
Abstract
Arthropod - and vertebrate-associated jingmenviruses (arbo-JMV) have segmented positive-strand RNA genomes and are provisional members of the genus Orthoflavivirus (family Flaviviridae). Current investigations have described arbo-JMV infection in vertebrate hosts in proximity to humans. This raises concerns about the virus host range and public health implications. This review explores the genomic and evolutionary relationship between arbo-JMV and orthoflaviviruses and evaluates the potential of arbo-JMV to pose a public health threat.
Similar content being viewed by others


Introduction
Jingmenviruses (JMV) are a group of rapidly emerging viruses. They were first described in Rhipicephalus microplus ticks collected in 2010 near Jingmen City, China, during a survey for Huaiyangshan virus1. Currently, JMV has been identified in continental Asia, Africa, Oceania, Europe, North and South America2,3,4,5,6,7,8,9,10. The JMV are regarded as provisional members of the genus Orthoflavivirus (family Flaviviridae)11,12, largely because of their segmented genome structure and protein function1,13. Other orthoflaviviruses like West Nile virus (WNV), dengue virus (DENV) and Powassan virus/deer tick virus (POWV/DTV) are able to infect a wide variety of animal hosts and are associated with serious health consequences in human14,15.
The JMV genome is comprised of single-stranded, positive-sense RNA (ss (+) RNA) encoding structural and non-structural proteins (NS), similar to the orthoflaviviruses genome. However, unlike other orthoflaviviruses, the genome is in 4 or 5 segments; each containing at least one open reading frame and each flanked by 5’ and 3’untranslated regions (UTRs)16,17,18,19. Segments 1 and 3 contain sequences, respectively, homologous to orthoflavivirus NS5 and NS3-NS2B complex, while segments S2 and S4 are highly divergent and bear no obvious sequence similarity to known viral proteins1. Segment 5 is non-essential during virus replication and has so far been associated with only insect-restricted JMV, including Guaico Culex virus (GCXV) and Mole Culex virus (MoCV)17,20,21. The description of GCXV genomic segments represent the multipartitism nature of JMV genome, however, the mechanism and impact of this genome architecture on viral replication remains undetermined17,20,21,22. Nevertheless, the characteristic genome organization pattern provides an important insight into possible relationships with other orthoflaviviruses23, highlighting our limited understanding of the prevalence and evolution of these pervasive viral agents.
Another similarity between JMV and more typical orthoflaviviruses is the host range. By host preference range, the orthoflaviviruses are clustered into three broad phylogenetic groups (a) the no-known-vector flaviviruses (NKV) with no established vector involved in their transmission, (b) the vertebrate pathogenic group containing the mosquito- and tick-borne orthoflaviviruses, and (c) the arthropod-specific group which reproduces exclusively in arthropods and lacks any defined ability to infect vertebrates24,25,26. At present, the JMV can be broadly categorized into three broad phylogenetic groups (Fig. 1): (a) an arbo-JMV group that infects both arthropods and vertebrates represented by Jingmen tick virus (JMTV), Alongshan virus (ALSV), Mogiana tick virus (MGTV), Kindia tick virus (KITV) and Yanggou tick virus (YGTV)20, (b) an arthropod restricted group such as GCXV, MoCV, Carajing virus (CaJV), Inopus flavus jingmenvirus 1 (IFJV1) among others20 and (c) the Histiostoma jingmenvirus (HJMV) group, which clusters between arbo- and arthropod-associated JMV. This latter group has been identified in mites collected from home environments but has so far not been detected in an established vector or vertebrate27.

The phylogenetic tree based on JMV Segment 1 comprises arbo-JMV group infecting both arthropods and vertebrates, arthropod-restricted group, Histiostoma jingmenvirus (HJMV) group and tick-borne orthoflaviviruses.
The proper containment level of new viruses is determined by standard processes at most institutions and many orthoflaviviruses, such as POWV, Japanese encephalitis virus (JEV) and Louping ill virus (LIV), are classified as biosafety level 3 (BSL-3). However, the majority of the JMVs remain unclassified apart from the BSL-2 GCXV (https://www.cdc.gov/labs/BMBL.html). As additional information is acquired on the potential for these viruses to cause human and animal disease, it is very likely that more of the JMV will be formally classified.
The potential pathogenicity of the arbo-JMV for humans has been demonstrated by antibodies and by isolation of viable JMTV and ALSV from patients with a history of tick bite2,28,29. To date, there are no reports of vertebrate mortality associated with arbo-JMV infection; however, available data suggest that humans experience unique clinical symptoms. In JMTV and ALSV infection, the most common clinical symptoms in the patients include headache and fever2,28,29. In vitro experimental infections demonstrate that arbo-JMV can infect diverse mammalian cells30,31, however, pathogenicity for animals other than human and mice remains undetermined. Although reverse zoonosis transmission is still unclear, arbo-JMV zoonotic potential has been established by the detection of viral RNA and antibodies in animals as well as parasitizing ticks32,33,34,35. Furthermore, evidence of circulation within communities has been shown by the existence of JMTV RNA in wastewater and environmental samples36,37. Importantly, there is as yet no report of direct spread between humans. Actual isolations of the virus are rare, but arbo-JMV viral nucleic acids have been detected in a variety of animals in close proximity to humans. These include arthropods, reptiles and mammals, including cattle, sheep, goats and horses, all of which accentuate concerns about the public health significance and host range of arbo-JMV2,17,28,32,38.
