
Abstract
Giant viruses (GVs) play crucial roles in the global ocean microbial food web and biogeochemistry. Recent metagenomic advances have uncovered >1800 new GV genomes from the world’s oceans. While this rapid increase in genomic information is impressive, it is nowhere close to the extensive genomic information available for other marine entities—e.g., prokaryotes and their “virome”. We present 230 new high-quality GV genomes (genomes with 4 or more marker genes) and 398 partial genomes from nine global ocean datasets. Notably, we identified numerous GV genomes from the Baltic Sea, offering insights into their phylogenomics, metabolic potential, and environmental drivers in one of the largest brackish water ecosystems. We discovered new GV functions and identified a significant functional divide between the Imitervirales and Algavirales orders. Additionally, we evaluated factors affecting GV abundance through a case study on the Baltic Sea dataset. Our study significantly expands the marine GV genomic and functional diversity, broadening our understanding of their roles in the food web and biogeochemistry.
Similar content being viewed by others

Introduction
The discovery of giant viruses (GVs) belonging to the phylum Nucleocytoviricota represents a paradigm shift in the world of virology due to their remarkable genomic and functional complexities1. These viruses, with their large virion (up to 2 µM) and genome sizes (up to 2.5 Mbp)2,3 have redefined our understanding of viruses. Of note is their ability to encode numerous functionalities previously unknown to viruses, which has raised significant questions regarding their ecological and evolutionary roles in the biosphere4,5,6. Currently, six identified orders exist for GVs - including Imitervirales, Algavirales, Pimascovirales, Asfuvirales, Pandoravirales, and Chitovirales7. Members of several of these orders have a seemingly broad host range, consisting of mainly single-celled eukaryotes8,9, and are widespread in the global ocean and sediments10,11.
The impact of giant viruses is no doubt present across all environments, but special interest has been taken to uncover their impact on aquatic ecosystems due to the important contribution of single-celled eukaryotes to global biogeochemical cycles and aquatic food webs12,13. Giant viruses infecting these cells have been shown to modulate host metabolism during infection14,15,16 through the introduction of a diverse set of metabolic genes encoded in the GV genomes4,5. Giant viruses also have the potential to modulate how hosts acquire nutrients through the use of viral transporter proteins17,18. This impact on host metabolism has potential implications for marine nutrient cycling19 and food webs20.
Ever since the discovery of the first giant virus in the 1980s21 many methodological and technological strides have facilitated our understanding of the diversity and functional potential of these viruses. Much of the early understanding of GVs came from culture-based approaches22 and the first GV genome wasnot sequenced until 200323. While this culture-based approach is invaluable in the understanding of GV biology and there have been many advances in virus recovery methods24,25, to date, there are only around 200 genomes recovered from isolates26. Compared to the over 14,000 bacteriophage genomes27 and 600,000 bacterial genomes28, this number is minuscule.
In recent years, this culture-based approach has been complemented by large-scale metagenomics to find signatures of GVs in environmental data. Early metagenomic methods relied on the recovery of GV signatures such as polymerases and major capsid proteins10,29,30,31,32,33,34. These early surveys demonstrated the widespread distribution of GVs and suggested there was a huge pool of undiscovered diversity of these viruses35. The first giant virus Metagenome-Assembled Genome (GVMAG) was recovered in 2011 from Organic Lake, Antarctica36. This recovery was followed by an additional four genomes recovered from Yellowstone Lake37 as well as a few more genomes derived from wastewater, soil, lake, and deep-sea sediment samples11,38,39,40. The power of metagenomic-based approaches became most evident with the discovery of 2074 GVMAGs from diverse environments41, and the unveiling of 501 GVMAGs from mostly marine environments42. In the years to follow, only one other major study has recovered a large diversity of additional GVMAGs from marine environments, focusing on the TARA global oceans data to build the Global Ocean Eukaryotic Viral database (GOEV)43. In total, Gaïa et al. discovered over 400 additional GVMAGs as well as expanding diversity to include the putative new class Proculvirales and the new phylum Mirusviricota, which reveals evolutionary connections of giant viruses to herpesviruses.
