
Abstract
Background
Multiple sulfatase deficiency (MSD) is an exceptionally rare neurodegenerative disorder due to the absence or deficiency of 17 known cellular sulfatases. The activation of all these cellular sulfatases is dependent on the presence of the formylglycine-generating enzyme, which is encoded by the SUMF1 gene. Disease-causing homozygous or compound heterozygous variants in SUMF1 result in MSD. Other than symptomatic treatment, no curative therapy exists as of yet for MSD. Eight out of these 17 sulfatases are primarily localized in the lysosome.
Methods
Two siblings with attenuated MSD underwent hematopoietic cell transplantation (HCT), evaluating the possibility of lysosomal enzymatic cross-correction from the donor cells.
Results
There is evidence of correction of currently available biomarkers within 3 months post-HCT. Untargeted metabolomics also shows continued correction of multiple biochemical abnormalities in the post-HCT period. Furthermore, this article also presents the neuropsychological outcomes of these children as well as the results of untargeted metabolomics analysis in this condition.
Conclusions
These data suggest biochemical benefits post-transplant along with slowing of disease progression. Long-term follow-up is necessary to fully evaluate the therapeutic benefit of HCT in MSD.
Plain language summary
Multiple sulfatase deficiency (MSD) is a genetic disorder in which body is unable to produce certain enzymes which are a type of protein. MSD affects multiple body systems, primarily the brain. The current treatments available are catered toward easing the symptoms and cannot cure the disease. In this study, two siblings with a mild form of MSD received bone marrow transplantation (HCT) that replaced their bone marrow with stem cells containing the healthy version of the gene responsible for MSD. The children were monitored for two years, during which time the siblings continued to produce the enzymes that are normally absent in MSD and did not have mental or behavioral deterioration. This study suggests that HCT might help slow the progress of MSD and be an improved treatment. However, further and longer-term studies are required to investigate this further.
Similar content being viewed by others


Introduction
Multiple sulfatase deficiency (MSD; MIM# 272200) is an extremely rare autosomal recessive disorder characterized by the deficiency of formylglycine generating enzyme (FGE), encoded by SUMF1, affecting the activation of 17 different sulfatases known to date. In sulfatases, FGE aids in the oxidation of cysteine residues to formylglycine, hence activating it via post-translational modification in the endoplasmic reticulum1,2. Ten out of the seventeen human sulfatases have been linked to human diseases which include arylsulfatase A (metachromatic leukodystrophy, MLD), arylsulfatase B (Maroteaux-Lamy syndrome/MPS VI), arylsulfatase C (X-linked ichthyosis), arylsulfatase L (chondrodysplasia punctata, X-linked), arylsulfatase K (MPS X), iduronate-2-sulfatase (Hurler syndrome/MPSII), heparan-N-sulfatase (Sanfilippo syndrome IIIA/MPSIIIA), N-acetyglucosamine-6-sulfatase ((Sanfilippo syndrome IIID/ MPS IIID), arylsulfatase G (Usher syndrome), and N-acetylgalactosamine-6-sulfatase (Morquio disease/MPS IVA)3.
The characteristic phenotype is a combination of the above-mentioned disorders. Affected individuals could present with primarily a neurodegenerative disorder, accompanied by additional features such as hepatosplenomegaly, dysostosis multiplex, chondrodysplasia punctata, cardiomegaly, and valvular heart disease, and ichthyosis4. The phenotype has been broadly divided into severe and attenuated forms based on the age of onset, early motor development and severity of symptoms5,6.
Currently, in the absence of any standardized therapies for MSD, treatment is symptom-based and supportive7. The scope of hematopoietic cell transplantation (HCT) has been studied in various disorders and has now emerged as the treatment of choice for certain lysosomal storage disorders, including MPS IH and MLD, although gene therapies are being explored. HCT has been shown to reduce hepatosplenomegaly and stabilize the neurodevelopmental phenotype in MPS IH8,9,10. Even though in comparison to MPS IH, the benefit of HCT in MPS types II, III, IV, and VI is limited, it has been shown to improve clinical status in selected cases11,12,13,14. Similarly, HCT may have a neuroprotective and immunomodulatory role in MLD, improving clinical outcomes in certain subgroups, including juvenile MLD15,16. Cross-correction of the enzyme activity by HCT for steroid sulfatase and chondrodysplasia punctata has not been documented.
In regard to biomarkers, apart from reduced levels of the affected enzymes, these individuals also have increased excretion of multiple glycosaminoglycans (GAG) in the urine including heparan sulfate (associated with iduronate-2-sulfatase, heparan sulfatase, and N-acetyglucosamine-6-sulfatase deficiency), dermatan sulfate (reflective of iduronate sulfatase and arylsulfatase B deficiency), chondroitin sulfate (associated with N-acetyl-galactosamine-6-sulfatase deficiency) and keratan sulfate (associated with N-acetyl-galactosamine-6-sulfatase deficiency)17. Urine sulfatide excretion may be increased but may vary based on the level of arylsulfatase A enzyme activity. Recently, Adang et al. reported that among the currently available biomarkers of this disease, heparan sulfate GAG non-reducing ends (NRE) would be a reliable diagnostic biomarker as well as a biomarker that could be used in the interpretation of disease severity18.
