from 64% (51–72%) (plasma p-tau217) to 88% (81–90%) (tau-PETMTL; Fig. 3) or to 85% (76–89%) (tau-PETNEO; Extended Data Fig. 4) when using the mPACC5 as the outcome measure. As another example in the population with plasma p-tau217 concentrations in Q4, selecting participants with tau-PET in Q3 and Q4 would further reduce the sample size 82% (77–87%) (plasma p-tau217) to 88% (82–94%) (tau-PETMTL; Fig. 4) or to 94% (92–97%) (tau-PETNEO; Supplementary Fig. 5) when using clinical progression to MCI as the outcome measure. The estimated sample size reductions for all plasma p-tau217 and tau-PET quartile combinations are presented in Supplementary Table 14. Repeating the same set of analyses but now restricted to Aβ-positive CU individuals showed that similarly large sample size reductions can be achieved in this population (Extended Data Fig. 5). Finally, we considered a scenario in which we tested the sequence plasma p-tau217, followed by amyloid-PET and then tau-PET when using change in the mPACC as an outcome measure (Extended Data Fig. 6). The results suggest that, after initial plasma p-tau217 screening (step 1), incorporating amyloid-PET positivity (step 2) further reduces the number of trial participants needed. This number can in turn subtly be lowered further by implementing tau-PET (step 3). Changing the outcome measure from the mPACC5 to progression to MCI yielded very similar results (Supplementary Fig. 6).

a, Conceptual framework of a sequential two-step recruitment strategy of a clinical trial in preclinical AD using a cognitive endpoint. b, The obtained sample size reduction using sample selection based on different percentiles (75th, 50th and 25th) of baseline plasma p-tau217 levels in step 1 followed by selection based on the same percentiles (75th, 50th and 25th) of the tau-PETMTL measurement in step 2 with the mPACC5 as the primary endpoint. Note that 100% in step 2 refers to the participants selected by plasma p-tau217 in step 1. Error bars represent the 95% CI around the mean derived from linear effects models. c, The calculated sample size reductions for various plasma p-tau217 and tau-PETMTL quartile combinations. Red lines represent step 1 with plasma p-tau217, and green lines represent step 2 with tau-PETMTL. Different line styles represent different quartiles of tau-PETMTL from those participants already selected from step 1. Dashed black lines represent 100% of participants needed without that step. Calculations in b,c are based on the assumption of 80% power to detect a 30% change in the mPACC5 in a 4-year trial. The analyses presented in this figure are based on 1,376 CU individuals.

a, The obtained sample size reduction using sample selection based on different percentiles (75th, 50th and 25th) of baseline plasma p-tau217 levels in step 1 followed by selection based on the same percentiles (75th, 50th and 25th) of the tau-PETMTL measurement in step 2 with clinical progression to MCI as the primary endpoint. Note that 100% in step 2 refers to the participants selected by plasma p-tau217 in step 1. Error bars represent the 95% CI around the mean. b, The calculated sample size reductions for various plasma p-tau217 and tau-PETMTL quartile combinations. Red lines represent step 1 with plasma p-tau217, and green lines represent step 2 with tau-PETMTL. Different line styles represent different quartiles of tau-PETMTL from those participants already selected from step 1. Dashed black lines represent 100% of participants needed without that step. The analyses presented in this figure are based on 1,426 CU individuals.
Characterization of combined plasma p-tau217 and tau-PET groups
Finally, we aimed to characterize the groups resulting from combining different plasma p-tau217 and tau-PETMTL quartiles in terms of rates of mPACC5 decline, Aβ positivity and the proportion of participants who would be included in the trial as well as the proportion of ‘non-progressors’ based on each combination relative to the full dataset. For this purpose, we created four different groups based on increasingly restrictive tau biomarker combinations: (1) a ‘liberal’ group consisting of participants with plasma p-tau217 levels in Q2–Q4 and then tau-PETMTL uptake in Q2–Q4 of those selected by plasma, (2) a ‘moderate’ group consisting of plasma p-tau217 levels in Q3 and Q4 and then tau-PETMTL uptake in Q3 and Q4, (3) a ‘plasma p-tau217 Q4-only’ group consisting of individuals with plasma p-tau217 levels in Q4 regardless of tau-PET and (4) a ‘conservative’ group consisting of plasma p-tau217 levels in Q4 and then tau-PETMTL uptake in Q4. Fig. 5 indicates a progressively worse outcome for individuals from approach (1) to (4), with more rapid decline on the mPACC5 from the liberal (1) to the conservative (4) threshold approach (from standardized β = −0.06 ± 0.09 to −0.17 ± 0.13; Fig. 5a), an increasing proportion of Aβ-positive individuals (from 41% to 97%; Fig. 5b) and a smaller proportion of ‘non-progressors’ on the mPACC5 (from 38% to 10%; Fig. 5d). Furthermore, the proportion of participants who would be included in a hypothesized clinical trial with the mPACC5 as an outcome measure decreased from 54% (of the entire population) when using the liberal threshold approach to only 6% when using the conservative threshold approach (Fig. 5c). Notably, there were no group differences between the plasma p-tau217 Q4-only approach versus the moderate combined threshold approach for mPACC5 decline (standardized β = −0.09 ± 0.10 versus −0.10 ± 0.11), the proportion of participants selected (24.0% versus 24.0%) and the proportion of ‘non-progressors’ (24.4% versus 23.8%) and only a modest difference in Aβ positivity (that is, 73.0% versus 62.2%). The same set of analyses using tau-PETNEO instead of tau-PETMTL yielded similar results (Supplementary Fig. 7). Detailed group characterizations for all the other possible combinations are presented in Supplementary Table 15 (that is, Aβ positivity, mPACC5 decline and non-progressors) and Supplementary Table 16 (that is, age, sex, APOE status and years of education). Additionally, in the BioFINDER-2 study, we applied predefined cutoffs for both plasma p-tau217 and tau-PET and found that tau biomarker-positive individuals showed characteristics very similar to those of individuals in Q4 concerning mPACC5 slopes (plasma p-tau217 positive, −0.22 ± 0.15; tau-PETMTL positive, −0.19 ± 0.15), percent of Aβ-positive individuals (plasma p-tau217, 90%; tau-PETMTL, 100%), percent of participants included in the trial (plasma p-tau217, 4%; tau-PETMTL, 8%) and percent of non-progressors (plasma p-tau217, 10%; tau-PETMTL, 13%; Supplementary Fig. 8).

This figure shows how different group compositions based on their baseline plasma p-tau217 and tau-PETMTL levels are related to various relevant trial metrics, including the annual mPACC5 slope (a, n = 1,376), the proportion of Aβ+ individuals (b, n = 1,473), the proportion of individuals from the entire population who would be included in a clinical trial based on the group definitions described on the x axis (c, all participants) and the proportion of ‘non-progressors’ on the mPACC5 (defined as slope > −0.016, see the Methods for details) (d, n = 1,376). Error bars in a represent the 95% CI. More efficient trials are expected with lower mPACC slopes and higher percentages of Aβ+ individuals and trial participants but lower percentages of non-progressors.