While much progress has been made in recovering GVMAGs from metagenomic datasets, we still have far to go in terms of recovering additional diversity and functional potential of giant viruses, especially their diversity in the oceans as only ~1800 genomes exist from marine datasets43. Further efforts towards understanding GV diversity and functional potential in the global ocean are necessary for a comprehensive assessment of their impact on marine protist ecology and biogeochemical cycles. To this end, we leveraged the BEREN tool44 and reported 230 novel high-quality marine GVMAGs and 398 partial GVMAGs from 9 different datasets throughout the global oceans. Analysis of these data reveals novel functional potential encoded by giant viruses and their ecological constraints and provides insights into best practices in methodologies for GVMAG recovery. We also reveal a large number of GVMAGs from the Baltic Sea, an ecologically unique, large brackish water body for which little data exists on the phylogenetic diversity and ecology of GVs.
Results
Biogeography of 230 novel giant virus genomes
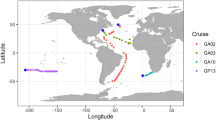
After applying BEREN to the metagenomic assemblies, 230 high-quality giant virus metagenome-assembled genomes (GVMAGs) were recovered from 9 different datasets (See methods) (Table S1). These datasets encompassed 4 of the 5 major oceans with a special focus on colder bodies of water (Fig. 1a). The most high-quality GVMAGs were recovered from the Baltic Sea (n = 108), followed by the Antarctic (n = 65).

a Giant virus metagenome-assembled genomes (GVMAGs) of every major giant virus order were acquired from datasets spanning pole to pole. The pie charts represent the proportion of genomes recovered from each order from each locale. b For sequencing projects with available depth data, this information was used to show recovery success at various depths.
We recovered GVs from all major orders, with the majority of them from either Algavirales (n = 135) or Imitervirales (n = 81) orders. Two novel Mirusviricota genomes were also recovered from the Sargasso Sea and North Atlantic datasets. Although not all datasets had accompanying depth data, the ones that did showed that most of these GVMAGs originated from the upper 100 m except for 2 coming from a hydrothermal vent environment (Fig. 1b).
In addition to the 230 high-quality GVMAGs, 398 partial GVMAGs were also recovered from the datasets (Fig. S1). The majority of these came from the Baltic Sea (n = 203) and were classified as either Algavirales (n = 127) or Imitervirales (n = 102).
Genome statistics
Recovered GVMAGs (both high quality and partial) ranged in length from 50 kbp to 1.3 Mbp, averaging 211 kbp (Fig. 2). Broken down by order, Algavirales GVMAGs were significantly larger than other orders with an average length of 362 kbp.

Violin plots representing length, GC percentage, average nitrogen per sidechain, and average sulfur per sidechain were constructed to show differences between GV phylogenetic orders. For each order with more than 2 recovered genomes, a t-test was performed to see statistically significant differences in these genomic characteristics. FDR corrected p-values are reported here to account for multiple comparisons (*= p < 0.05, **= p < 0.01, ***= p < 0.001, ****= p < 0.0001).
The average GC percentage for all recovered GVMAGs was 38.37%. This average did have significant differences between orders, as Pandoravirales had the highest average GC percentage at 55%. Algavirales had on average the lowest GC percentage with 31.5%.
To obtain further insights into the protein makeup of the GVMAGs, we calculated the average nitrogen per sidechain and average sulfur per sidechain for all proteins in the GVMAGs. On average these numbers varied only slightly between genomes with an average of 0.395 nitrogen molecules and 0.044 sulfur molecules per sidechain. Significant differences did exist between orders as Pandoravirales had the highest average nitrogen per sidechain (0.44) and Mirusviricota genomes had the highest average sulfur per sidechain (0.052).
Phylogenetic analysis
A phylogenetic reconstruction of the newly discovered high-quality and partial GVMAGs based on the DNA Polymerase B marker gene (PolB) showed that many of these GVMAGs clustered within known GV families (Fig. 3). Phylogenetic analysis did not reveal biogeography to be a key driver of the evolutionary history of these genomes, as genomes from diverse orders were recovered from different oceanic regions without any ___location-specific clustering on the tree.

A phylogenetic tree was constructed using the PolB marker gene from our recovered GVMAGs as well as PolB genes from a curated set of reference genomes representing all major GV orders and families. The type of sequence as well as the ___location of recovery are represented as colorstrips on the tree. The tree was constructed using IQ-TREE with the “LG + F + R10” model and 1000 bootstraps. It was visualized using iTOL.
Phylogeny-informed metabolic potential of Giant viruses
While GVs are known to sometimes have highly divergent versions of host metabolic genes4, many GV auxiliary metabolic genes share close homology to cellular variants of the gene, making it possible to annotate these genes with known databases and pipelines16,42.