Metabolomics, an emerging field among “omics” investigations, analyzes the endogenous and exogenous small molecule metabolites that comprise the human metabolome and has thus enabled the discovery of novel biomarkers for many rare diseases19,20,21; however, the utility of untargeted metabolomics in MSD has not been explored.
Here, as a part of the non-prespecified interim analysis, we report the outcome of HCT in two siblings with mild late infantile form of MSD who underwent HCT at 10 and 8 years of age respectively. The rationale behind bone marrow transplantation in these siblings was that there would be enzymatic cross-correction of M6P-containing enzymes by the donor cells, resulting in alleviation of symptoms, particularly from disorders including MPS II, III, IV, VI, and MLD.
The clinical and biochemical outcomes of these siblings two years after HCT are presented here.
The results show continued correction of the currently available biomarkers at 3 years post-HCT. Untargeted metabolomics also reveals continued correction of multiple biochemical abnormalities post-HCT. Additionally, neuropsychology evaluation shows no evidence of regression and continued slow gain of milestones. These data highlight differences between siblings at the time of presentation and support the use of untargeted metabolomics for the detection of significant metabolites and pathways that may aid in the diagnosis and surveillance of MSD.
Methods
This HCT was done as a part of the clinical trial at the University of Minnesota registered under ClinicalTrials.gov ID NCT00176904. The study was conducted in accordance with the principles of the Declaration of Helsinki and followed IRB-approved protocols approved by the University of Minnesota (IRB ID: CR00014502). The interim analysis was published due to the ultra-rare nature of MSD and the absence of any available approved therapies. Informed consent was obtained from both parents prior to this treatment. These siblings were the first to receive HCT as a potential treatment option. Signed parental consent was obtained to publish the transplant-related data, acknowledging that the children’s identities might be discernible from the data presented in this paper.
Patient I (P1) is an 11-year-old female who was born at 38 weeks gestation via spontaneous vaginal delivery to a 31-year-old G2P1 mother after an uncomplicated pregnancy. Following birth, the patient had jaundice and poor feeding. Her birth weight was 3.43 kg (65th centile), and her length was 19.25 in. Her Apgar scores were 8 and 9 at 1 and 5 min, respectively.
She presented with ankyloglossia within the first month of life and struggled to gain weight, which resolved without supplementation around 6 weeks. An umbilical hernia was also noted before 1 year of age. She sat independently at 9 months, crawled at 10 months, said her first words at 11 months, and walked independently at 14 months. Her parents raised concerns for developmental delay, mostly in the form of speech delay around 15–18 months of age. At 2.5 years of age, she was diagnosed with an expressive speech delay and began speech therapy. Parents noted behavioral difficulties, sensory integration and sleep issues, and poor social interaction at the age of 4 years. She was evaluated and subsequently diagnosed with autism at age 5 for which she started receiving physical, occupational, speech, and applied behavior analysis (ABA) therapies regularly. She was diagnosed with ichthyosis with dry and scaly skin on the neck, back, and scalp at 5 years of age.
The patient was referred to genetics at 8 years of age for evaluation of short stature and behavioral concerns. During evaluation, she was noted to have thick alveolar ridges, hypotonia, and overlapping toes. Her chromosomal microarray identified a 297 kb de novo duplication of uncertain clinical significance at 13q13.3. An autism/intellectual disability panel was then ordered which identified compound heterozygous, likely pathogenic variants in SUMF1 (please see results section). Subsequent urine glycosaminoglycans (GAGs) analysis showed mild elevation of heparan sulfate and trace sulfatides. Leukocyte sulfatase activity testing revealed low iduronate-2-sulfatase, N-acetylgalactosamine-6-sulfatase, and arylsulfatase A levels. These results are consistent with a diagnosis of MSD. Since her diagnosis, the patient has had a normal EKG, echocardiogram, and abdominal ultrasound. Skeletal survey showed 18-degree scoliosis but was otherwise normal. Brain MRI revealed agenesis of the corpus callosum and no evidence of leukodystrophy.
She currently wears glasses and has bilateral astigmatism that has been progressing over the past few years. There is no evidence of keratoconus, corneal clouding, or retinal abnormalities. She also has a history of frequent otitis media and mild conductive hearing loss requiring bilateral pressure-equalizing (PE) tubes placed at age 5. Subsequent audiology evaluations have been normal.
After informed consent, she underwent a myeloablative 8/8 HLA-matched unrelated donor HCT at 10 years of age. Her early post-transplant course was complicated by mucositis, intermittent fevers, and BK-virus-related hemorrhagic cystitis. At the time of latest evaluation, 2 years post-HCT, her ichthyosis is still persisting in the areas mentioned before, however, no worsening has been noted as she is only needing emollients once a week. Her brain MRI done 2 years post HCT, remains stable without any significant white matter signal abnormalities. The degree of engraftment in her myeloid and T cell components began to decline approximately two years after the hematopoietic cell transplantation (HCT) and had dropped to 43% and 70%, respectively, as they approached the three-year mark post-HCT. As a result, close monitoring of her clinical condition and biochemical markers is ongoing.