We also calculated the projected costs that could be saved in a hypothetical trial with mPACC5 or MCI progression rates as an outcome measure using the same four groups as above. When assuming a 1:15 ratio (that is, cost of one tau-PET scan equals 15 plasma p-tau217 assessments) and using tau-PETMTL, both the plasma p-tau217 Q4-only (3) group (96.9%) and the conservative combined (4) group (96.6% cost reduction) yielded substantially higher cost reductions than the moderate (2) group (85.6%) and especially the liberal (1) group (54.4%) when using mPACC as an endpoint (Supplementary Fig. 9a, tau-PETNEO results are presented in Supplementary Fig. 9b). Using the same ratio (1:15) but now using clinical progression as the endpoint, the plasma p-tau217 Q4-only (3) group yielded the highest cost reduction (88.6%), followed by the conservative combined (4) group (70.9%), the moderate (2) group (53.4%) and the liberal (1) group (25.1%; Supplementary Fig. 10a, tau-PETNEO results are presented in Supplementary Fig. 10b).
Discussion
In this multicohort study, we investigated whether tau-PET, a marker of aggregated tau pathology, or plasma p-tau217, a marker of soluble hyperphosphorylated tau, is more strongly associated with future cognitive decline among 1,474 CU individuals and whether they would provide complementary information in screening approaches for clinical trials. This is a timely research question, as both tau biomarkers are frequently incorporated into clinical trials and often in combination, as they reflect different aspects of tau pathophysiology. According to the revised AD criteria by the Alzheimer’s Association Workgroup, plasma p-tau217 is a core 1 biomarker (T1) of phosphorylated and secreted AD tau, while tau-PET is a core 2 biomarker (T2) of AD tau proteinopathy32. In this study, we observed comparably strong associations between plasma p-tau217 and tau-PET with cognitive decline on a sensitive global cognitive composite test (that is, mPACC5) and with clinical progression to MCI in CU individuals. Importantly, models including both plasma p-tau217 and tau-PET consistently outperformed single-biomarker models, suggesting that plasma p-tau217 and tau-PET provide complementary information. Simulations showed that a two-step approach (that is, plasma p-tau217 first, followed by tau-PET in individuals with high plasma p-tau217 only) could substantially reduce the number and the cost of recruiting participants for a preclinical AD clinical trial with change in mPACC5 or progression to MCI as the primary endpoint. We conclude that plasma p-tau217 and tau-PET showed similar associations with future cognitive decline in a CU population, that both tau biomarkers provide complementary information and that their sequential use (that is, plasma p-tau217 followed by tau-PET in a subset with high plasma p-tau217) is useful for screening in clinical trials in preclinical AD.
A main finding of this study is that there were no statistical differences between plasma p-tau217 concentrations and tau-PET uptake in their associations with future cognitive changes in a CU population. This is in contrast with cognitively impaired populations, in which tau-PET generally outperforms plasma p-tau217 in terms of prognostic accuracy23,24. This discrepancy between disease stages might be explained by the differences in underlying pathophysiology and subsequent temporal dynamics of the two tau biomarkers. Plasma p-tau217 measures the hyperphosphorylated tau protein in soluble forms and has been shown to change very early in the disease process33. This was also observed in the lower range of our population, where plasma p-tau217 z scores started to increase while levels of tau-PET retention remained unchanged (Extended Data Fig. 1). Post-mortem studies indicated that ante-mortem plasma p-tau217 levels are associated with the density of both neurofibrillary tau tangle and Aβ plaque pathology in the brain21,34. It is conceivable that this mix of Aβ- and tau-related signals reflected by plasma p-tau217 levels contributed to its non-inferiority versus tau-PET for predicting cognitive change over time in this early population. For instance, Aβ toxicity might impact synaptic function or neuroinflammation, which could subsequently influence cognitive performance35. By contrast, tau-PET signal largely represents the presence of aggregated paired helical filaments of the tau protein forming insoluble neurofibrillary tangles36. In vivo studies have shown that positive tau-PET scans are relatively rare in CU individuals (that is, ~5–10% among Aβ-positive CU individuals in a temporal meta-region of interest)3,4, and post-mortem studies have indicated that a positive [18F]flortaucipir tau-PET scan (quantitatively or by visual read) represents tau pathology in Braak stages IV and above37,38. This may also explain why the relative contribution to cognitive decline of tau-PET versus plasma p-tau217 was greater in Aβ-positive individuals than in Aβ-negative CU individuals. Altogether, this indicates that substantial changes on a tau-PET scan occur in rather advanced clinical and biological stages of AD, which may explain why tau-PET in the current mix of Aβ-positive and Aβ-negative CU individuals did not outperform plasma p-tau217 in predicting future cognitive decline.
Another important finding of the current study was that we did not observe marked differences between tau-PETMTL and tau-PETNEO in this CU population, while previous studies consistently showed that tau-PET signal in neocortical areas is a better predictor of future clinical progression than tau-PET signal in medial temporal lobe regions, especially in cognitively impaired individuals5,39,40,41,42,43. This result may also appear contradictory with our previous study in which the tau-PETNEO-positive group exhibited a considerably worse prognosis than the tau-PETMTL-positive group29. This discrepancy can be explained by the different group definitions used in the current study (quartile based) compared to those of the previous study (binary cutoffs). In the previous study, the tau-PETNEO-positive group comprised a relatively small proportion of the overall CU population (only ~4%). As a result, even in the highest quartile (Q4) of the current study, most individuals are tau-PET negative (that is, 56% for tau-PETMTL and 84% for tau-PETNEO). The scarcity of tau-PETNEO-positive individuals in the current study (and in CU populations in general) attenuates the overall association between tau-PET and cognitive decline and dilutes the association even further for the tau-PETNEO group. We conclude that the ‘negative’ result for the tau-PETNEO group is a valid finding but only within the specific context of this study (that is, evaluating a CU population using a quartile-based approach).
Because there were no clear distinctions between the two tau biomarkers in individual models, plasma p-tau217 should be prioritized over tau-PET as a standalone screening tool in CU populations due to its major practical advantages. An exception could be when the goal is to demonstrate target engagement of an anti-tau agent, in which case tau-PET might be the preferred tau biomarker20. Compared to single-biomarker models, simultaneous modeling of plasma p-tau217 and tau-PET consistently led to a more accurate forecast of subsequent cognitive decline. We therefore proposed a conceptual framework of a two-step sequential approach for clinical trial design (Fig. 3a), starting with participant selection based on plasma p-tau217 followed by tau-PET, and tested a simplified version of this strategy. This approach decreased the cost of selecting appropriate participants for clinical trials by drastically reducing the number of required tau-PET scans and participants for screening. This was mainly achieved by selecting, from the entire CU study population, the subset of individuals that are at highest risk for short-term cognitive decline due to their elevated levels of both baseline plasma p-tau217 and tau-PET. In such a workflow, the participants’ eligibility in terms of inclusion (for example, age, cognitive status) and exclusion (for example, the absence of major neurological or psychiatric disorders) criteria would be assessed first. Next, participants would be further triaged based on their plasma p-tau217 levels, where ‘low or negative’ participants would not undergo a tau-PET scan as their risk for future cognitive decline would be low, while ‘high or positive’ participants would undergo a tau-PET scan to further refine their risk profile44. The degree of tau-PET uptake can then be used to assign CU individuals on a continuum ranging from intermediate (that is, ‘high or positive’ plasma p-tau217, ‘low or negative’ tau-PET) to high (that is, ‘high or positive’ results on both plasma p-tau217 and tau-PET) risk for future progression. The final prescreening decision will be based on factors such as the acceptable rate of screening failures, trial duration, expected effect size of the drug or intervention and whether the trial is a primary or secondary intervention strategy. The framework depicted in Fig. 3a is primarily conceptual, and we recognize that numerous crucial decisions need to be made during its implementation. This includes, among other factors, determining the criteria for ‘high or positive’ versus ‘low or negative’ for both plasma p-tau217 and tau-PET, deciding whether to use p-tau217 alone or in combination with other plasma biomarkers (for example, Aβ42/40), identifying the target region of interest for tau-PET quantification, selecting a quantitative threshold and/or visual read metric for tau-PET and potentially adding amyloid-PET to the proposed workflow (Extended Data Fig. 6)45,46.