Genes homologous to cellular genes functioning in central carbon metabolism (Glycolysis, TCA cycle, and the pentose phosphate pathway) were commonly found in Imitervirales genomes with some genomes even having multiple genes involved in the pathway. Despite larger genome sizes of recovered Algavirales GVMAGs, central carbon metabolism genes were nearly absent from these genomes with glycolysis genes being present in only 3 genomes, and phosphate pentose genes being present in 7 genomes (Fig. 4a).

a A heatmap showing the presence(red)/absence(blue) of common giant virus metabolic genes in our recovered GVMAGs. These genes are separated into pathways based on cellular function and are organized into phylogenetic groups based on the phylogeny of their PolB (see previous figure). b Functional differences between Imitervirales and Algavirales genomes visualized in Anvi’o. Each ring represents a genome, and each column is an individual COG functional annotation. The plot is broken up into sections based on functions present only in Imitervirales genomes, those present only in Algavirales genomes, and those shared between the two as core genes. A complete list of these genes and their enrichment can be found on the Figshare for this paper.
The light-harvesting Chl a/b-binding (LHCB) protein was found across orders at similar rates of occurrence except for recovered Pimascovirales genomes. On the other hand, rhodopsins were found only in recovered Imitervirales genomes, showing a stark phylogenetic separation. These proteins were quite common within this order, being found in 53% of recovered genomes (Fig. 4a). Genes involved in beta-oxidation also showed order-level patterns, only found in recovered Pimascovirales (20%) and Imitervirales genomes (25%).
Certain genes involved in nutrient metabolism and transport also showed order-specific bias in genomic distribution. Specifically, no Algavirales or Pandoravirales genomes contained genes for sulfite export while Imitervirales (25%) and Pimascovirales (20%) recovered genomes had these genes. Genes for nutrient metabolism such as Glutaminase, Glutamine synthetase, and Pho regulon protein were only found in recovered Imitervirales and Pimascovirales genomes with Glutamine synthetase being found in 22% of Imitervirales genomes and Pho regulon protein being found in 40% of Pimascovirales genomes.
Genes involved in DNA processing were found almost exclusively in recovered Imitervirales genomes. A surprising 95% of these genomes were found to contain a DNA mismatch repair gene (MutS) while all other orders had much less representation of this gene (5% in Algavirales; 20% in Pimascovirales; 11% in Pandoravirales). Histone acetyltransferase genes were also found in 20% of Imitervirales and 20% of Pimascovirales genomes.
Beyond differences in key metabolic genes, the two major orders of GV recovered in this study show broad functional differences encoded in their genomes (Fig. 4b). Out of the 575 COG (clusters of orthologous genes) annotations from both groups of genomes, 383 (67%) were found to be exclusive to Imitervirales, 40 (7%) to Algavirales, and 152 (26%) were shared between the two groups.
New functionality found in GVMAGs and partial MAGs
A survey of the functional landscape in our newly recovered GVMAGs and partial GVMAGs revealed 569 novel proteins not found previously in GVs after dereplication of protein clusters and annotations from both the Global Ocean Eukaryotic Virus (GOEV) database and the Giant Virus Database (GVDB) (Fig. 5a). Most of these proteins have unknown GO functional annotations, with many also having a role in protein binding, membrane interactions, and ATP binding (Fig. 5b). The vast majority of the new functions are encoded within genomes from the Arctic, Antarctic, and Baltic Sea, contributing to 88% of the total new proteins.

a Proteins that have never been found in giant viruses (dereplicated from the GOEV and GVDB databases) were annotated to expand the functional potential of known GVs. b A heatmap of unique functional potential in new GVMAGs. Each row represents a genome and columns are separated by gene ontology (GO) categories. The ___location of the recovered genome is also shown on the right. c Specific unique genes involved in the photosynthesis pathway are highlighted here and combined with proteins found in previous studies, such as those by Schultz et al.41 and Gaia et al.43.
This survey also revealed many new proteins putatively involved in photosynthesis such as those involved in photosystem I (PsaK, PsaG, PsaH, Ferredoxin reductase), photosystem II (PsbQ, PsbY, PsbP), cytochrome b6f (PetM), and ATP synthase (Fig. 5c). While contamination from other cellular sources cannot be completely ruled out, we note that these photosynthesis proteins were found on contigs with multiple other known giant virus genes (Fig. S2). In addition, these contigs also harbored many genes with unknown function, which is typical of viral genomes (Fig. S2). This finding would greatly expand the number of known photosynthetic genes encoded in GV genomes as reported in other studies41,42.