Considering the diverse potential hosts, it is likely that many arbo-JMV remain undiscovered in various ecologies. Therefore, this review summarizes existing information on the arbo-JMV discovery and genome organization. Further, to provide insight into arbo-JMV transmission dynamics, the review seeks to address questions such as what is the host range of arbo-JMV? Do arbo-JMV have specific animal host reservoirs? Is there an optimal cell line for isolation and stable growth of arbo-JMV? In addition, is there evidence of arbo-JMV onward spillover?
Because of the concerns about host range and public health significance, we review investigations focusing on JMTV and ALSV, both of which have been associated with human illness. In addition, we propose the development of arbo-JMV animal models to improve understanding of transmission dynamics. Our key message is the need to investigate newly emerging arboviruses and evaluate their potential public health threats.
Isolation and geographic distribution of arbo-JMV
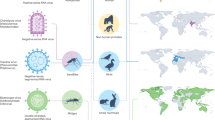
The first evidence of JMV was RNA of Jingmen tick virus (JMTV) identified in Rhipicephalus microplus ticks collected in the Jingmen region of China in 20101. JMTV was later isolated from Amblyomma javanense ticks collected from pangolins in China and from R. bursa ticks collected from sheep in Turkey 2,3. Alongshan virus (ALSV) was isolated from Ixodes persulcatus ticks collected in Russia39,40. Another arbo-JMV, YGTV, was isolated from Dermacentor reticulatus, D. marginatus, I. persulcatus and D. nuttalli ticks collected in Russia39,41,42. Current evidence indicates that arbo-JMV are present world-wide as depicted in Fig. 2 and detailed in Supplementary Table 1, the geographic distribution of arbo-JMV isolates is also summarized in Table 1. In parts of Asia and Europe arbo-JMV distribution overlaps with that of other tick-borne orthoflaviviruses such as tick-borne encephalitis virus (TBEV) complex43,44. In these regions, I. ricinus and I. persulcatus are known vectors of TBEV. In areas without Ixodid ticks, TBEV transmission relies on ticks of Haemaphysalis and Dermacentor species43,44, potentially sharing vector species with arbo-JMV (Supplementary Table 1). Similarly, both arbo-JMV and TBEV are capable of infecting the same vertebrate host, as shown by the detection of viral RNA and antibodies in livestock and deer43. The distribution overlap of arbo-JMV and other tick-borne orthoflaviviruses and the association with the same vector species and vertebrate host implies that arbo-JMV origin and the segmented genomic structure may be a result of genetic recombination and reassortment during co-infection with other tick-borne orthoflaviviruses1,45. However, the exact genetic recombination events and evolutionary patterns remain undetermined.

The arbo-JMV viruses have been detected worldwide; more genetically diverse strains have been observed in China and Russia than in any other geographic region. The map was created in https://mapchart.net/world.html and modified by authors.
Genome structure and function
Advanced nucleic acid sequencing and analytical techniques have enabled the characterization of the arbo-JMV genome structure and organization, sometimes even in the absence of actual virus isolates1,2,28,29,46. Thus, full genome sequences of several arbo-JMV have been obtained and others such as Newport tick virus (NTV) are partially characterized47. The genomes of the arbo-JMV resemble those of orthoflaviviruses. In every case, the results indicate a multipartite genome with four segments, arbitrarily denoted as S1–S42,17,28,29. Based on the available data, the complete genome is most likely single-stranded, positive-sense RNA (ss (+) RNA) encoding both structural and non-structural (NS) proteins1. Because the length of each segment ranges from 2800 bp to 3700 nucleotides, the full genome size exceeds the 11 kb size of a typical orthoflavivirus (Fig. 3A).

A Genomic organization of arbo-JMV. The coding region and translational direction are indicated by means of a different colour for each segment. The virus strain is indicated in the bracket. Segments 1 and 3 in all arbo-JMV code one protein, while segments 2 and 4 code for either one or two proteins depending on the virus strain. B Jingmen tick virus (JMTV), Alongshan virus (ALSV) and Tick-borne encephalitis virus (TBEV) NS3 helicase structural models. i JMTV NS3 helicase structure predicted by Alphafold. ii JMTV NS3 helicase structure superimposed onto experimentally determined ALSV NS3 helicase structure (PDB code: 6M40)49. iii JMTV NS3 helicase structure superimposed onto experimentally determined TBEV NS3 helicase (PDB code: 7OJ4)51. The functional domains are shown in ii and iii. C JMTV, ALSV and dengue virus methyltransferase (MTase) structural models. i JMTV MTase structure predicted by Alphafold. ii JMTV MTase structure superimpositioned onto experimentally determined ALSV MTase structure (PDB code: 8GY9)55. iii JMTV MTase structure superimpositioned onto experimentally determined Dengue virus MTase structure (PDB code: 3P97)83. The position of S-adenosyl-l-methionine (SAM) methyl donor during viral cap formation is shown in ii and iii.