The main strength of this study is the multicenter approach that yielded a sufficient sample size for a robust head-to-head comparison between tau-PET and plasma p-tau217 as well as a thorough assessment of their potential complementary value. The main limitations of the study are related to the inherent challenges of a multicenter study that was not co-designed from the start. First and foremost, we did not have plasma p-tau217 cutoffs available from all cohorts and instead used either continuous (z scores) or categorical (quartiles) data for the analyses. Also, we aimed to optimize pooling of data across cohorts by standardizing biomarker values using CU Aβ-negative participants. We acknowledge, however, that there may still be residual heterogeneity caused by use of different amyloid-PET and tau-PET tracers and different p-tau217 assays as well as the use of different neuropsychological tests to generate mPACC5 scores. To mitigate this, we present both the pooled (main report) and cohort-specific (supplementary) results. Additionally, tau-PET may have been at a slight disadvantage compared to plasma p-tau217, as tau-PET data were more often analyzed locally, whereas plasma p-tau217 was predominantly analyzed centrally at Lund University. In one of the cohorts (that is, BioFINDER-2) in which we had predefined cutoffs available for both plasma p-tau217 and tau-PET, we found that tau biomarker-positive individuals showed characteristics very similar to those of individuals in Q4 concerning, for example, mPACC5 slopes and percent of Aβ-positive individuals (Supplementary Fig. 8). However, future large-scale studies using predefined cutoffs for both plasma p-tau217 and tau-PET, preferentially in a more diverse population in terms of ethnicity, socioeconomic status and medical comorbidities, are of importance to establish the generalizability of our findings. Another potential threat to the generalizability of our findings is the intentional focus on CU individuals. This may have introduced several biases, including survival bias (that is, the CU individuals in this study may systematically differ from the general population in terms of genetics or lifestyle, especially among those with elevated tau biomarkers) and selection bias (that is, CU individuals were recruited through varying methods across cohorts). Related to the latter, inspection of demographic data (Table 1) suggests that this multicenter study population is enriched for AD risk factors. For example, the proportion of APOE ε4 carriers in our study was higher (37%) than in the general population (15–30%, depending on ethnic group variations)47,48. This pattern was also observed within the Aβ-positive group, in which 57% were APOE ε4 carriers, compared to 51% in an independent multicenter study49.
In summary, our data suggest that plasma p-tau217 and tau-PET show similar associations with future cognitive decline and clinical progression in a CU population. We also showed that plasma p-tau217 and tau-PET provide complementary information and that a two-step approach (that is, plasma p-tau217 followed by tau-PET) substantially reduces the number of required tau-PET scans and screened participants. Altogether, our data support the feasibility of a clinical trial design in which all participants undergo screening with plasma p-tau217, but only a subset with high or abnormal plasma p-tau217 will undergo tau-PET.
Methods
Participants
We included 1,474 participants from the Swedish BioFINDER-1 (n = 39, NCT01208675) and BioFINDER-2 (n = 481, NCT03174938) studies at Lund University7,43, the MCSA50 (n = 363), the AIBL51 (n = 180), the Knight ADRC at Washington University (n = 109), TRIAD (n = 124) and PREVENT-AD (n = 52) at McGill University, the WRAP (n = 82) at the University of Wisconsin–Madison and the SCIENCe project52, which is part of the ADC (n = 44). The overlap of CU participants between the current and our previous multicenter studies where we assessed the prognostic performance of tau-PET and plasma p-tau217 individually was 630 of 1,474 (43%) and 170 of 1,474 (12%), respectively. A brief description of each cohort is provided in Supplementary Table 17. All participants were (1) CU at baseline, defined by neuropsychological test scores within the normative range given an individual’s age, sex and educational background, (2) had amyloid-PET available to determine Aβ status, (3) underwent a tau-PET scan and blood sampling within a maximum interval of 1 year and (4) had at least one clinical follow-up visit available. Follow-up data were collected until 1 November 2023. Written informed consent was obtained from all participants and local institutional review boards for human research approved in the study. This includes the Mayo Clinic and Olmsted Medical Center institutional review boards for the MSCA, the regional ethics committee at Lund University for BioFINDER-1 and BioFINDER-2, institutional human research ethics committees of Austin Health, St. Vincent’s Health, Hollywood Private Hospital and Edith Cowan University for the AIBL, the medical ethics review committee of the Amsterdam University Medical Center for the ADC, the McGill Research Ethics Board for PREVENT-AD and TRIAD, the Washington University School of Medicine in St. Louis for the Knight ADRC and the University of Wisconsin Institutional Review Board for the WRAP.
Amyloid-PET status
Aβ status was determined using center-specific cutoffs or visual read metrics using [18F]flutemetamol PET for BioFINDER-1 and BioFINDER-2, [11C]Pittsburgh compound B PET was used for the MCSA, the Knight ADRC and the WRAP, [18F]florbetapir PET was used for the ADC, and [18F]NAV4694 was used for TRIAD, PREVENT-AD and the AIBL; see Supplementary Table 18 for details.
Tau positron emission tomography
Tau-PET was performed using [18F]flortaucipir for BioFINDER-1, MSCA, Knight ADRC and ADC cohorts using [18F]MK6240 for TRIAD, the AIBL and the WRAP and using [18F]RO948 for BioFINDER-2. Data were centrally processed at Lund University for BioFINDER-1, BioFINDER-2 and the ADC and locally processed for the other cohorts following previously described procedures (Supplementary Table 19). In line with our previous study29, we generated tau-PET standardized uptake value ratios (SUVRs) for a medial temporal lobe (unweighted average of the bilateral entorhinal cortex and the amygdala) and a neocortical temporal (weighted average of bilateral middle temporal and inferior temporal gyri) region of interest3,53.
Plasma p-tau217
Plasma p-tau217 levels were measured using an immunoassay developed by Lilly Research Laboratories on a Meso Scale Discovery platform at Lund University for BioFINDER-1, BioFINDER-2, the ADC, the WRAP, PREVENT-AD and the Knight ADRC54 and at the Mayo Clinic for the MSCA15. For the AIBL and TRIAD, a plasma p-tau217+ assay developed by Janssen R&D on a Single Molecule Array (Simoa) HD-X platform was used25. The correspondence between the two plasma p-tau217 assays used in this study has been shown to be high in a direct head-to-head comparison study55.