Environmental factors associated with Baltic Sea GV community composition
Due to the large number of Baltic Sea samples, as well as the experimental design utilizing multiple filter sizes, we were able to utilize this data to perform a case study on factors affecting genome recovery, size, and abundance. A Mantel test and canonical correspondence analysis (CCA) were used to determine environmental factors correlated with GV community composition in the Baltic Sea. When all GVs were considered together, bacterial production, depth, and salinity were predicted to be significantly correlated with GV abundance (p <.05) (Fig. 6a). Genomes were also separated by order-level classification to see specific phylogenetic differences in factors affecting community composition. Genomes belonging to the Algavirales and Imitervirales orders were only significantly correlated with depth. Asfuvirales were positively correlated with dissolved organic nitrogen and dissolved organic carbon. Pandoravirales were only correlated with bacterial production, and Pimascovirales were correlated with bacterial production, dissolved oxygen, and salinity. The CCA analysis confirmed salinity, bacterial production, and depth as the driving factors of the combined community (Fig. 6b).

Both (a) Mantel test and (b) CCA analysis were performed to see correlations with GVMAG abundance in the Baltic Sea. The Mantel test was conducted on all combined GVMAGs as well as each order that had more than 2 recovered GVMAGs, and the correlation coefficient is displayed (* = p < 0.05).
Discussion
In an era of metagenomic research, discoveries on giant virus diversity and functional potential have greatly been spurred on by a number of recent large-scale GV genome recovery approaches from publicly available metagenomic datasets41,42,43. These sweeping studies greatly increased the number of genomes available for GVs, but much work is still to be done to resolve their immense diversity in the marine ecosystem. Our 230 new genomes reported in this study provide additional resources for accessing GV diversity and functional potential.
Analysis of our recovered genomes confirmed what was known broadly about differences between GV orders such as the higher GC percentage of Pandoravirales genomes (>60%)3 compared to other orders45. Other metrics such as Nitrogen and Sulfur content of GV genomes have never been reported. The proteomic nitrogen stores have been hypothesized to give insight into the connection between ecology and genomics as lower nitrogen content can be a proteomic signature of environmental nitrogen scarcity46. In the marine environment, it is estimated that minimization of proteomic nitrogen can reduce the cellular nitrogen budget by up to 10% for marine microbes47. While viruses donot directly have a nitrogen budget, during infection they have the potential to shift the host’s nitrogen budget through its own replication. Changing the host’s genomic and proteomic pool could be yet another way that these viruses influence host metabolism and survival in the ocean as GVs with higher N per sidechain could have a greater toll on the host nitrogen budget17.
The findings of phylogeny-associated metabolic potential were largely consistent with previous studies looking at GV metabolic potential. Moniruzamman et al.42, Ha et al.5, and Farzad et al.45 report the largest number of metabolic genes associated with the Imitervirales order, with the Algavirales order having much less DNA processing and nutrient transport genes. We also observed the scarcity of rhodopsins inside the Algavirales order, consistent with these studies. Many of the metabolic genes are functional during infection. For example, MutS and HAT proteins were shown to be packed inside the capsid48,49 and the presence of these genes more frequently in the Imitervirales order could imply a phylogenetic pattern in DNA packaging strategy within Nucleocytoviricota50.
In addition to more metabolic genes, we also found the functional landscape between Imitervirales and Algavirales genomes to be quite different with Imitervirales genomes encoding a large array of genes not found in Algavirales. The repeated finding of more auxiliary metabolic genes as well as the broad functional capacity within the genomes of Imitervirales could be indicative of a different “life strategy” as Imitervirales viruses most likely infect a broad range of hosts, including slow-growing hosts, where metabolic augmentation may be necessary for productive infection51.
In addition to uncovering known GV-encoded functional patterns, we found many proteins previously unknown to GVs to be found in our new high-quality and partial GVMAGs. This finding shows that the protein universe for GVs remains largely open as large amounts of novel proteins and functional capacity remain to be discovered, expanding our understanding of what functions virus genomes can encode and perform. A large number of these novel functions coming from polar or Baltic Sea GVMAGs could be further evidence of environment-specific infection strategies or functional repertoires as is seen in other nutrient and light regimes52,53. Previous analysis of polar giant viruses also found high levels of adaptation and unique gene content compared to temperate and tropical counterparts54, showing these colder environments could be reservoirs of novel GV-encoded functions.