Patient 2 (P2) is a full sibling to Patient 1 and a 9-year-old male born at 38 weeks gestation via spontaneous vaginal delivery to a 33-year-old G3P2 mother after an uncomplicated pregnancy. His birth weight was 2.92 kg (18th centile). There were no postnatal complications.
He was found to have an umbilical hernia at 6 weeks of age. Evaluation for short stature led to the diagnosis of growth hormone deficiency from an outside hospital, and he was started on growth hormone supplementation at 3 years of age. This was following an abnormal growth hormone stimulation test, though he had normal insulin-like growth factor 1 (IGF-1) and IGF-binding protein 3 (IGFBP-3). The actual results were not available for review.
His developmental milestones were appropriate for his age; he sat independently at 6 months, crawled at 9 months, had 2–5 words at 12 months, and walked independently at 16 months. At age 3.5, he was diagnosed with attention deficit- hyperactivity disorder. He had a neuropsychology evaluation at age 5, documenting below-average verbal and nonverbal intellect with impaired auditory working memory and processing speed. He was referred for speech therapy and occupational therapy and was started on guanfacine for his hyperactive behavior.
Follow-up evaluations demonstrated clear cornea, mild astigmatism of both eyes and normal optic nerves with no evidence of hypoplasia. He was seen for recurrent otitis media infections in the first few years of life. He was initially evaluated by genetics at age 2 years due to short stature. During the exam, he was noted to have a flat occiput, short forehead, right esotropia, prominent and broad nasal root, wide uvula, short and webbed neck, wide-spaced nipples, hypotonia, lordosis, and ichthyosis mostly around the pelvic girdle. He had a normal chromosomal microarray. At 6-month follow-up, his muscle tone was apparently normal, and he was discharged from the clinic.
Brain MRI at 5 years demonstrated agenesis of the corpus callosum with underdevelopment of the genu and the rostrum, with slightly decreased hemispheric white matter volume posteriorly.
The patient was first evaluated for “loud, snorty breathing” around 6 months of age, and a sleep study documented severe obstructive sleep apnea. He underwent tonsillectomy and adenoidectomy and ultimately was placed on a continuous positive airway pressure (CPAP) machine.
Following his sister’s diagnosis with multiple sulfatase deficiency, the patient had targeted testing for the familial SUMF1 variants and was found to carry both variants. Subsequent urine glycosaminoglycans (GAGs) analysis showed mild elevation of heparan sulfate; sulfatides were normal. Sulfatase activity testing revealed low N-acetylgalactosamine-6-sulfatase and arylsulfatase A levels. The iduronate-2-sulfatase level was normal. He has had normal skeletal surveys, EKGs, and echocardiograms. At the time of initial evaluation by endocrinology at our institution, he was on 0.4 mg Norditropin (0.09 mg/kg/week) was 133 cm tall (78th centile), and had normal IGF-1 and IGFBP-3 levels. As a result, GH supplementation was held. Following the HCT for his sibling, he underwent a myeloablative 8/8 HLA-matched unrelated donor HCT at 8 years of age. Post-HCT complications included episodes of ectopic atrial tachycardia, mucositis, pericardial effusion, and hemorrhagic cystitis. At the time of the last follow-up, 2 years post-HCT, he continues to have mild ichthyosis but remains off of growth hormone supplementation, with growth appropriate for age. A brain MRI obtained 2 years post-HCT did not show any white matter abnormalities or any evidence of leukodystrophy. Since he was able to maintain normal growth velocity after HCT, he remains off of GH supplementation with close monitoring. At the time of his last visit, 2 years post-HCT, he is 141.4 cm (63rd centile) and has a growth velocity of 4.8 cm/year (31st centile), with low-normal IGF-1 at 150 (reference: 100-449 ng/mL), normal IGFBP-3 at 5.5 (reference: 2.3-6.3 ng/mL) and normal bone age. Though mildly delayed, he continues to attain milestones. He remains 100% engrafted in the myeloid fraction (CD33/66+) and T cell fraction (CD3+).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Molecular
Chromosomal microarray
Whole genome array-based comparative genomic hybridization (aCGH) and genotype analyses are performed on a custom-designed oligonucleotide microarray (GenomeDx v5). The array design is based on the human genome build GrCh37.
Gene panel
Genomic DNA from the submitted sample was enriched for complete coding regions and splice site junctions using a proprietary capture system with next generation sequencing with CNV calling. The enriched targets were simultaneously sequenced with paired end-reads on an Illumina platform. Bi-directional sequence reads were assembled and aligned to reference sequences based on NCBI RefSeq transcripts and human genome build GRCh37/UCSC hg19. Sequence and copy number variants were reported according to the Human Genome Variation Society or International System for Cytogenetic Nomenclature guidelines, respectively.