Clinical outcome measures
We used both continuous and binary measures of clinical progression. First, we examined cognitive trajectories using a sensitive composite measure specifically developed to detect cognitive changes in preclinical stages of AD (that is, the mPACC5 (refs. 31,56)). The mPACC5 consists of tests capturing episodic memory, executive function, semantic memory and global cognition31. Individual neuropsychological tests were z transformed using the baseline test scores of Aβ-negative participants in each cohort as the reference group and then averaged to obtain a composite z score. The composition of the mPACC5 for each cohort is described in Supplementary Table 20. Second, we examined progression from CU to MCI. MCI was established using the Petersen criteria57 and is defined as notable cognitive symptoms as assessed by a physician, in combination with cognitive impairment in one or multiple domains (for example, memory, executive functioning, attention, language), that is, below the normative range given an individual’s age, sex and educational background but not sufficiently severe to meet diagnostic criteria for dementia58.
Statistics and reproducibility
All statistical analyses were performed in R version 4.3.1. Statistical significance for all models was set at P < 0.05, two sided, without correction for multiple comparisons. To enable pooling of the data across cohorts, we z transformed both tau-PET SUVRs and plasma p-tau217 concentrations based on the mean and the standard deviation of Aβ-negative participants within each cohort. We conducted two sets of main analyses, in which we examined (1) the individual and combined utility of tau biomarkers for their associations with longitudinal cognitive decline on the mPACC5 and with clinical progression from CU to MCI and (2) a sequential two-step approach for participant selection in a preclinical AD trial with a cognitive endpoint.
Within analyses assessing tau-PET and plasma p-tau217 versus mPACC5 change and progression to MCI, we used three different statistical models, including (1) only plasma p-tau217 concentration as a (continuous) predictor, (2) only a tau-PET measure as a (continuous) predictor (that is, tau-PETMTL or tau-PETNEO SUVR) or (3) including plasma p-tau217 and one of the tau-PET measures (that is, tau-PETMTL or tau-PETNEO SUVR) simultaneously as predictors. Additionally, we tested two basic models: model 1 including age, sex, years of education and cohort and model 2 consisting of the model 1 variables and APOE ε4 carriership. We calculated change in mPACC5 using linear mixed models with random time slopes and a random intercept using the lme4 package. Subsequently, these slopes were entered as the dependent variable in linear regression models with plasma p-tau217 and/or tau-PET as predictors, while adjusting for age, sex, education, cohort and APOE ε4 carriership. Differences in model performance were assessed by comparing differences in R2 by bootstrapping (1,000 repetitions with resample, boot package). Partial R2 values were calculated to assess the specific contribution of each predictor in the combined plasma p-tau217 and tau-PET models (rsq package).
Next, we examined progression from CU to MCI using Cox proportional hazard models, adjusting for age, sex, years of education, APOE ε4 carriership and cohort using the continuous variables as predictors. For individuals who progressed to MCI, we used the respective times at diagnostic progression to MCI for the analyses. To compare the predictive value of tau-PET versus plasma p-tau217 for diagnostic conversion, we compared the difference in AICc using bootstrapping procedures (1,000 repetitions with resample). In secondary analyses, all the above-mentioned analyses were repeated in Aβ-positive participants.
To assess potential differences between cohorts, we performed several sensitivity analyses. First, for the associations between the tau biomarkers and mPACC5 decline, we examined the effect sizes in all the previous models for each cohort independently (Figs. 1b and 2b). To that end, we applied the same linear regression models used in the main analyses to each cohort separately, without including cohort as a covariate. Similarly, we used Cox proportional hazard models in all cohorts independently to calculate the HRs and C-indices (Supplementary Table 11). Finally, we calculated the root mean square error of the predictions for each cohort when it was excluded from the training set. To do this, we calculated the cognition slopes for all cohorts combined, regressed out the cohort variable using a linear regression model and then trained a model (with age, sex and APOE as covariates) on all cohorts except one, subsequently predicting cognitive decline in the excluded cohort. We then calculated the root mean square error from the difference between the predicted data versus the observed data. We also performed additional analyses for both mPACC5 and progression to MCI when including only individuals with a minimum of 4 and 5 years of follow-up data available.
To derive optimal sample size reduction for a clinical trial in the two-step approach, we generated a data-driven estimate of the complementary value of tau-PET and plasma p-tau217 when implementing a sequential two-step approach (that is, plasma p-tau217 first, followed by tau-PET). First, we calculated the obtained sample size reduction when assuming 80% power to detect a 30% change in cognitive change (mPACC5) in a 4-year clinical trial (with annual repeated testing). Sample size was then defined by using different percentiles (75th, 50th and 25th) of the participants’ baseline plasma p-tau217 levels using the lmmpower function in the longpower package. The approach was repeated, selecting the 75th, 50th and 25th percentiles of the new participants’ tau-PET measures. We additionally tested a three-step approach in which, after initial plasma p-tau217 screening (step 1), amyloid-PET positivity (step 2) was incorporated, in turn followed by tau-PET (step 3). The percentage of participants needed compared to the entire study population and the plasma-selected sample are reported. Similar analyses were performed for progression to MC, using the ssizeCT.default function from the powerSurvEpi package. In this analysis, we aimed to detect a 30% reduction of events (that is, progression to MCI) at 80% power.
Next, we compared the characteristics of the sample included and excluded from the hypothetical clinical trial based on the different approaches presented. We focused on four combinations based on the participants selected on their plasma p-tau217 (step 1) and tau-PETMTL (step 2) levels, that is, (1) a ‘liberal’ group comprising Q2–Q4 of plasma p-tau217 levels and Q2–Q4 of tau-PET of those selected by plasma, (2) a ‘moderate’ group consisting of individuals within Q3 and Q4 of plasma p-tau217 levels and Q3 and Q4 of tau-PET of those selected by plasma, (3) plasma p-tau217 Q4-only and (4) a ‘conservative’ group consisting of individuals within Q4 of plasma p-tau217 levels and Q4 of tau-PET of those selected by plasma. Based on these selection criteria, we compared mPACC slopes, the proportion of Aβ positivity, the final number of participants included and the proportion of ‘non-progressors’ between the plasma p-tau217 and tau-PET groups. We defined ‘non-progressors’ based on the mean and s.d. of the mPACC5 slope in the Aβ-negative CU participants from BioFINDER-2 (the largest cohort). By extracting the s.d. from the mean, we accounted for natural variation and/or fluctuations in (longitudinal) cognitive testing and potential learning effects. Based on this approach, every participant with a slope > −0.016 on the mPACC5 was classified as a ‘non-progressor’. As a sensitivity analysis, we repeated this analysis in BioFINDER-2 in which we had predefined cutoffs available for both plasma p-tau217 (0.499)30 and tau-PET (medial temporal lobe, 1.34 SUVR; temporal neocortex, 1.36 SUVR)29 and could classify individuals as ‘tau positive’ instead of using the quartile approach. Finally, we investigated the percentage of cost reductions of such approaches for participant selection in a hypothetical clinical trial using either mPACC5 or progression to MCI as the outcome measure assuming 80% power to detect a 30% change in mPACC5 or progression to MCI in a 4-year clinical trial (with annual repeated clinical assessments). We provided percentage cost reductions using different ratios of costs for tau-PET versus plasma p-tau217: 1:5 (that is, cost of one tau-PET scan resembling five plasma p-tau217 assessments), 1:10, 1:15 and 1:20.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Due to the multicenter design of the study, access to individual participant data from each cohort will have to be made available through the PIs of the respective cohorts (that is, [email protected] for the Swedish BioFINDER-1 and BioFINDER-2 studies, C.R.J. for the MCSA, C.R. for the AIBL, T.L.S.B. for the Knight ADRC, P.R.-N. for TRIAD, S.V. for the PREVENT-AD, S.J. for the WRAP and W.M.v.d.F. for the ADC). Generally, anonymized data can be shared by request from qualified academic investigators for the purpose of replicating procedures and results presented in the article, if data transfer is in agreement with data protection regulation at the institution and is approved by the local ethics review board.