A well-explored and interesting area of GV functional potential is the ability of GVs to influence host photosynthetic potential through the introduction of rhodopsins and light-harvesting proteins55,56,57. Here, we show that not only are these genes present in the new genomes recovered in this study, but additional genes involved in all stages of photosynthetic light harvesting are present as well. It is well known that some GV infections depend on light and functioning photosynthetic machinery during the early stages of infection58,59, so GVs may be using auxiliary genes to keep host machinery operational during infection as the host tries to shut off its transcriptional machinery60,61. Photosystem 1 and 2 components have also been found previously in bacteriophages where they are hypothesized to be used during infection to alter host metabolism to favor phage reproduction62,63. Here we report many new proteins involved in the photosynthetic pathway, adding evidence to the claim that GVs can modulate this pathway in their host.
Our study also revealed GV genomes from the Baltic Sea, one of the largest bodies of brackish water in the world. Within the context of this unique ecosystem, recovered GVMAGs demonstrated differential abundance correlating with various environmental parameters. Previous work on prokaryotic viruses has shown the high explanatory power these parameters can have on viral abundance across coastal waters64, and our data confirm this conclusion as factors such as salinity and depth showed significant correlation with GV abundance. Despite these factors being significant for the total GV population, factors affecting GV community composition differed by phylogeny, possibly reflecting the different hosts of viruses within these phylogenetic orders9.
Overall, our work provides new insights into the diversity and functional potential of GVs in the world’s oceans through our addition of 230 genomes with an expanded set of photosynthesis proteins as well as many other metabolic genes. We hope that these new genomes along with protein annotations, will be useful in the expansion of insights into GVs from further metagenomic datasets across all aquatic ecosystems.
Methods
Data Acquisition
To recover GV genomes, we downloaded raw sequencing data (Illumina) from 9 publicly available BioProjects which represent 8 different bodies of water spanning from pole to pole (Table S1). These projects were chosen based on their sampling of the cellular size fraction (>0.2 µM) and the high number of hits to giant virus major capsid proteins (MCPs) that we assessed using a pre-screening tool we developed (https://github.com/BenMinch/PIGv/tree/main/viral_screening). We also avoided large datasets that had already been searched for GVs such as the Tara Oceans data43. All raw metagenomic reads from these projects were downloaded and trimmed with trimGalore (v0.6.10) using default parameters65. The raw reads were then assembled using Megahit (v1.2.9) with default settings66 and reads were mapped back onto the assemblies to generate coverage using CoverM (v0.7.0)(https://github.com/wwood/CoverM) in coverage mode.
Giant virus genome recovery
Metagenomes were run through the recently developed BEREN tool44 using the “NCLDV_bins” module to recover GV genomes. Briefly, this tool utilizes genome binning67 and screens bins for NCLDV marker genes as well as positive ViralRecall scores68. This tool recovers both “high quality” and “partial” GV bins depending on the number of identified marker genes (≥4 for high quality and ≥1 for partial). All bins are purged of potential bacterial and eukaryotic contamination within the tool.
All genomes were dereplicated at 98% average nucleotide identity (ANI) from the Global Ocean Eukaryotic Virus database (GOEV) with a 25% minimum coverage threshold using dRep (v3.4.2)69 as was utilized in ref. 43. Cleaned partial bins were dereplicated at 99% ANI with a 25% minimum coverage threshold to get rid of duplicate bins. This resulted in a total of 230 new GV genomes and 398 cleaned partial bins for downstream analysis.
Basic genome statistics such as length and GC percentages were calculated using Seqkit (v2.3)70 and nitrogen/sulfur per sidechain were calculated using a script adapted from ref. 71(github.com/BenMinch/NutriVi). Comparisons of nitrogen and sulfur per sidechain, average length, and GC content between orders were done using a two-sample t-test with FDR correction. All plots were made using ggplot2 (v3.5.1)47 in R (v4.4.2).