Biochemical
Urine GAGs were analyzed by liquid chromatography–tandem mass spectrometry (LC–MS/MS). Lipid extraction and thin layer chromatography of the urine was used in sulfatide analysis. Leukocyte enzyme assays were measured by incubating the leukocytes with an enzyme-specific artificial substrate containing a 4-methylumbelliferone (4-MU) and measuring the fluorescence when the 4-MU residue is removed by endogenous enzyme activity. The amount of 4-MU released is determined by comparing the fluorescence to a standard curve.
Untargeted metabolomics
Metabolomic profiling of plasma and urine was conducted by Baylor Genetics Laboratories (Houston, TX) and Metabolon, Inc. (Durham, NC) as previously described22,23,24. Small molecules were extracted from 100 µl of sample in an 80% methanol solution and analyzed using four independent platforms. Positive ionization was performed with Waters BEH C18 chromatographic separation for hydrophilic compounds (LC/MS/MS Pos Polar) and hydrophobic compounds (LC/MS/MS Pos Lipid). Negative ionization was carried out with Waters BEH C18 optimized conditions (LC/MS/MS Neg) and Waters BEH Amide (HILIC) chromatography (LC/MS/MS Polar). Chromatography utilized a Waters Acquity UPLC held at 40–50 °C. Metabolites were identified based on ion chromatographic retention index, accurate mass, and mass spectral fragmentation signatures, matching with reference library entries created from authentic standard metabolites under identical experimental conditions.
The raw spectral intensity values of biochemical compounds were first normalized to the median intensity of corresponding compounds in the anchor samples. Subsequently, these normalized values were transformed into z-scores using the mean and standard deviation derived from the median-normalized reference population. Z-scores indicate how many standard deviations a compound in the patient sample deviates from the mean of the reference population, providing a precise measure of its relative abundance or significance.
Neuropsychological evaluation
P1 underwent neuropsychological evaluations prior to HCT and annually thereafter as part of her clinical multidisciplinary assessment of outcomes. Her pre-HCT assessment was completed in her home state, and those records were reviewed. Fine motor testing was completed at the University of Minnesota to supplement home-state testing prior to HCT. All neuropsychological assessments were completed at the University of Minnesota thereafter. Methods for measurement of function adhered to international guidelines for neuropsychological assessment of related rare conditions (mucopolysaccharidosis disorders) in terms of instrument selection, testing approach and accommodations, and interpretation of scores25. The measure of cognitive function was the Wechsler Intelligence Scale for Children, Fifth Edition26, and the measure of fine motor function was the Purdue Pegboard27. Tracking the neurocognitive course of rare conditions involves analysis of how the individual compares themselves over time10. For the set of instruments appropriate for P1, this was accomplished by examining the number of points earned on tests at each assessment (known as raw scores)28. Neuropsychological assessment scheduling and methodology were the same for P2 as for P1, except his pre-HCT assessment was also completed at the University of Minnesota. For both siblings, a measure of adaptive functioning, the Vineland Adaptive Behavior Scales, Third Edition (VABS-3)29, was administered at a subset of time points. The latest VABS-3 is reviewed to reflect post-HCT outcomes.
Results
Molecular
P1 had a chromosomal microarray that showed a 297 kb de-novo duplication on chromosome 13q13.3: arr[GRCh37] 13q13.3(37308701-37605739 ×3 that was classified as a variant of uncertain significance. An autism/intellectual disability (ID) Xpanded panel showed compound heterozygous variants in SUMF1(NM_182760.4): c.726-1_726delGA; a maternally inherited pathogenic variant and c.640 G > A; p.Ala214Thr; a paternally inherited likely pathogenic variant. P2 had a normal chromosomal microarray. The p.A214T variant has been previously reported to be associated with milder phenotype with in silico modeling showing minimal strain on secondary structure5. Known familial variant testing in SUMF1 detected the same variants in P2 confirming the diagnosis.
Biochemical
Following the diagnosis of MSD, both siblings had enzyme assays that showed reduced enzyme activity of more than 3 sulfatases (Table 1). P1 had reduced levels of arylsulfatase A, arylsulfatase B, iduronate-2-sulfatase, N acetyl galactosamine 6 sulfatase, and heparan-N-sulfatase levels pre-HCT. P2 had reduced enzymatic levels of arylsulfatase A, arylsulfatase B, iduronate-2-sulfatase, heparan-N-sulfatase, and N-acetyl glucosamine-6-sulfatase. Compared to the pre-HCT values biochemical correction of all the previously deficient enzyme activity in the leukocytes was evident within 3 months post-HCT. Urine heparan sulfate was mildly elevated initially in P1 normalized after HCT. Urine dermatan sulfate, along with urine sulfatides, were normal in the siblings pre- and post-HCT due to their milder phenotype (Table 1).