Code availability
The codes used for data analyses can be found at https://github.com/OssenKoppeLab.
References
Hansson, O. Biomarkers for neurodegenerative diseases. Nat. Med. 27, 954–963 (2021).
Strikwerda-Brown, C. et al. Association of elevated amyloid and tau positron emission tomography signal with near-term development of Alzheimer disease symptoms in older adults without cognitive impairment. JAMA Neurol. 79, 975–985 (2022).
Ossenkoppele, R. et al. Discriminative accuracy of [18F]flortaucipir positron emission tomography for Alzheimer disease vs other neurodegenerative disorders. JAMA 320, 1151–1162 (2018).
Jack, C. R. et al. The bivariate distribution of amyloid-β and tau: relationship with established neurocognitive clinical syndromes. Brain 142, 3230–3242 (2019).
Ossenkoppele, R. et al. Accuracy of tau positron emission tomography as a prognostic marker in preclinical and prodromal Alzheimer disease: a head-to-head comparison against amyloid positron emission tomography and magnetic resonance imaging. JAMA Neurol. 78, 961–971 (2021).
Pascoal, T. A. et al. 18F-MK-6240 PET for early and late detection of neurofibrillary tangles. Brain 143, 2818–2830 (2020).
Leuzy, A. et al. Diagnostic performance of RO948 F 18 tau positron emission tomography in the differentiation of Alzheimer disease from other neurodegenerative disorders. JAMA Neurol. 77, 955–965 (2020).
Ossenkoppele, R. & Hansson, O. Towards clinical application of tau PET tracers for diagnosing dementia due to Alzheimer’s disease. Alzheimers Dement. 17, 1998–2008 (2021).
Hansson, O., Blennow, K., Zetterberg, H. & Dage, J. Blood biomarkers for Alzheimer’s disease in clinical practice and trials. Nat. Aging 3, 506–519 (2023).
Dore, V. et al. Plasma p217+tau versus NAV4694 amyloid and MK6240 tau PET across the Alzheimer’s continuum. Alzheimers Dement. 14, e12307 (2022).
Ashton, N. J. et al. Differential roles of Aβ42/40, p-tau231 and p-tau217 for Alzheimer’s trial selection and disease monitoring. Nat. Med. 28, 2555–2562 (2022).
Palmqvist, S. et al. Prediction of future Alzheimer’s disease dementia using plasma phospho-tau combined with other accessible measures. Nat. Med. 27, 1034–1042 (2021).
Barthelemy, N. R. et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer’s disease. Nat. Med. 26, 398–407 (2020).
Mila-Aloma, M. et al. Plasma p-tau231 and p-tau217 as state markers of amyloid-β pathology in preclinical Alzheimer’s disease. Nat. Med. 28, 1797–1801 (2022).
Mielke, M. M. et al. Performance of plasma phosphorylated tau 181 and 217 in the community. Nat. Med. 28, 1398–1405 (2022).
Thijssen, E. H. et al. Plasma phosphorylated tau 217 and phosphorylated tau 181 as biomarkers in Alzheimer’s disease and frontotemporal lobar degeneration: a retrospective diagnostic performance study. Lancet Neurol. 20, 739–752 (2021).
Sperling, R. A. et al. Amyloid and tau prediction of cognitive and functional decline in unimpaired older individuals: longitudinal data from the A4 and LEARN studies. J. Prev. Alzheimers Dis. 11, 802–813 (2024).
Palmqvist, S. et al. Blood biomarkers to detect Alzheimer disease in primary care and secondary care. JAMA 332, 1245–1257 (2024).
Lista, S. et al. A critical appraisal of blood-based biomarkers for Alzheimer’s disease. Ageing Res. Rev. 96, 102290 (2024).
Ossenkoppele, R., van der Kant, R. & Hansson, O. Tau biomarkers in Alzheimer’s disease: towards implementation in clinical practice and trials. Lancet Neurol. 21, 726–734 (2022).
Salvado, G. et al. Specific associations between plasma biomarkers and postmortem amyloid plaque and tau tangle loads. EMBO Mol. Med. 15, e17123 (2023).
Therriault, J. et al. Association of phosphorylated tau biomarkers with amyloid positron emission tomography vs tau positron emission tomography. JAMA Neurol. 80, 188–199 (2023).
Smith, R. et al. Tau-PET is superior to phospho-tau when predicting cognitive decline in symptomatic AD patients. Alzheimers Dement. 19, 2497–2507 (2023).
Mundada, N. S. et al. Head-to-head comparison between plasma p-tau217 and flortaucipir-PET in amyloid-positive patients with cognitive impairment. Alzheimers Res. Ther. 15, 157 (2023).
Feizpour, A. et al. Two-year prognostic utility of plasma p217+tau across the Alzheimer’s continuum. J. Prev. Alzheimers Dis. 10, 828–836 (2023).
Sperling, R. A. et al. Trial of solanezumab in preclinical Alzheimer’s disease. N. Engl. J. Med. 389, 1096–1107 (2023).
van Dyck, C. H. et al. Lecanemab in early Alzheimer’s disease. N. Engl. J. Med. 388, 9–21 (2023).
Sims, J. R. et al. Donanemab in early symptomatic Alzheimer disease: the TRAILBLAZER-ALZ 2 randomized clinical trial. JAMA 330, 512–527 (2023).
Ossenkoppele, R. et al. Amyloid and tau PET-positive cognitively unimpaired individuals are at high risk for future cognitive decline. Nat. Med. 28, 2381–2387 (2022).
Mattsson-Carlgren, N. et al. Prediction of longitudinal cognitive decline in preclinical Alzheimer disease using plasma biomarkers. JAMA Neurol. 80, 360–369 (2023).
Papp, K. V., Rentz, D. M., Orlovsky, I., Sperling, R. A. & Mormino, E. C. Optimizing the preclinical Alzheimer’s cognitive composite with semantic processing: the PACC5. Alzheimers Dement. 3, 668–677 (2017).
Jack, C. R. Jr. et al. Revised criteria for diagnosis and staging of Alzheimer’s disease: Alzheimer’s Association Workgroup. Alzheimers Dement. 20, 5143–5169 (2024).
Mattsson-Carlgren, N. et al. Longitudinal plasma p-tau217 is increased in early stages of Alzheimer’s disease. Brain 143, 3234–3241 (2020).
Montoliu-Gaya, L. et al. Optimal blood tau species for the detection of Alzheimer’s disease neuropathology: an immunoprecipitation mass spectrometry and autopsy study. Acta Neuropathol. 147, 5 (2023).
Salvado, G. et al. Cerebral amyloid-β load is associated with neurodegeneration and gliosis: mediation by p-tau and interactions with risk factors early in the Alzheimer’s continuum. Alzheimers Dement. 17, 788–800 (2021).
Marquie, M. et al. Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann. Neurol. 78, 787–800 (2015).
Fleisher, A. S. et al. Positron emission tomography imaging with [18F]flortaucipir and postmortem assessment of Alzheimer disease neuropathologic changes. JAMA Neurol. 77, 829–839 (2020).