Taxonomy and phylogeny
Taxonomy was predicted for all genomes using TIGTOG (v1)72, a recently developed tool that classifies GV genomes or partial genomes based on protein family trademarks. In addition to classifying genomes, a phylogenetic tree was made using the PolB marker gene. Briefly, PolB genes were gathered from all the high-quality and partial genomes (n = 351) and dereplicated at 98% using cd-hit (v4.8.1)73. These proteins were aligned with a curated set of reference PolB representing all major GV families from the GOEV database43. Proteins were aligned using MAFFT (--auto) (v7.453)74 and alignments were trimmed using trimAL (v1.4) with the “gt 0.1” parameter75. A maximum-likelihood phylogenetic tree was made using IQ-TREE (v2.0.3)76 with the “LG + F + R10” model with 1000 bootstraps using an ultrafast bootstrap. The tree was visualized in iTOL77.
Protein annotation
All proteins from both high-quality and partial bins were predicted using prodigal-gv (v.2.10.0-gv)78. These predicted proteins were then annotated with the PFAM79, GVOG7, VOG80, and COG81 databases using Annomazing (github.com/BenMinch/AnnoMazing). This tool performs annotations based on hmm searches against requested databases and compiles them all together. For all searches, an e-value cutoff of 1e-5 was used. Genomes in the entire GOEV database were also annotated similarly.
Proteins that are unique to the genomes generated in this study were identified by first clustering our new genome-encoded proteins using mmseqs2 (v13.45111) with 50% identity (coverage mode 5, -c 0.4)82. After clustering, representative sequences from each cluster were searched against a database of all GOEV (Global Ocean Eukaryotic Virus) and giant virus database (GVDB)7 proteins using BLASTp (v2.9.0). Proteins without a significant hit (e-value greater than 1e-3) were chosen for further analysis. These representative proteins were then screened for unique functional annotations by looking for unique PFAM accession numbers not present in GOEV-annotated genomes. This yielded a total of 569 novel protein clusters in our genomes.
Metabolic genes for high-quality genomes were annotated using NuMP (Nucleocytoviricota metabolic profiler) (github.com/BenMinch/NuMP), which looks for specific metabolic genes that have been found previously in GVs. These were displayed as a heatmap using pheatmap (v1.0.12) (github.com/raivokolde/pheatmap) based on presence/absence. Functional differences between Imitervirales and Algavirales genomes were displayed using the “anvi-display-functions” program in Anvi’o (v8) with COG annotations81,83.
Assessment of GV abundance in the Baltic Sea metagenomes
Abundance profiles of high-quality GVMAGs recovered from the Baltic Sea (n = 108) were acquired through mapping trimmed reads from the Baltic Sea metagenomes to the genomes at 95% ANI using CoverM with minimap2 (v2.28)84. Read counts were normalized by library and genome size and used to assess GV abundance in different size fractions as well as correlations with various environmental variables (depth, temperature, salinity, chlorophyll, turbidity, bacterial production, dissolved organic nitrogen, dissolved oxygen, dissolved organic carbon, and nitrite).
To assess correlations between GV genome relative abundance and environmental variables, a Mantel test was performed using Spearman correlation and 9999 bootstraps in the vegan R package (v2.6) (github.com/vegandevs/vegan). A canonical correlation analysis (CCA) was also done on individual genomes using the same environmental variables.
Data availability
All metagenomes used for this analysis are publicly available on NCBI using the project numbers in Table S1. The GVMAGs, Genome statistics, taxonomy, phylogenetic tree, PolB proteins, Anvi’o database, and protein annotations are available on figshare (https://figshare.com/projects/Expansion_of_the_genomic_and_functional_diversity_of_global_ocean_giant_viruses/214120). GVMAGs are also available on Zenodo for convenience: (https://zenodo.org/records/14782687). The data and analysis are also available from the authors upon request.
References
Aylward, F. O. & Moniruzzaman, M. Viral complexity. Biomolecules 12, 1061 (2022).
Abrahão, J. et al. Tailed giant Tupanvirus possesses the most complete translational apparatus of the known virosphere. Nat. Commun. 9, 749 (2018).
Philippe, N. et al. Pandoraviruses: amoeba viruses with genomes up to 2.5 Mb reaching that of parasitic eukaryotes. Science 341, 281–286 (2013).
Aherfi, S. et al. Incomplete tricarboxylic acid cycle and proton gradient in Pandoravirus massiliensis: is it still a virus?. ISME J. 16, 695–704 (2022).
Ha, A. D., Moniruzzaman, M. & Aylward, F. O. High transcriptional activity and diverse functional repertoires of hundreds of giant viruses in a coastal marine system. mSystems 6, e0029321 (2021).