Global untargeted metabolomics
Clinical global untargeted metabolomics (Global Metabolomic Assisted Pathway Screen, Global MAPS, Baylor Genetics) was performed as previously described on plasma from patients P1 and P2 to investigate the broader impact of multiple sulfatase deficiency (MSD) and as a means to monitor the effectiveness of treatment30,31,32,33.
Untargeted metabolomics identified a mean (SD) of 755 (32) analytes in each individual plasma sample. An enrichment analysis was conducted on pre-HCT samples from both siblings to determine the key molecules and sub-pathways disturbed in MSD patients. While single, unique biomarkers were not identified for MSD, multiple consistent metabolic perturbations across patients were noted. Specifically, glycerophospholipids constituted the predominant category of perturbed sub-pathways in both siblings (Fig. 1 and Supplementary Data 1b, c). Arylsulfates and fatty acids and derivatives exhibited substantial perturbations, representing the second or third largest proportion of perturbed metabolites across the siblings. Additionally, energy-related sub-pathways such as bile acids, cholesterols, sterols, nucleotides, and various amino acids were disrupted. Notably, the relative levels of disruption across these sub-pathways between the siblings are consistent. These results indicate that MSD induces a broad state of metabolic dysregulation.

Metabolomic data underwent enrichment analysis to identify key molecules and sub-pathways perturbed in MSD patients before and after HCT (Supplementary Data). Z-scores were utilized to indicate the significance of metabolite alterations, with a z-score greater than +1.7 and lower than −1.7 considered significantly perturbed. Non-z-scored metabolites in pre- and post-treatment samples were evaluated by ranking raw spectral intensity values across previously analyzed patient cohorts, identifying metabolites with a Rank (%)_Raw (see Supplementary data legend for detailed information). Ranking scores above 20% are identified as significantly elevated, while those below this threshold were considered within normal ranges. This metabolomic analysis reveals multiple altered metabolic pathways, highlighting lipids, including glycerophospholipid metabolism and fatty acids and derivatives-related analytes, and arylsulfates as the predominant perturbed pathways in both patients at disease status (left side of the x-axis, also see Supplementary Data 1b, c). The right side of the x-axis indicates the number of perturbed molecules in each sub-pathway post-HCT, illustrating the effectiveness of the treatment in correcting most of the metabolic abnormalities.
The impact of HCT was monitored by comparing metabolomic profiles pre- and post-HCT. An abrupt neutralization effect was observed following HCT (Fig. 1, right side of the x-axis). More than 70% of the biochemical abnormalities observed in the initial sample were normalized in the post-HCT period. This correction persisted in subsequent post-HCT samples (Fig. 2A, B), suggesting sustained biochemical correction induced by HCT.

A Rate of abnormal metabolites being corrected by HCT treatment. Abnormal metabolites are considered “corrected” if the levels of Z-scored metabolites fall within the ranges between −1.7 and +1.7, and the Rank (%)_Raw of non-z-scored metabolites fall below 20% by ranking previously tested patient cohort. P1 showed 142 abnormal metabolites in pre-HCT plasma samples, and the post-HCT samples at corresponding collection times revealed 26, 28, 31, 25, and 17 abnormal compounds, respectively. P2 displayed 193 abnormal metabolites in pre-HCT plasma samples, and in the post-HCT samples, 55, 38, and 51 abnormal compounds were observed at corresponding collection times (See detailed data in Supplementary Data 1b, c). B Heatmap of abnormal (pre-HCT) metabolites identified in two MSD siblings and their normalization in the subsequent samples post-HCT. Metabolites (rows) are grouped by sub-pathways. Samples (columns) are grouped in the order of collection time. Higher z-scores are red in color, and lower z-scores are green in color, as shown in the legend bar. Analytes that were not detected are depicted in gray.
Neuropsychological evaluation
Neuropsychological evaluation results from before HCT until the 2-year HCT anniversary (Supplementary Table 1) do not indicate regression, as the patterns of score changes broadly show either forward developmental gains or maintenance of skills for each sibling. Both pre-HCT and 2 years post-HCT, P1’s overall cognitive scores (i.e., full-scale IQ scores) were in the impaired qualitative range (≤3 SD) when using the norm-referenced scoring method. Among the Index scores of the cognitive test, P1 showed the most significant impairments in speed of processing and retaining information for short-term recall (i.e., working memory, such as holding a phone number in mind before writing it down). When comparing P1’s points earned on the cognitive subtests at 2 years post-HCT (Supplementary Table 1), her scores are either the same as, or higher than, the points she earned pre-HCT. Significant gains were measured on a single subtest of quantitative and analogical reasoning (Figure Weights). At her 2-year HCT anniversary, P1 was in the 6th grade, and testing of academic skills placed her broadly at the kindergarten achievement level for reading comprehension, spelling, and math calculation. P1’s mother’s ratings of her independence in daily life (i.e., adaptive skills) at her 2-year HCT anniversary indicated generally below-average levels of independence, as assessed by a standardized rating measure of adaptive functioning.