Lowe, V. J. et al. Tau-positron emission tomography correlates with neuropathology findings. Alzheimers Dement. 16, 561–571 (2020).
Cho, H. et al. Tau PET in Alzheimer disease and mild cognitive impairment. Neurology 87, 375–383 (2016).
Sperling, R. A. et al. The impact of amyloid-β and tau on prospective cognitive decline in older individuals. Ann. Neurol. 85, 181–193 (2019).
Biel, D. et al. Tau-PET and in vivo Braak-staging as prognostic markers of future cognitive decline in cognitively normal to demented individuals. Alzheimers Res. Ther. 13, 137 (2021).
Jack, C. R. Jr. et al. Associations of amyloid, tau, and neurodegeneration biomarker profiles with rates of memory decline among individuals without dementia. JAMA 321, 2316–2325 (2019).
Ossenkoppele, R. et al. Associations between tau, Aβ, and cortical thickness with cognition in Alzheimer disease. Neurology 92, e601–e612 (2019).
Brum, W. S. et al. A blood-based biomarker workflow for optimal tau-PET referral in memory clinic settings. Nat. Commun. 15, 2311 (2024).
Mattsson-Carlgren, N. et al. Plasma biomarker strategy for selecting patients with Alzheimer disease for antiamyloid immunotherapies. JAMA Neurol. 81, 69–78 (2024).
Villemagne, V. L. et al. CenTauR: toward a universal scale and masks for standardizing tau imaging studies. Alzheimers Dement. 15, e12454 (2023).
Farrer, L. A. et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. JAMA 278, 1349–1356 (1997).
Yamazaki, Y., Zhao, N., Caulfield, T. R., Liu, C. C. & Bu, G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol. 15, 501–518 (2019).
Mattsson, N. et al. Prevalence of the apolipoprotein E ε4 allele in amyloid β positive subjects across the spectrum of Alzheimer’s disease. Alzheimers Dement. 14, 913–924 (2018).
Roberts, R. O. et al. The Mayo Clinic Study of Aging: design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology 30, 58–69 (2008).
Fowler, C. et al. Fifteen years of the Australian Imaging, Biomarkers and Lifestyle (AIBL) Study: progress and observations from 2,359 older adults spanning the spectrum from cognitive normality to Alzheimer’s disease. J. Alzheimers Dis. Rep. 5, 443–468 (2021).
Slot, R. E. R. et al. Subjective Cognitive Impairment Cohort (SCIENCe): study design and first results. Alzheimers Res. Ther. 10, 76 (2018).
Jack, C. R. Jr. et al. Defining imaging biomarker cut points for brain aging and Alzheimer’s disease. Alzheimers Dement. 13, 205–216 (2017).
Palmqvist, S. et al. Discriminative accuracy of plasma phospho-tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA 324, 772–781 (2020).
Groot, C. et al. Diagnostic and prognostic performance to detect Alzheimer’s disease and clinical progression of a novel assay for plasma p-tau217. Alzheimers Res. Ther. 14, 67 (2022).
Donohue, M. C. et al. The preclinical Alzheimer cognitive composite: measuring amyloid-related decline. JAMA Neurol. 71, 961–970 (2014).
Petersen, R. C. Mild cognitive impairment as a diagnostic entity. J. Intern. Med. 256, 183–194 (2004).
Petersen, R. C. et al. Practice guideline update summary: mild cognitive impairment: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology 90, 126–135 (2018).
Acknowledgements
Work at Lund University was supported by the National Institute of Aging (R01AG083740), the European Research Council (ADG-101096455), the Alzheimer’s Association (ZEN24-1069572, SG-23-1061717), the GHR Foundation, the Swedish Research Council (2022-00775, 2018-02052), ERA PerMed (ERAPERMED2021-184), the Knut and Alice Wallenberg Foundation (2022-0231), the Strategic Research Area MultiPark (multidisciplinary research in Parkinson’s disease) at Lund University, the Swedish Alzheimer Foundation (AF-980907, AF-994229, AF-994075), the Swedish Brain Foundation (FO2021-0293, FO2023-0163, FO2022-0204), the Parkinson foundation of Sweden (1412/22), Cure Alzheimer’s Fund, the Rönström Family Foundation (FRS-0003), Konung Gustaf V:s och Drottning Victorias Frimurarestiftelse, the Skåne University Hospital Foundation (2020-O000028), Regionalt Forskningsstöd (2022-1259), the Swedish federal government under the ALF agreement (2022-Projekt0080, 2022-Projekt0107), WASP and DDLS joint call for research projects (WASP/DDLS22-066), the BrightFocus Foundation (A2021013F) and the Fonds de Recherche en Santé Québec (298314). The precursor of [18F]flortaucipir was provided by Avid Radiopharmaceuticals, and the precursor of [18F]RO948 was provided by Roche. The precursor of [18F]flutemetamol was sponsored by GE Healthcare. The MCSA is supported by the National Institute on Aging, U01 AG006786, GHR and the Alzheimer’s Association. G.S. received funding from the European Union’s Horizon 2020 Research and Innovation Programme under Marie Sklodowska-Curie action grant agreement number 101061836, an Alzheimer’s Association Research Fellowship (AARF-22-972612), the Alzheimerfonden (AF-980942), Greta och Johan Kocks research grants and travel grants from the Strategic Research Area MultiPark at Lund University. R.O. has received research funding from the European Research Council, ZonMw, NWO, the National Institutes of Health, the Alzheimer Association, Alzheimer Nederland, the Stichting Dioraphte, Cure Alzheimer’s Fund, Health Holland, ERA PerMed, the Alzheimerfonden and the Hjarnfonden. Research at Alzheimer Center Amsterdam is part of the neurodegeneration research program of Amsterdam Neuroscience. Alzheimer Center Amsterdam is supported by the Stichting Alzheimer Nederland and the Stichting Steun Alzheimercentrum Amsterdam. The chair of W.M.v.d.F. is supported by the Pasman Stichting. The SCIENCe project receives funding from the Stichting Dioraphte and the Noaber Foundation. Funding for amyloid-PET scans was provided by Avid. C.E.T.’s research is supported by the European Commission (Marie Curie International Training Network, grant agreement no. 860197 (MIRIADE), Innovative Medicines Initiatives 3TR (Horizon 2020, grant no. 831434), EPND (IMI 2 Joint Undertaking, grant no. 101034344) and JPND (bPRIDE), the National MS Society (Progressive MS Alliance), the CANTATE project funded by the Alzheimer Drug Discovery Foundation, the Alzheimer Association, Health Holland, the Dutch Research Council (ZonMw), the Alzheimer Drug Discovery Foundation, the Selfridges Group Foundation and Alzheimer Netherlands. C.E.T. is recipient of ABOARD, which is a public–private partnership receiving funding from ZonMw (73305095007) and Health~Holland, Topsector Life Sciences & Health (PPP allowance, LSHM20106). C.E.T. is a recipient of TAP-dementia, a ZonMw-funded project (10510032120003) in the context of the Dutch National Dementia Strategy. E.v.d.G. has received research support from the NWO, ZonMw, the Hersenstichting, Alzheimer Nederland, Health~Holland and the KWF. The data contributed from the Wisconsin Registry for Alzheimer’s Prevention was supported by grant funding from the National Institute on Aging to the University of Wisconsin (R01 AG021155 and R01 AG027161 to S.C.J.). Funding for data from Washington University was supported by National Institute on Aging grants R01AG070941 (S.E.S.), P30AG066444 (J.C. Morris), P01AG003991 (J.C. Morris) and P01AG026276 (J.C. Morris). Blood plasma measurements were supported by RF1AG061900 (R.J.B), R56AG061900 (R.J.B.) and the Tracy Family SILQ Center (R.J.B.). The data contributed from the PREVENT-AD was supported by public–private partnership funds provided by McGill University, the Fonds de Recherche du Québec — Santé, an unrestricted research grant from Pfizer Canada, the Levesque Foundation, the Douglas Hospital Research Centre and Foundation, the Government of Canada, the Canada Fund for Innovation, the Canadian Institutes of Health Research, the Alzheimer Society of Canada, the National Institutes of Health of the United States, the Alzheimer Association and Brain Canada Foundation. J.T. is funded by a Colin J. Adair Charitable Foundation fellowship. Funding for the TRIAD cohort comes from the Weston Brain Institute, the Canadian Institutes of Health Research (MOP-11-51-31; RFN 152985, 159815, 162303), the Canadian Consortium of Neurodegeneration and Aging (MOP-11-51-31, team 1), the Alzheimer’s Association (NIRG-12-92090, NIRP-12-259245), Brain Canada Foundation (CFI project 34874; 33397), the Fonds de Recherche du Québec — Santé (Chercheur Boursier, 2020-VICO-279314) and the Colin J. Adair Charitable Foundation. Data were obtained from the AIBL flagship study of aging and the Australian Dementia Network funded by the National Health and Medical Research Council of Australia (grants APP1132604, APP1140853 and APP1152623), a grant from Enigma Australia and support from the Commonwealth Scientific and Industrial Research Organization. The funding sources had no role in the design and conduct of the study; in the collection, analysis or interpretation of the data; or in the preparation, review or approval of the manuscript.