Wilson, W. H. et al. Genomic exploration of individual giant ocean viruses. ISME J. 11, 1736–1745 (2017).
Aylward, F. O., Moniruzzaman, M., Ha, A. D. & Koonin, E. V. A phylogenomic framework for charting the diversity and evolution of giant viruses. PLOS Biol. 19, e3001430 (2021).
Moniruzzaman, M. et al. Virus-host relationships of marine single-celled eukaryotes resolved from metatranscriptomics. Nat. Commun. 8, 16054 (2017).
Sun, T.-W. et al. Host range and coding potential of eukaryotic giant viruses. Viruses 12, 1337 (2020).
Hingamp, P. et al. Exploring nucleo-cytoplasmic large DNA viruses in Tara Oceans microbial metagenomes. ISME J. 7, 1678–1679 (2013).
Schulz, F. et al. Hidden diversity of soil giant viruses. Nat. Commun. 9, 4881 (2018).
Benoiston, A.-S. The evolution of diatoms and their biogeochemical functions. Philos. Trans. R. Soc. Lond. B Biol. Sci. 372, 20160397 (2017).
Sherr, E. B. & Sherr, B. F. Bacterivory and herbivory: key roles of phagotrophic protists in pelagic food webs. Microb. Ecol. 28, 223–235 (1994).
Blanc-Mathieu, R. et al. A persistent giant algal virus, with a unique morphology, encodes an unprecedented number of genes involved in energy metabolism. J. Virol. 95, e02446–20 (2021).
Ku, C. Giant virus-eukaryote interactions as ecological and evolutionary driving forces. mSystems 6, e0073721 (2021).
Moniruzzaman, M. et al. Virologs, viral mimicry, and virocell metabolism: the expanding scale of cellular functions encoded in the complex genomes of giant viruses. FEMS Microbiol. Rev. 47, fuad053 (2023).
Monier, A. et al. Host-derived viral transporter protein for nitrogen uptake in infected marine phytoplankton. Proc. Natl. Acad. Sci. 114, E7489–E7498 (2017).
Plugge, B. et al. A potassium channel protein encoded by Chlorella virus PBCV-1. Science 287, 1641–1644 (2000).
Kaneko, H. et al. Eukaryotic virus composition can predict the efficiency of carbon export in the global ocean. iScience 24, 102002 (2021).
Wilhelm, S. W. & Suttle, C. A. Viruses and nutrient cycles in the sea: viruses play critical roles in the structure and function of aquatic food webs. BioScience 49, 781–788 (1999).
Meints, R. H., Van Etten, J. L., Kuczmarski, D., Lee, K. & Ang, B. Viral infection of the symbiotic chlorella-like alga present in Hydra viridis. Virology 113, 698–703 (1981).
Scola, B. L. et al. A giant virus in amoebae. Science 299, 2033–2033 (2003).
Raoult, D. et al. The 1.2-megabase genome sequence of mimivirus. Science 306, 1344–1350 (2004).
Francis, R., Ominami, Y., Bou Khalil, J. Y. & La Scola, B. High-throughput isolation of giant viruses using high-content screening. Commun. Biol. 2, 216 (2019).
Khalil, J. Y. B. et al. High-throughput isolation of giant viruses in liquid medium using automated flow cytometry and fluorescence staining. Front. Microbiol. 7, 26 (2016).
Schulz, F., Abergel, C. & Woyke, T. Giant virus biology and diversity in the era of genome-resolved metagenomics. Nat. Rev. Microbiol. 20, 721–736 (2022).
Cook, R. et al. INfrastructure for a PHAge REference database: identification of large-scale biases in the current collection of cultured phage genomes. PHAGE 2, 214–223 (2021).
Blackwell, G. A. et al. Exploring bacterial diversity via a curated and searchable snapshot of archived DNA sequences. PLOS Biol. 19, e3001421 (2021).
Andreani, J., Verneau, J., Raoult, D., Levasseur, A. & La Scola, B. Deciphering viral presences: two novel partial giant viruses detected in marine metagenome and in a mine drainage metagenome. Virol. J. 15, 66 (2018).
Colson, P., Scola, B. L. & Raoult, D. Giant viruses of amoebae as potential human pathogens. Intervirology 56, 376–385 (2013).
Ghedin, E. & Claverie, J.-M. Mimivirus relatives in the Sargasso sea. Virol. J. 2, 62 (2005).