Each segment has a putative coding region for at least one protein and is flanked by both 5’ and 3’ untranslated regions (UTRs), findings that support a bonafide segmented genome. There is sequence conservation at the 5’- and 3’-UTRs among the segments16,17,18,19. There is a conserved 5’-CAAGUG-3’ 3’-UTR sequence in all the segments of ALSV and JMTV16,17,18. As with typical orthoflaviviruses, these conserved sequences are likely involved in the RNA structure formation and virus replication. In contrast, sequence differences in the 5’ UTRs across members of the JMV group might reflect differences in the replication processes or host tropism18.
Nonstructural protein similarities
The open reading frames (ORF) of 2 segments have homology to orthoflavivirus NS proteins. The orthoflavivirus NS3 has protease and helicase activities crucial in virus replication, RNA polyprotein processing, and viral cap formation, respectively48. Segment 3 (S3) ORF codes for non-structural protein 2 (NSP2) homologous to the orthoflavivirus NS3-NS2B complex both in the N-terminal helicase portion and the C-terminal protease1. NS3-NS2B complex structure and function are well studied in orthoflaviviruses but, with the exception of ALSV remain less investigated in arbo-JMV. Biochemical and biophysical experiments show that ALSV NS3-like helicase ATPase activity and overall folding are comparable to those of orthoflaviviruses49. These findings are further illustrated by the similarity of folding among structure models of JMTV, generated by the artificial intelligence-based algorithm AlphaFold50, and crystal structure comparisons of ALSV (root mean square deviation, RMSD 1.002 Å) and tick-borne encephalitis virus (TBEV, RMSD 1.137 Å) (Fig. 3B)49,51. However, in spite of these shared topological features, arbo-JMV and orthoflaviviruses NS3 helicase have low sequence similarities, 15–28%, between themselves49.
Arbo-JMV segment 1 (S1) ORF shows homology to the orthoflavivirus NS5, which contains the RNA-dependent RNA polymerase (RdRp) and methyltransferase (MTase) activities1,52. The RdRp of arbo-JMV and the more typical orthoflaviviruses have topological similarity and superimpose well on each other, indicating similar functions in virus replication53,54. Crystal structures of ALSV MTases show great structural similarity with its Orthoflavivirus homologues55, but the MTase gene structure and activity are uninvestigated for other arbo-JMV. Nevertheless, although a comparison of arbo-JMV MTases shows very low amino acid sequence identity with closely related orthoflaviruses, 15–28% (Supplementary Fig. 1), similarities are further supported by AlphaFold generated models of JMTV and crystal structure comparisons of ALSV (RMSD 0.671 Å) and dengue virus (DENV, RMSD 1.133 Å) (Fig. 3C)50,55.
The comparability of these 2 arbo-JMV and orthoflavivirus NS protein structures (RMSD 1.133 and 1.137 Å) in the face of low sequence similarity suggests that promising antiviral compounds targeting orthoflaviviruses NS proteins may not show comparable activity against arbo-JMV NS proteins. Therefore, arbo-JMV structures should be included when modelling new drug compounds against orthoflaviviruses and related emerging infectious diseases.
Structural proteins divergence
The coding sequences for arbo-JMV S2 and S4 show no similarity to known orthoflaviviruses in protein sequence databases1. Computational analysis of arbo-JMV S2 amino acid sequences predicts that protein is a capsid and membrane protein56. However, in spite of the fact that the S2 arbo-JMV proteins have substantial amino acid similarities to membrane proteins with multiple transmembrane domains, there is no similarity to the structural proteins of other orthoflaviviruses56.
Similarly, S4 is suspected to encode envelope glycoproteins referred to as VP2 and VP31. Once again, arbo-JMV glycoproteins share substantial amino acid similarities among themselves but lack similarity to the glycoproteins of other orthoflaviviruses. Nevertheless, prediction suggests that arbo-JMV glycoproteins have traits associated with class II viral fusion proteins important for virus entry into host cells56. Although S2 and S4 have been shown to be related to Toxocara canis larval cDNA library transcripts, it is intriguing that S2 and S4 have definite features of viral structural proteins but fail to resemble other viruses1. Clearly, much study is needed.
5’ and 3’untranslated regions (UTRs) sequence conservation similarities
All the four genome segments have 5’ and 3’ UTRs and there is sequence conservation in 5’ and 3’ among the segments (Fig. 3A)1,18. In the 3’ UTR, ALSV and JMTV have conserved 5’-CAAGUG-3’ sequences in all the segments18,19. By analogy with typical orthoflaviviruses, the conserved sequences are likely involved in the RNA structure formation and regulation of virus replication57. In contrast, there are differences in the 5’ UTR conserved sequences across members of the JMV group, perhaps reflecting differences in the replication processes and host tropism18. In addition, in other arbo-JMV such as KITV, 5’ and 3’ UTRs modelling and functional annotation revealed the presence of viral replication and translation regulatory elements such as multiple UAG sites and 5’/3’ downstream AUG region (DAR), which have also been described in orthoflaviviruses19,58,59. In contrast to orthoflaviviruses where 3′ UTR structure facilitates the formation of subgenomic flavivirus RNA (sfRNA) attributed to host immune evasion and pathogenicity, the structuring and function of sfRNA in arbo-JMV remain uninvestigated18,60,61,62.