Funding
Open access funding provided by Lund University.
Author information
Authors and Affiliations
Consortia
Contributions
R.O., G.S. and O.H. designed the study. R.O. and G.S. had full access to raw data and carried out the statistical analyses. R.O., G.S. and O.H. wrote the manuscript and had the final responsibility to submit for publication. All other authors contributed demographic, clinical, biomarker and neuroimaging data, contributed to the interpretation of the results and critically reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
R.O. has received research support from Avid Radiopharmaceuticals, Janssen Research & Development, Roche, Quanterix and Optina Diagnostics. He has given lectures in symposia sponsored by GE Healthcare, received speaker fees from Springer and serves on advisory boards and/or steering committees for Asceneuron, Bristol Myers Squibb and Biogen. G.S. has received speaker fees from Springer and Adium. S.P. has acquired research support (for the institution) from ki elements (ADDF) and Avid. In the past 2 years, he has received consultancy and/or speaker fees from BioArctic, Biogen, Esai, Lilly and Roche. Research programs of W.M.v.d.F. have been funded by ZonMw, the NWO, EU-JPND, EU-IHI, Alzheimer Nederland, the Hersenstichting CardioVascular Onderzoek Nederland, Health~Holland, Topsector Life Sciences & Health, the Stichting Dioraphte, Gieskes-Strijbis Fonds, the Stichting Equilibrio, Edwin Bouw Fonds, the Pasman Stichting, the Stichting Alzheimer & Neuropsychiatrie Foundation, Philips, Biogen MA, Novartis-NL, Life-MI, Avid, Roche, Fujifilm, Eisai and Combinostics. W.M.v.d.F. holds the Pasman chair. W.M.v.d.F. is a recipient of ABOARD, which is a public–private partnership receiving funding from ZonMw (73305095007) and Health~Holland, Topsector Life Sciences & Health (PPP allowance, LSHM20106). W.M.v.d.F. is a recipient of TAP-dementia (http://www.tap-dementia.nl), receiving funding from ZonMw (10510032120003) in the context of the Onderzoeksprogramma Dementie, part of the Dutch National Dementia Strategy. TAP-dementia receives cofinancing from Avid Radiopharmaceuticals and Amprion. Gieskes-Strijbis Fonds also contributes to TAP-dementia. W.M.v.d.F. has been an invited speaker at Biogen MA, Danone, Eisai, WebMD Neurology (Medscape), Novo Nordisk, Springer Healthcare and the European Brain Council. W.M.v.d.F. is a consultant to the Oxford Health Policy Forum CIC, Roche, Biogen MA and Eisai. W.M.v.d.F. participated in advisory boards of Biogen MA, Roche and Eli Lilly. W.M.v.d.F. is member of the steering committee of EVOKE and EVOKE+ (Novo Nordisk). All funding is paid to her institution. W.M.v.d.F. is member of the steering committee of PAVE and Think Brain Health. W.M.v.d.F. was an associate editor of Alzheimer’s Research and Therapy in 2020 and 2021. W.M.v.d.F. is an associate editor at Brain. E.v.d.G. has performed contract research for Heuron and Roche. E.v.d.G. has a consultancy agreement with IXICO and Life Molecular Imaging for reading PET scans. C.E.T. has research contracts with Acumen, ADx Neurosciences, AC Immune, Alamar, Aribio, Axon Neurosciences, Beckman-Coulter, BioConnect, Bioorchestra, Brainstorm Therapeutics, Celgene, Cognition Therapeutics, EIP Pharma, Eisai, Eli Lilly, Fujirebio, Instant NanoBiosensors, Novo Nordisk, Olink, PeopleBio, Quanterix, Roche, Toyama and Vivoryon. She is the editor in chief of Alzheimer’s Research and Therapy and serves on editorial boards of Medidact Neurologie/Springer and Neurology: Neuroimmunology & Neuroinflammation. She had consultancy and/or speaker contracts for Eli Lilly, Merck, Novo Nordisk, Olink and Roche. S.E.S. has served as a consultant to Eisai and Novo Nordisk and has received speaker fees from Eli Lilly and Medscape. She has analyzed biomarker data provided by C2N Diagnostics. J.T. has served as a consultant for the Neurotorium educational platform and for Alzheon. P.R.-N. has served on scientific advisory boards and/or as a consultant for Roche, Novo Nordisk, Eisai and Cerveau Radiopharmaceuticals. C.R. has received research grants from the National Health and Medical Research Council, Enigma Australia, Biogen, Eisai and AbbVie. He is on the scientific advisory board for Enigma/Meilleur Technologies and has consulted for Prothena, Eisai, Roche and Biogen Australia. R.J.B. has received laboratory research funding from the National Institutes of Health, the Alzheimer’s Association, the BrightFocus Foundation, the Rainwater Foundation, the Association for Frontotemporal Degeneration FTD Biomarkers Initiative, Avid Radiopharmaceuticals, Janssen, the Tau Consortium, Novartis, Centene, the Association for Frontotemporal Degeneration, the Cure Alzheimer’s Fund, Coins for Alzheimer’s Research Trust Fund, the Foundation for Barnes-Jewish Hospital, the Good Ventures Foundation, the DIAN-TU Pharma Consortium, the Tau SILK Consortium (AbbVie, Biogen, Eli Lilly and an anonymous organization), the NfL Consortium (AbbVie, Biogen, Bristol Meyers Squibb, Hoffmann-La Roche) and the Tracy Family SILQ Center; has equity ownership interest in C2N Diagnostics and receives income based on technology licensed by Washington University to C2N Diagnostics; and receives income from C2N Diagnostics for serving on the scientific advisory board. O.H. has acquired research support (for the institution) from Avid Radiopharmaceuticals, Biogen, C2N Diagnostics, Eli Lilly, Eisai, Fujirebio, GE Healthcare and Roche. In the past 2 years, he has received consultancy and/or speaker fees from AC Immune, ALZpath, BioArctic, Biogen, Bristol Meyers Squibb, Cerveau, Eisai, Eli Lilly, Fujirebio, Merck, Novartis, Novo Nordisk, Roche, Sanofi and Siemens. S.C.J. has served in the past 2 years on advisory boards for Enigma Biomedical and ALZpath. The other authors report no competing interests.