P2’s overall cognitive scores (i.e., full-scale IQ scores) show a numeric decline in norm-referenced scores over time, which reflects a slowed pace of forward development that created a widening gap between P2 and the normative reference group. His performance was in the below-average qualitative range before HCT and in the impaired qualitative range 2 years after HCT. Unlike his sister, P2 did not have a significant relative weakness in his scores for speed of processing and working memory. When comparing P2’s points earned on the cognitive subtests at 2 years post-HCT (Supplementary Table 1), his scores are either the same as, or higher than, the points he earned pre-HCT. On fine motor testing, P2’s scores were in the impaired range before HCT, but at 2 years post-HCT, his performance improved to the below-average range for each hand individually, while staying in the impaired range when coordinating both hands together. At his 2-year HCT anniversary, P2 was in the 4th grade, and testing of academic skills placed him in the kindergarten-grade equivalent range for word decoding, reading comprehension, and spelling, while in math he was at a grade one equivalent. P2’s mother’s ratings of his independence in daily life (i.e., adaptive skills) at his 2-year HCT anniversary indicated generally age-typical average levels of independence overall, with slightly below-average skills related to communication and daily living skills.
Discussion
Due to the heterogeneous nature of MSD, as observed in these siblings, who presented with mild development delay and autism, the diagnosis would have been much delayed had next-generation sequencing (NGS) not been utilized. Hence, these instances emphasize the importance of genetic evaluation and pursuing cytogenetic and molecular testing in individuals with development delay and autism spectrum disorder as such approaches are crucial in uncovering ultrarare diseases like MSD.
HCT has been demonstrated to be an effective therapeutic strategy for certain inborn errors of metabolism primarily involving peroxisomal and lysosomal storage diseases34. When compared to enzyme replacement therapy (ERT), in addition to preventing/slowing down the progression of the disease, HCT also has the capacity to improve neurocognitive outcomes, thought to be due to the migration of donor-derived cells across the blood–brain barrier and subsequent “cross-correction”34,35. In the subset of conditions for which HCT is effective, the timing of transplant early in the disease is critical for positive neurocognitive outcomes, as neurocognitive deficits have been shown to be irreversible10,36. The utility of HCT in MSD has not been previously evaluated.
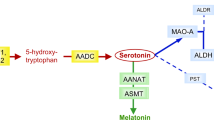
Sardiello et al. in 2005 described the different sulfatases affected by MSD3. Among these, the sulfatases localized in the lysosomes include—arylsulfatases A, B, G, and K, iduronate-2-sulfatase, heparan-N-sulfatase, and N-acetylglucosamine-6-sulfatase (Fig. 3). The rationale behind HCT was that it could slow down/prevent the progression of part of this disease by cross correction of the lysosomal enzymes associated with known disease phenotypes (iduronate-2-sulfatase, arylsulfatase B, N-acetyl-galactosamine-6-sulfatase, N-acetyl-glucosamine-6-sulfatase, Heparan-N-sulfatase, and arylsulfatase A). As expected, biochemical correction of these enzymes in leukocytes was detected within 3 months post-HCT and continues to remain corrected at 2 years post-HCT. Due to the attenuated phenotype of these patients, urine sulfatide excretion has always been normal and was not helpful in evaluating the post-HCT course in the siblings. Steroid sulfatase, a membrane-bound microsomal enzyme, has been localized to various tissues, including the placenta, liver, kidney, adrenal glands, ovaries, fibroblasts, and leukocytes37. There was mild improvement in the ichthyosis after HCT for both siblings, mostly for P1, even though the mechanism for this is currently unknown.

N/A: not associated with any human diseases to date. In addition to these sulfatases, the sub-cellular localization or any known human or animal disease association of arylsulfatase H, arylsulfatase I, and arylsulfatase J are currently unknown. Created with BioRender.com
We conducted a comprehensive assessment of the biochemical features associated with MSD through clinical metabolomic profiling of plasmas obtained from both siblings. This in-depth metabolomic analysis augmented our understanding of the critical role sulfatases play in maintaining normal cellular homeostasis. Our investigation of pre-HCT samples revealed notable perturbations in multiple sub-pathways. Particularly striking was the downregulation observed in lipids including glycerophospholipids, fatty acids and their derivatives, and sphingolipids. Additionally, disruptions in bile acids, cholesterols, sterols, nucleotides, various amino acids, and carbohydrates were identified. These findings shed light on the impairment of lipid and energy metabolism in MSD.
In direct comparison with the post-HCT period, our metabolomic profiling demonstrated a profound correction of distributed molecules across the majority of sub-pathways. This correction was notably observed beginning with the initial post-3-month samples in both siblings and persisted consistently in subsequent post-HCT samples, highlighting the efficacy of HCT in correcting the metabolic anomalies associated with MSD. These insights not only expand our understanding of the broader scope of metabolic dysregulation beyond previous knowledge but also yield valuable insight into potential therapeutic interventions for this condition.