Polymerase gene integration similarities
Another similarity between arbo-JMV and orthoflaviviruses is the integration of the RdRp gene into the invertebrate host genome. For example, the study of Morozkin et al. demonstrate the integration of JMTV and ALV RdRp gene into I. ricinus genome63. The observed virus-derived DNA integration in the host genome is so far limited to vectors and has also been demonstrated by the presence of orthoflaviviruses-like sequences of Cell Fusing Agent virus (CFAV) and Kamiti River virus (KRV) in Aedes mosquitoes64,65,66. While in mosquitoes, the significance of orthoflaviviruses-derived gene integration in the host genome has been associated with improved tolerance to viral infection and survival67, the importance of arbo-JMV gene integration is undetermined in ticks.
Evolutionary lineage
Evolutionary virology analyses have identified endogenous viral elements (EVES) homologous to arbo-JMV suggesting that orthoflaviviruses are ancient by hundreds of years and that arbo-JMTV originated several years ago68. The identification of arbo-JMTV genome organization and the ancestral relationship with unsegmented orthoflaviviruses represent a rare occurrence in virus emergence and evolution1. Although the exact mechanism remains undescribed, it is suspected that arbo-JMV emerged from unsegmented orthoflavivirus and an unrelated virus coinfecting the same host, which resulted in recombination and reassortment of structural and nonstructural proteins as observed in RNA-DNA hybrid virus1,69. The impact of this genomic structuring is poorly described. Computational analyses suggest that arbo-JMV genome organization into shorter segments might improve virion stability70,71. Further, the genomic packaging enhances arbo-JMV genetic recombination and reassortment, leading to the emergence of new viral strains72. However, genomic components of segmented genomes might have different infection rates during host invasion lowering successful infection73. These findings may be supported by arbo-JMV maintenance in diverse hosts and the challenge of stably growing arbo-JMV in cell culture.
Protein evolution and time-direct molecular clock analysis complement each other. Beyond evolution details, protein evolutionary analysis provides additional information such as protein function. Several arbo-JMV genome sequences are available through public databases and recent studies have reconstructed time-trees demonstrating arbo-JMV evolutionary relationships and potential genetic recombination during virus transmission45. However, arbo-JMV protein structures and functions are less studied. To track the evolutionary lineage of arbo-JMV, Alphafold was used to predict structural models of JMTV proteins, including until now S2 and S4 structural models (Fig. 4A)50. The predicted protein structures were comparable to those obtained using EMSFold and showed well-predicted domains comprising α-helices, β-sheets and unstructured tails74. There were more than 200 sequences in the AlphaFold multiple sequence alignment (MSA) generated for S1 and S3. Maximum likelihood (ML) analysis of MSA files revealed a close relationship of arbo-JMV S1 to NS5 from a clade comprising diverse strains of bovine viral diarrhoea viruses (BVDVs), whereas arbo-JMV S3 was closely related to NS3 clade comprising Tamana bat virus (TBV) (Fig. 4B). These observations support earlier findings demonstrating homology of S1 and S3 with the orthoflavivirus NS5 and NS3-NS2B complex, respectively1,17.

A Jingmen tick virus (JMTV) AlphaFold predicted protein structures. The colours indicate confidence scores shown by the predicted local distance difference test (pLDDT). B The maximum-likelihood phylogenetic trees inferred by PhyML v. 2.2.4-embedded in Geneious prime with tree reliability assessed over 1000 bootstraps.
Notably, there were only 16–30 sequences in the MSA generated for S2 and S4, so, in contrast to the clear relationships to other orthoflaviviruses for S1 and S3, the presumed proteins from S2 and S4 have no virus proteins homologues from protein databases (Fig. 4B). This is a puzzling and very interesting finding, because although the arbo-JMV are phylogenetically related to orthoflaviviruses by several measures, the structural and nonstructural proteins come from completely different virus lineages.
Arbo-JMV maintenance cycle in ticks and vertebrate hosts
All of the available information suggests that arbo-JMV is maintained in a natural cycle featuring alternating replication in competent arthropod vectors and susceptible vertebrate hosts. The candidate vectors include hard ticks of genera Rhipicephalus, Amblyomma and Ixodes. While arbo-JMV has also been detected in mosquitoes, transmission has not been demonstrated in this arthropod75. Moreover, despite arbo-JMV being associated with vertebrate hosts, such as humans, livestock and wildlife, the precise animal host or reservoir for these viruses remains speculative but the actual cycle likely involves hard ticks and vertebrate hosts.
Several examples confirm this idea, including the detection of replicating JMTV by fluorescence in-situ hybridisation (FISH) in Amblyomma javanense midgut and salivary glands2. In A. javanense, JMTV infection is established in the salivary gland, indicating the virus spreads from the midgut to the salivary gland after blood feeding2. In addition, JMTV RNA has been identified in nymphs and unfed larvae of A. testudinarium implying that ticks can get infected vertically through eggs from an infected adult female (transovarial transmission) as well as by feeding on infected hosts8. Transovarial transmission has also been shown by JMTV infection in newly hatched Haemaphysalis longicornis larvae76. Together, the findings showing arbo-JMV infection in animals as well as parasitizing ticks point to A. javanense as a potential JMTV reservoir and vector. However, insufficient data on vertical transmission efficiency and other potential tick species vectors hinders the implication of ticks as amplifying hosts.