Peer review
Peer review information
Nature Aging thanks Takeshi Iwatsubo, Qianhua Zhao, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 The association between plasma p-tau217 and Tau-PETMTL/Tau-PETNEO across cohorts.
Spearman correlation analyses between plasma p-tau217 and Tau-PETMTL (left panel) and between plasma p-tau217 and Tau-PETNEO (right panel), color coded by cohort.
Extended Data Fig. 2 Plasma p-tau217 and Tau-PET prediction of future cognitive decline on the mPACC5 in Aβ+ participants.
a, Scatterplots showing the association between cognitive change over time on the mPACC5 and the tau biomarkers (Quartile 1-3 vs Quartile 4) across Aβ+ participants. The shadow area indicated the 95% confidence interval around the mean derived from linear regression models. Note that the standardized β-coefficients and R2 statistics relate to the tau biomarker as a continuous variable and that classification into quartiles was performed for visualization purposes only. b, Explained variance (R2, inside the bar plot) and model fit (corrected Akaike criterion, outside the bar plot) for various models predicting longitudinal change on the mPACC5 across all participants. Errorbars represent the 95% CI around the mean derived from linear regression models. c, Partial explained variance (R2) for combined biofluid and neuroimaging models predicting longitudinal change on the mPACC5 across all participants. “Shared” in panel c refers to the explained variance shared between Tau-PET and plasma p-tau217 that could not be attributed to a single tau biomarker in the model. Note that we computed cohort-specific z-scores for plasma p-tau217 and Tau-PET using amyloid-PET negative CU individuals from the same cohort as the reference group. *p<0.05, **p<0.01, ***p<0.001. Exact p-values can be found in Supplementary Tables 6 and 7.
Extended Data Fig. 3 Plasma p-tau217 and Tau-PET prediction of future progression to MCI in Aβ+ participants.
Note that all analyses are performed in Aβ+ participants only. a, Survival curves for progression to mild cognitive impairment (Quartile 1-3 vs Quartile 4) across in Aβ+ participants, including a table of total number of participants available at each time point. The shadow area indicated the 95% confidence interval around the mean derived from Cox proportional hazard models. Note that the HR and AIC statistics relate to the tau biomarker as a continuous variable, and that classification into quartiles was only done for visualization purposes. b, Model fit (corrected Akaike criterion) for various models predicting future clinical progression to mild cognitive impairment across all participants. Error bars represent the 95% CI around the mean derived from Cox proportional hazard models. c, Hazard ratios and 95% confidence intervals around the mean derived from Cox proportional hazard models in simple models (that is, modeling one tau biomarker at the time, top three HR’s) and combined models (that is, modeling plasma and PET biomarkers simultaneously, bottom four HR’s). Note that we computed cohort-specific z-scores for plasma p-tau217 and Tau-PET using amyloid-PET negative CU individuals from the same cohort as the reference group. * p<0.05, **p<0.01, ***p<0.001. Exact p-values can be found in Supplementary Tables 12 and 13.
Extended Data Fig. 4 A two-step recruitment approach for clinical trials in preclinical Alzheimer’s disease using the mPACC as outcome measure with plasma p-tau217 and Tau-PETNEO.
Note that all analyses are performed using Tau-PETNEO only. a, the obtained sample size reduction using sample selection based on different percentiles (75th, 50th and 25th) of baseline plasma p-tau217 levels in step 1 followed by the selection based on the same percentiles (75th, 50th and 25th) of the Tau-PETNEO measurement in step 2 with mPACC5 as the primary endpoint. Note that 100% in step 2 refers to the participants selected by plasma p-tau217 in step 1. Error bars represent the 95% CI around the mean derived from mixed effects models. b, shows the calculated sample size reductions for various plasma p-tau217 and Tau-PETNEO quartile combinations. Calculations are based on the assumption of 80% power to detect a 30% change on the mPACC5 in a 4-year trial. Red lines represent step 1 with plasma p-tau217 and blue lines represent step 2 with Tau-PETNEO. Different line styles represent different quartiles of Tau-PETNEO from those subjects already selected from step 1. Dotted black lines represent 100% participants needed without that step.
Extended Data Fig. 5 A two-step recruitment approach for clinical trials in preclinical Alzheimer’s disease using the mPACC as outcome measure in Aβ+ participants.
Note that all analyses are performed in Aβ+ participants only. a, the obtained sample size reduction using sample selection based on different percentiles (75th, 50th and 25th) of baseline plasma p-tau217 levels in step 1 followed by the selection based on the same percentiles (75th, 50th and 25th) of the Tau-PETMTL measurement in step 2 with mPACC5 as the primary endpoint. Note that 100% in step 2 refers to the participants selected by plasma p-tau217 in step 1. Error bars represent the 95% CI around the mean. b, shows the calculated sample size reductions for various plasma p-tau217 and Tau-PETMTL quartile combinations. Calculations are based on the assumption of 80% power to detect a 30% change on the mPACC5 in a 4-year trial. Red lines represent step 1 with plasma p-tau217 and green lines represent step 2 with Tau-PETMTL. Different line styles represent different quartiles of Tau-PETMTL from those subjects already selected from step 1. Dotted black lines represent 100% participants needed without that step. c,d same as above, but now for Tau-PETNEO instead of Tau-PETMTL.
Extended Data Fig. 6 A three-step recruitment approach for clinical trials in preclinical Alzheimer’s disease, now also implementing Amyloid-PET.
a, the obtained sample size reduction using sample selection based on different percentiles (75th, 50th and 25th) of baseline plasma p-tau217 levels in step 1, followed by adding Amyloid-PET positivity (step 2) followed by the selection based on the same percentiles (75th, 50th and 25th) of the Tau-PETMTL measurement in step 3 with mPACC5 as the primary endpoint. Error bars represent the 95% CI around the mean. Red lines represent step 1 with plasma p-tau217, light blue lines represent step 2 with Amyloid PET and green lines step 3 with Tau-PETNEO. Different line styles represent different quartiles of Tau-PETNEO from those subjects already selected from steps 1 and 2. b, shows the calculated sample size reductions for various plasma p-tau217 (quartiles), Amyloid-PET (status) and Tau-PETMTL (quartiles) combinations. Calculations are based on the assumption of 80% power to detect a 30% change on the mPACC5 in a 4-year trial. c,d same as above, but now for Tau-PETNEO instead of Tau-PETMTL.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ossenkoppele, R., Salvadó, G., Janelidze, S. et al. Plasma p-tau217 and tau-PET predict future cognitive decline among cognitively unimpaired individuals: implications for clinical trials. Nat Aging 5, 883–896 (2025). https://doi.org/10.1038/s43587-025-00835-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43587-025-00835-z