Periodic neuropsychological evaluation did not reveal any additional loss of skills in the 2 years following HCT. Both siblings were either maintaining or gaining gain skills at a slower pace than the reference population. As the natural history of the neurocognitive course of this disease is not well quantitated due to its rarity, it is not yet clear if HCT has modified its neurodevelopmental trajectories. Longer-term follow-up may provide insights into treatment response.
The two-year follow-up of these siblings shows continued enzymatic correction and essentially no decline in neurocognitive status. Long-term follow-up of these siblings is needed to better understand the clinical effect of HCT in MSD. The outcome of HCT in these siblings with MSD must be interpreted cautiously, as they cannot be readily extrapolated to other patients with the same condition due to their milder phenotype. Therefore, while the study provides valuable insights into HCT as a potential therapy for MSD, it underscores the need for additional experience across a wider spectrum of disease severity to establish what benefits may be achieved with HCT in MSD. It is also equally imperative to explore alternative genetic treatment modalities for addressing non-lysosomal sulfatases that remain uncorrected by HCT in MSD.
Conclusion
MSD is an ultra-rare genetic disorder for which no curative therapy exists to date. In the siblings who underwent HCT, we observed continued normalization of the biochemical parameters and neuropsychological stabilization at 2 years post-HCT. These data suggest biochemical benefits post-transplant, although the clinical implication of these corrections is unknown, particularly in the context of the siblings’ attenuated phenotype. Long-term follow-up is necessary to fully evaluate the therapeutic benefit of HCT in MSD.
References
Cosma, M. P. et al. The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell 113, 445–456 (2003).
Dierks, T. et al. Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(alpha)-formylglycine generating enzyme. Cell 113, 435–444 (2003).
Sardiello, M., Annunziata, I., Roma, G. & Ballabio, A. Sulfatases and sulfatase modifying factors: an exclusive and promiscuous relationship. Hum. Mol. Genet. 14, 3203–3217 (2005).
Schlotawa, L., Adang, L. A., Radhakrishnan, K. & Ahrens-Nicklas, R. C. Multiple sulfatase deficiency: a disease comprising mucopolysaccharidosis, sphingolipidosis, and more caused by a defect in posttranslational modification. Int. J. Mol. Sci. 21, 3448 (2020).
Adang, L. A. et al. Natural history of multiple sulfatase deficiency: retrospective phenotyping and functional variant analysis to characterize an ultra-rare disease. J. Inherit. Metab. Dis. 43, 1298–1309 (2020).
Cappuccio, G., Alagia, M. & Brunetti-Pierri, N. A systematic cross-sectional survey of multiple sulfatase deficiency. Mol. Genet Metab. 130, 283–288 (2020).
Ahrens-Nicklas, R. et al. Complex care of individuals with multiple sulfatase deficiency: clinical cases and consensus statement. Mol. Genet. Metab. 123, 337–346 (2018).
Whitley, C. B. et al. Long-term outcome of Hurler syndrome following bone marrow transplantation. Am. J. Med Genet 46, 209–218 (1993).
Shapiro, E. G., Lockman, L. A., Balthazor, M. & Krivit, W. Neuropsychological outcomes of several storage diseases with and without bone marrow transplantation. J. Inherit. Metab. Dis. 18, 413–429 (1995).
Shapiro, E. G. & Eisengart, J. B. The natural history of neurocognition in MPS disorders: a review. Mol. Genet. Metab. 133, 8–34 (2021).
Barth, A. L. et al. Early hematopoietic stem cell transplantation in a patient with severe mucopolysaccharidosis II: a 7years follow-up. Mol. Genet. Metab. Rep. 12, 62–68 (2017).
Kubaski, F. et al. Hematopoietic stem cell transplantation for patients with mucopolysaccharidosis II. Biol. Blood Marrow Transplant. 23, 1795–1803 (2017).
Yabe, H. et al. Hematopoietic stem cell transplantation for Morquio A syndrome. Mol. Genet. Metab. 117, 84–94 (2016).
Wang, J. et al. Allogeneic hematopoietic stem cell transplantation in thirty-four pediatric cases of mucopolysaccharidosis—a ten-year report from the China Children Transplant Group. Biol. Blood Marrow Transplant. 22, 2104–2108 (2016).
Boucher, A. A. et al. Long-term outcomes after allogeneic hematopoietic stem cell transplantation for metachromatic leukodystrophy: the largest single-institution cohort report. Orphanet J. Rare Dis. 10, 94–94 (2015).
Wolf, N. I. et al. Metachromatic leukodystrophy and transplantation: remyelination, no cross-correction. Ann. Clin. Transl. Neurol. 7, 169–180 (2020).
Schlotawa, L., Adang, L. A., Radhakrishnan, K. & Ahrens-Nicklas, R. C. Multiple sulfatase deficiency: a disease comprising mucopolysaccharidosis, sphingolipidosis, and more caused by a defect in posttranslational modification. Int. J. Mol. Sci. https://doi.org/10.3390/ijms21103448 (2020).