Arbo-JMTV infection during tick blood feeding has been demonstrated by the detection of JMTV RNA and IgG antibodies against JMTV in humans and I. persulcatus collected from humans2. Sequencing of human-associated JMTV and tick-associated JMTV revealed up to 99.9% genome sequence homology, reinforcing this revelation2. Similarity in vector and host JMTV genome sequence has also been observed in rodents and parasitizing ticks. Further, the majority of JMTV has been described in engorged tick species from known animal hosts. JMTV RNA has also been detected in non-human primates, rodents, reptiles, cattle and bats17,38.
A similar transmission dynamic has been established for ALSV following virus detection in I. persulcatus and humans with a history of tick bite28. ALSV viral RNA and anti-ALSV antibodies have also been reported in deer and parasitizing I. ricinus34. ALSV reports in livestock include viral IgG antibodies and RNA detection in cattle and sheep by enzyme-linked immunosorbent assay (ELISA), viral neutralization test (VNT) and qRT-PCR32. As demonstrated for JMTV, sequencing of animal-associated ALSV and tick-associated ALSV show very high genome sequence homology suggesting tick transmission32,34.
Generally, the definition of the host range and susceptibility of arbo-JMV is so far limited to a few viruses, including JMTV and ALSV. Some arbo-JMV represented by Sichuan tick virus (SCTV) and Mogiana tick virus (MGTV) have been reported only in ticks. Together, although sampling bias has not yet been excluded, these findings clearly infer that variants of arbo-JMV can be maintained in nature by diverse tick species and vertebrate hosts and that arbo-JMV may represent an emerging tick-borne zoonosis. The most likely arbo-JMV maintenance cycle in ticks and vertebrate hosts is illustrated in Fig. 5.

Vertebrate hosts can get infected through a tick bite during an infectious blood-meal while ticks can get infected vertically through eggs from an infected adult female.
Non-viraemic transmission in ticks during co-feeding with infected ticks observed in other orthoflaviviruses such as POWV/DTV, TBEV and LIV77,78,79, or transmission in vertebrates through contact with infected vertebrates or their products such as blood and unpasteurized milk remains unestablished in arbo-JMV 80,81. These observations suggest that specific tick species and vertebrate hosts are key component in the maintenance system of these viruses in nature, however, transmission characteristics remain poorly investigated.
In vitro tick and vertebrate host range
Experimental infection studies with tick and vertebrate cells show mixed outcomes but suggest a restricted in vitro host range for the arbo-JMV. JMTV isolated from A. javanense and R. microplus stably replicates in BME/CTVM23 (R. microplus) with the isolate showing no obvious cytopathic effects (CPE); however, virus is only detectable up to the second passage in BME26 (R. microplus), DH82 (Canis familiaris), Vero (Cercopithecus sabaeus) and BHK-21 (Mesocricetus auratus) cells2,38,82. ALSV isolated from humans and livestock stably replicates in Vero cells28,32. Stable replication of I. persulcatus-associated ALSV in HAE/CTVM8 (Hyalomma anatolicum anatolicum) and IRE/CTVM19 (I. ricinus) cells does not show obvious CPE42. In addition, ALSV proteins have been successfully expressed in human embryonic kidney (HEK293T) and liver cancer (HepG2) cells using expression vectors30,31. Experimental infection studies of other arbo-JMV variants, such as KITV, Takachi virus (TAKV) and MGTV have been sparse.
These findings indicate that in vitro arbo-JMV are likely to infect cells derived from hosts from which they were detected, however, the determinants of cellular tropism mechanism are not well defined. Furthermore, ideal cell lines for virus isolation and stable growth remain to be fully characterized.
Evidence of arbo-JMV onward spillover
Phylogenetic analyses revealed greater genetic divergence of reptile-associated JMTV than livestock-associated JMTV. This would be consistent with JMTV in tortoise populations being subjected to relaxed selection or existing for a longer time than in livestock38. If JMTV existed in reptiles previous to mammals then JMTV might have spread from reptiles to mammals. Even so, evidence for and implications of onward transmission or spillover are uncertain. While blood and tissues from infected vertebrates in a population might also serve as a potential direct transmission route through contact with infected vertebrates or their products, there is no experimental data supporting this possibility. In addition, the isolated reports of JMTV RNA in wastewater and environmental samples remain difficult to evaluate36,37, although these findings have been taken to infer infection patterns within human and animal populations. Future studies will doubtless clarify this important area.
Concluding remarks and future perspective
Arbo-JMV are significant new emerging tick-borne viruses. Considering the diverse potential arthropods and vertebrate hosts, it seems probable that many arbo-JMV variants exist in various geographical locations. This review article outlines the relationship between arbo-JMV and orthoflaviviruses, illustrates a potential transmission system and presents several unresolved questions. Virus identification in humans, ticks and vertebrate hosts in proximity clearly suggests public health importance. Risk may be highest among humans with close contact with livestock and animals, however, reservoir host and sources of variant emergence remains unconfirmed. Another issue is the lack of studies characterizing arbo-JMV genotypes associated with spillover. Future arbo-JMV studies designed to determine animal disease models would greatly enhance our perspective of arbo-JMV transmission and inform the development of countermeasures against these and other enigmatic arboviruses.
Data availability
The analysed data in the current study are available within the article, Supplementary file, or public databases.
References
Qin, X. C. et al. A tick-borne segmented RNA virus contains genome segments derived from unsegmented viral ancestors. Proc. Natl Acad. Sci. USA 111, 6744–6749 (2014).
Jia, N. et al. Emergence of human infection with Jingmen tick virus in China: a retrospective study. EBioMedicine 43, 317–324 (2019).
Dinçer, E. et al. Survey and characterization of jingmen tick virus variants. Viruses 11, 1071 (2019).
Vandegrift, K. J. et al. Presence of segmented flavivirus infections in North America. Emerg. Infect. Dis. 26, 1810–1817 (2020).
Pascoal, J. et al. Detection and molecular characterization of Mogiana tick virus (MGTV) in Rhipicephalus microplus collected from cattle in a savannah area, Uberlândia, Brazil. Ticks Tick. Borne Dis 10, 162–165 (2019).
Sameroff, S. et al. Viral diversity of tick species parasitizing cattle and dogs in Trinidad and Tobago. Sci. Rep. 9, 1–10 (2019).
Gondard, M. et al. RNA viruses of Amblyomma variegatum and Rhipicephalus microplus and cattle susceptibility in the French antilles. Viruses 12, 144 (2020).
Kobayashi, D. et al. Detection of Jingmenviruses in Japan with evidence of vertical transmission in ticks. Viruses 13, 2547 (2021).
Kiwan, P. et al. First detection and molecular characterization of Jingmen Tick Virus with a high occurrence in Rhipicephalus (Boophilus) microplus collected from livestock in Cameroon. Parasit. Vectors 18, 41 (2025).
Cicculli, V. et al. First detection of Jingmen tick virus in Corsica with a new generic RTqPCR system. npj Viruses 2, 44 (2024).
Simmonds, P. et al. ICTV virus taxonomy profile: Flaviviridae. J. Gen. Virol. 98, 2–3 (2017).
Postler, T. S. et al. Renaming of the genus Flavivirus to Orthoflavivirus and extension of binomial species names within the family Flaviviridae. Arch. Virol. 168, 224 (2023).
Mifsud, J. C. O. et al. Mapping glycoprotein structure reveals Flaviviridae evolutionary history. Nature 633, 695–703 (2024).
Naeem, A. et al. Recurrent West Nile virus outbreak in the United States in 2022: current challenges and recommendations. J. Biosaf. Biosecur. 5, 146–152 (2023).
Siegel, E., Xu, G., Killinger, P., Brown, C. M. & Rich, S. M. Passive surveillance of Powassan virus in human-biting ticks and health outcomes of associated bite victims. Clin. Microbiol. Infect. 30, 1332–1334 (2024).
Shi, M. et al. Divergent viruses discovered in arthropods and vertebrates revise the evolutionary history of the Flaviviridae and related viruses. J. Virol. 90, 659–669 (2016).
Ladner, J. T. et al. A multicomponent animal virus isolated from mosquitoes. Cell Host Microbe 20, 357–367 (2016).
Litov, A. G., Okhezin, E. V., Kholodilov, I. S., Belova, O. A. & Karganova, G. G. Conserved sequences in the 5′ and 3′ untranslated regions of jingmenvirus group representatives. Viruses 15, 971 (2023).
Tsishevskaya, A. A., Alkhireenko, D. A., Bayandin, R. B. & Yu, M. Untranslated regions of a segmented Kindia tick virus genome are highly conserved and contain multiple regulatory elements for viral replication. Microorganisms 12, 239 (2024).
Colmant, A. M. G., Charrel, R. N. & Coutard, B. Jingmenviruses: Ubiquitous, understudied, segmented flavi-like viruses. Front. Microbiol. 13, 997058 (2022).
Chen, R. Y. et al. The infection kinetics and transmission potential of two Guaico Culex viruses in Culex quinquefasciatus mosquitoes. Virol. Sin. 39, 228–234 (2024).
Zhang, X., Wang, N., Wang, Z. & Liu, Q. The discovery of segmented flaviviruses: implications for viral emergence. Curr. Opin. Virol. 40, 11–18 (2020).
Holmes, E. C. The Evolution and Emergence of RNA Viruses (Oxford University Press, 2009).
Blitvich, B. J. & Firth, A. E. Insect-specific flaviviruses: a systematic review of their discovery, host range, mode of transmission, superinfection exclusion potential and genomic organization. Viruses 7, 1927–1959 (2015).
Blitvich, B. J. & Firth, A. E. A review of flaviviruses that have no known arthropod vector. Viruses 9, 154 (2017).
Calisher, C. H. & Higgs, S. The discovery of arthropod-specific viruses in hematophagous arthropods: an open door to understanding the mechanisms of arbovirus and arthropod evolution? Annu. Rev. Entomol. 63, 87–103 (2018).
Guo, L. et al. Metatranscriptomic analysis reveals the virome and viral genomic evolution of medically important mites. J. Virol. 95, e01686–20 (2021).
Wang, Z. D. et al. A new segmented virus associated with human febrile illness in China. N. Engl. J. Med. 380, 2116–2125 (2019).
Zhang, J. et al. Skin infectome of patients with a tick bite history. Front. Cell. Infect. Microbiol. 13, 1113992 (2023).
Zhao, Y. et al. The segmented flavivirus Alongshan virus reduces mitochondrial mass via degrading 2 STAT2 to suppress innate immune response. J. Virol. 99, e01301–e01324 (2025).