Adang, L. A. et al. Biochemical signatures of disease severity in multiple sulfatase deficiency. J. Inherit. Metab. Dis. 47, 374–386 (2024).
Monteiro, M. S., Carvalho, M., Bastos, M. L. & Guedes de Pinho, P. Metabolomics analysis for biomarker discovery: advances and challenges. Curr. Med. Chem. 20, 257–271 (2013).
Kohler, I., Hankemeier, T., van der Graaf, P. H., Knibbe, C. A. J. & van Hasselt, J. G. C. Integrating clinical metabolomics-based biomarker discovery and clinical pharmacology to enable precision medicine. Eur. J. Pharm. Sci. 109s, S15–S21 (2017).
Tolstikov, V., Moser, A. J., Sarangarajan, R., Narain, N. R. & Kiebish, M. A. Current status of metabolomic biomarker discovery: impact of study design and demographic characteristics. Metabolites 10, 224 (2020).
Ford, L. et al. Precision of a clinical metabolomics profiling platform for use in the identification of inborn errors of metabolism. J. Appl. Lab. Med. 5, 342–356 (2020).
Kennedy, A. D. et al. Metabolomics in the clinic: a review of the shared and unique features of untargeted metabolomics for clinical research and clinical testing. J. Mass Spectrom. 53, 1143–1154 (2018).
Liu, N. et al. Comparison of untargeted metabolomic profiling vs traditional metabolic screening to identify inborn errors of metabolism. JAMA Netw. Open 4, e2114155 (2021).
van der Lee, J. H. et al. Therapy development for the mucopolysaccharidoses: updated consensus recommendations for neuropsychological endpoints. Mol. Genet. Metab. 131, 181–196 (2020).
Wechsler, D. Wechsler intelligence scale for children-fifth edition. (Pearson, Bloomington, MN, 2014).
Tiffen J. Purdue Pegboard Test. (Science Research Associates, Chicago, IL, 1968).
Eisengart, J. B. et al. Increasing precision in the measurement of change in pediatric neurodegenerative disease. Mol. Genet. Metab. 137, 201–209 (2022).
Sparrow, S., Cicchetti, D. V. & Saulnier, C. A. Vineland Adaptive Behavior Scales (Vineland-3). (Psychological Corporation, Antonio, 2016).
Alaimo, J. T. et al. Integrated analysis of metabolomic profiling and exome data supplements sequence variant interpretation, classification, and diagnosis. Genet. Med. 22, 1560–1566 (2020).
Liu, K. H. et al. Reference standardization for quantification and harmonization of large-scale metabolomics. Anal. Chem. 92, 8836–8844 (2020).
Miller, M. J. et al. Untargeted metabolomic analysis for the clinical screening of inborn errors of metabolism. J. Inherit. Metab. Dis. 38, 1029–1039 (2015).
Pillai, N. R. et al. Hematologic presentation and the role of untargeted metabolomics analysis in monitoring treatment for riboflavin transporter deficiency. Am. J. Med. Genet. A 182, 2781–2787 (2020).
Boelens, J. J. & van Hasselt, P. M. Neurodevelopmental outcome after hematopoietic cell transplantation in inborn errors of metabolism: current considerations and future perspectives. Neuropediatrics 47, 285–292 (2016).
Fratantoni, J. C., Hall, C. W. & Neufeld, E. F. Hurler and Hunter syndromes: mutual correction of the defect in cultured fibroblasts. Science 162, 570–572 (1968).
Pierpont, E. I. et al. Neurocognitive trajectory of boys who received a hematopoietic stem cell transplant at an early stage of childhood cerebral adrenoleukodystrophy. JAMA Neurol. 74, 710–717 (2017).
Hirato, K., Suzuki, T., Hondo, T., Saito, H. & Yanaihara, T. Steroid sulfatase activities in human leukocytes: biochemical and clinical aspects. Endocrinol. Jpn. 38, 597–602 (1991).
Acknowledgements
No funding was secured for this study. We sincerely thank the siblings and their family for their support and willingness to contribute to this study.
Author information
Authors and Affiliations
Contributions
N.R.P. conceptualized the study, collected data, carried out analysis, and drafted the original paper. P.J.O. and S.H.E. conceptualized the study and reviewed and revised the paper. J.B.E., N.L. and X.L. collected data, carried out the analysis, and drafted, reviewed, and revised the original paper. R.A.N., L.A., A.G., T.L. and C.B.W. reviewed and revised the paper. All authors participated in a critical review of the paper for important intellectual content. All authors approved the final paper as submitted and agreed to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Disclosure
Informed consent was obtained from the parents, and written consent from the mother was obtained to publish this case series and any accompanying images.
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Medicine thanks Jagdeep S. Walia, Nicola Brunetti-Pierri, François Eyskens and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pillai, N.R., Liu, N., Li, X. et al. Bone marrow transplantation reverses metabolic alterations in multiple sulfatase deficiency: a case series. Commun Med 5, 12 (2025). https://doi.org/10.1038/s43856-024-00703-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43856-024-00703-8