Zhao, Y. et al. Characterization and subcellular localization of Alongshan virus proteins. Front. Microbiol. 13, 1000322 (2022).
Wang, Z. D. et al. Prevalence of the emerging novel Alongshan virus infection in sheep and cattle in Inner Mongolia, northeastern China. Parasit. Vectors 12, 450 (2019).
Kuivanen, S. et al. Detection of novel tick-borne pathogen, Alongshan virus, in Ixodes ricinus ticks, south-eastern Finland, 2019. Eurosurveillance 24, 1900394 (2019).
Ebert, C. L. et al. Detection and characterization of Alongshan virus in ticks and tick saliva from Lower Saxony, Germany with serological evidence for viral transmission to game and domestic animals. Microorganisms 11, 543 (2023).
Guo, J. J. et al. Diversity and circulation of Jingmen tick virus in ticks and mammals. Virus Evol. 6, veaa051 (2020).
Stockdale, S. R. et al. RNA-Seq of untreated wastewater to assess COVID-19 and emerging and endemic viruses for public health surveillance. Lancet Reg. Health—Southeast Asia 14, 100205 (2023).
Chen, Y. M. et al. RNA viromes from terrestrial sites across China expand environmental viral diversity. Nat. Microbiol 7, 1312–1323 (2022).
Ogola, E. O. et al. Jingmen tick virus in ticks from Kenya. Viruses 14, 1041 (2022).
Kholodilov, I. S. et al. Geographical and tick-dependent distribution of flavi-like Alongshan and Yanggou tick viruses in Russia. Viruses 13, 458 (2021).
Kholodilov, I. S. et al. Isolation and characterisation of Alongshan virus in Russia. Viruses 12, 1–17 (2020).
Amoa-Bosompem, M. et al. Entomological assessment of the status and risk of mosquito-borne arboviral transmission in Ghana. Viruses 12, 147 (2020).
Kholodilov, I. S. et al. Distribution and characterisation of tick-borne flavi-, flavi-like, and phenuiviruses in the Chelyabinsk Region of Russia. Viruses 14, 2699 (2022).
Im, J. H. et al. Geographic distribution of Tick-borne encephalitis virus complex. J. Vector Borne Dis 57, 14–22 (2020).
Süss, J. Tick-borne encephalitis 2010: epidemiology, risk areas, and virus strains in Europe and Asia—an overview. Ticks Tick-Borne Dis. 2, 2–15 (2011).
Li, W. et al. Genomics evolution of Jingmen viruses associated with ticks and vertebrates. Genomics 115, 110734 (2023).
Maruyama, S. R. et al. Characterisation of divergent flavivirus NS3 and NS5 protein sequences detected in Rhipicephalus microplus ticks from Brazil. Mem. Inst. Oswaldo Cruz 109, 38–50 (2014).
Gofton, A. W. et al. Metatranscriptomic profiling reveals diverse tick-borne bacteria, protozoans and viruses in ticks and wildlife from Australia. Transbound. Emerg. Dis. 69, e2389–e2407 (2022).
Van Den Elsen, K., Quek, J. P. & Luo, D. Molecular insights into the flavivirus replication complex. Viruses 13, 956 (2021).
Gao, X. et al. Crystal structure of the NS3-like helicase from Alongshan virus. IUCrJ 7, 375–382 (2020).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Chen, C. et al. Crystal structure of the NS3 helicase of tick-borne encephalitis virus. Biochem. Biophys. Res. Commun. 528, 601–606 (2020).
Zhou, Y. et al. Structure and function of flavivirus NS5 methyltransferase. J. Virol. 81, 3891–3903 (2007).
Liu, Z. et al. Crystal structures of RNA-dependent RNA polymerases from Jingmen tick virus and Alongshan virus. hLife 2, 18–31 (2024).
Wang, X. et al. A jingmenvirus RNA-dependent RNA polymerase structurally resembles the flavivirus counterpart but with different features at the initiation phase. Nucleic Acids Res 52, 3278–3290 (2024).
Chen, H. et al. Structural and functional basis of low-affinity SAM/SAH-binding in the conserved MTase of the multi-segmented Alongshan virus distantly related to canonical unsegmented flaviviruses. PLoS Pathog 19, e1011694 (2023).
Garry, C. E. & Garry, R. F. Proteomics computational analyses suggest that the envelope glycoproteins of segmented jingmen flavi-like viruses are class II viral fusion proteins (β-Penetrenes) with mucin-like domains. Viruses 12, 260 (2020).
Upstone, L., Colley, R., Harris, M. & Goonawardane, N. Functional characterization of 50 untranslated region (UTR) secondary RNA structures in the replication of tick-borne encephalitis virus in mammalian cells. PLoS Negl. Trop. Dis. 17, 1–16 (2023).
Friebe, P., Shi, P. Y. & Harris, E. The 5′ and 3′ Downstream AUG region elements are required for mosquito-borne flavivirus RNA replication. J. Virol. 85, 1900–1905 (2011).
Darai, N. et al. Theoretical studies on RNA recognition by Musashi 1 RNA-binding protein. Sci. Rep. 12, 1–12 (2022).
Filomatori, C. V. et al. A 5′ RNA element promotes dengue virus RNA synthesis on a circular genome. Genes Dev 20, 2238–2249 (2006).
Chapman, E. G. et al. The structural basis of pathogenic subgenomic flavivirus RNA (sfRNA) production. Science (1979) 344, 307–310 (2014).
Liu, Y. et al. Structures and functions of the 3′ untranslated regions of positive-sense single-stranded RNA viruses infecting humans and animals. Front. Cell. Infect. Microbiol. 10, 453 (2020).
Morozkin, E. S. et al. Integrated Jingmenvirus polymerase gene in Ixodes ricinus genome. Viruses 14, 1908 (2022).
Roiz, D. et al. Detection of a new insect flavivirus and isolation of Aedes flavivirus in Northern Italy. Parasit. Vectors 5, 1–9 (2012).
Crochu, S. et al. Sequences of flavivirus-related RNA viruses persist in DNA form integrated in the genome of Aedes spp. mosquitoes. J. Gen. Virol. 85, 1971–1980 (2004).
Spadar, A., Phelan, J. E., Clark, T. G. & Campino, S. Large-scale reference-free analysis of flavivirus sequences in Aedes aegypti whole genome DNA sequencing data. Parasit. Vectors 16, 265 (2023).
Goic, B. et al. Virus-derived DNA drives mosquito vector tolerance to arboviral infection. Nat. Commun. 7, 1–10 (2016).
Bamford, C. G., de Souza, W. M., Parry, R. & Gifford, R. J. Comparative analysis of genome-encoded viral sequences reveals the evolutionary history of flavivirids (Flaviviridae). Virus Evol. 8, veac085 (2022).
Diemer, G. S. & Stedman, K. M. A novel virus genome discovered in an extreme environment suggests recombination between unrelated groups of RNA and DNA viruses. Biol. Direct 7, 1–14 (2012).
Ojosnegros, S. et al. Viral genome segmentation can result from a trade-off between genetic content and particle stability. PLoS Genet 7, 1001344 (2011).
Bennett, H. M. Split reality for novel tick virus. Nat. Rev. Microbiol. 12, 464 (2014).
McDonald, S. M., Nelson, M. I., Turner, P. E. & Patton, J. T. Reassortment in segmented RNA viruses: mechanisms and outcomes. Nat. Rev. Microbiol. 14, 448–460 (2016).
Sicard, A. et al. A multicellular way of life for a multipartite virus. Elife 8, e43599 (2019).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science (1979) 379, 1123–1130 (2023).
Parry, R., James, M. E. & Asgari, S. Uncovering the worldwide diversity and evolution of the virome of the mosquitoes Aedes aegypti and Aedes albopictus. Microorganisms 9, 1653 (2021).
Wu, Z. et al. Molecular evidence for potential transovarial transmission of Jingmen tick virus in Haemaphysalis longicornis fed on cattle from Yunnan Province, China. J. Med. Virol. 95, e28357 (2023).
Obellianne, C., Norman, P. D., Esteves, E. & Hermance, M. E. Interspecies co-feeding transmission of Powassan virus between a native tick, Ixodes scapularis, and the invasive East Asian tick, Haemaphysalis longicornis. Parasit. Vectors 17, 1–15 (2024).
Labuda, M. et al. Non-viraemic transmission of tick-borne encephalitis virus: a mechanism for arbovirus survival in nature. Experientia 49, 802–805 (1993).
Kuehnert, P. A., Stefan, C. P., Badger, C. V. & Ricks, K. M. Crimean-Congo hemorrhagic fever virus (CCHFV): a silent but widespread threat. Curr. Trop. Med Rep. 8, 141–147 (2021).
Paulsen, K. M. et al. Tick-borne encephalitis virus in cows and unpasteurized cow milk from Norway. Zoonoses Public Health 66, 216–222 (2019).
Martello, E. et al. Systematic review on the non-vectorial transmission of tick-borne encephalitis virus (TBEv). Ticks Tick-Borne Dis 13, 102028 (2022).
Zhang, L. et al. Identification and characterization of Jingmen tick virus from Rhipicephalus microplus in Hunan, China. Acta Trop 260, 107378 (2024).
Lim, S. P. et al. Small molecule inhibitors that selectively block dengue virus methyltransferase. J. Biol. Chem. 286, 6233–6240 (2011).
Acknowledgements
This work was supported by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases (NIAID) and the National Institutes of Health (NIH). This project has been funded in part with Federal funds from the NIAID, NIH, Department of Health and Human Services under BCBB Support Services Contract HHSN316201300006W/75N93022F00001 to Guidehouse Digital. Analyses were performed using the Office of Cyber Infrastructure and Computational Biology (OCICB) High-Performance Computing (HPC) cluster at the NIAID, Bethesda, MD. The authors would like to thank the Visual and Medical Arts group of the RML Research Technologies Branch (RTB) for the graphic illustrating the arbo-JMV maintenance cycle in ticks and vertebrate hosts.
Funding
Open access funding provided by the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
E.O.O. and M.E.B. conceptualized the manuscript; E.O.O. original draft preparation; E.O.O., A. R. and K.W. software, data analysis and figures preparation; E.O.O., M.E.B., A.R., K.W. and M.O. manuscript review and editing. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ogola, E.O., Roy, A., Wollenberg, K. et al. Strange relatives: the enigmatic arbo-jingmenviruses and orthoflaviviruses. npj Viruses 3, 24 (2025). https://doi.org/10.1038/s44298-025-00106-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s44298-025-00106-z