Kristensen, D. M., Mushegian, A. R., Dolja, V. V. & Koonin, E. V. New dimensions of the virus world discovered through metagenomics. Trends Microbiol. 18, 11–19 (2010).
Loh, J. et al. Detection of novel sequences related to African Swine Fever virus in human serum and sewage. J. Virol. 83, 13019–13025 (2009).
Monier, A., Claverie, J.-M. & Ogata, H. Taxonomic distribution of large DNA viruses in the sea. Genome Biol. 9, R106 (2008).
Mihara, T. et al. Taxon richness of ‘megaviridae’ exceeds those of bacteria and archaea in the ocean. Microbes Environ. 33, 162–171 (2018).
Yau, S. et al. Virophage control of Antarctic algal host–virus dynamics. Proc. Natl. Acad. Sci. 108, 6163–6168 (2011).
Zhang, W. et al. Four novel algal virus genomes discovered from Yellowstone Lake metagenomes. Sci. Rep. 5, 15131 (2015).
Bäckström, D. et al. Virus genomes from deep sea sediments expand the ocean megavirome and support independent origins of viral gigantism. mBio 10, e02497–18 (2019).
Chen, H. et al. The genome of a prasinoviruses-related freshwater virus reveals unusual diversity of phycodnaviruses. BMC Genomics 19, 49 (2018).
Schulz, F. et al. Giant viruses with an expanded complement of translation system components. Science 356, 82–85 (2017).
Schulz, F. et al. Giant virus diversity and host interactions through global metagenomics. Nature 578, 432–436 (2020).
Moniruzzaman, M., Martinez-Gutierrez, C. A., Weinheimer, A. R. & Aylward, F. O. Dynamic genome evolution and complex virocell metabolism of globally-distributed giant viruses. Nat. Commun. 11, 1710 (2020).
Gaïa M. et al., “Mirusviruses link herpesviruses to giant viruses,” Nature. 1–7 https://doi.org/10.1038/s41586-023-05962-4 (2023).
Minch, B. and Moniruzzaman, M., BEREN: A bioinformatic tool for recovering Giant viruses, Polinton-like Viruses, and Virophages in metagenomic data. bioRxiv, pp.2024-10. (2024).
Farzad, R., Ha, A. D. & Aylward, F. O. Diversity and genomics of giant viruses in the North Pacific Subtropical Gyre. Front. Microbiol. 13, 1021923 (2022).
Acquisti, C., Kumar, S. & Elser, J. J. Signatures of nitrogen limitation in the elemental composition of the proteins involved in the metabolic apparatus. Proc. R. Soc. B Biol. Sci. 276, 2605–2610 (2009).
Valero-Mora, P. M. ggplot2: elegant graphics for data analysis. J. Stat. Softw. 35, 1–3 (2010).
Koonin, E. V. & Yutin, N. Origin and evolution of eukaryotic large nucleo-cytoplasmic DNA viruses. Intervirology 53, 284–292 (2010).
Legendre, M. et al. mRNA deep sequencing reveals 75 new genes and a complex transcriptional landscape in Mimivirus. Genome Res. 20, 664–674 (2010).
Chelikani, V., Ranjan, T. & Kondabagil, K. Revisiting the genome packaging in viruses with lessons from the ‘Giants. Virology 466–467, 15–26 (2014).
Ha, A. D., Moniruzzaman, M. & Aylward, F. O. Assessing the biogeography of marine giant viruses in four oceanic transects. ISME Commun. 3, 43 (2023).
Chase, E. E., Truchon, A. R., Creasey, B. A. & Wilhelm, S. W. Time of day of infection shapes development of a eukaryotic algal-Nucleocytoviricota virocell. FEMS Microb. Ecol. 100, https://doi.org/10.1093/femsec/fiae123 (2024).
Gilbert, N. E. et al. Giant virus infection signatures are modulated by Euphotic Zone Depth Strata and iron regimes of the Subantarctic Southern Ocean. mSystems 8, e01260–22 (2023).
Meng, L. et al. Genomic adaptation of giant viruses in polar oceans. Nat. Commun. 14, 6233 (2023).
Needham, D. M. et al. A distinct lineage of giant viruses brings a rhodopsin photosystem to unicellular marine predators. Proc. Natl. Acad. Sci. USA 116, 20574–20583 (2019).
Rozenberg, A. et al. Lateral gene transfer of anion-conducting channelrhodopsins between green algae and giant viruses. Curr. Biol. CB 30, 4910–4920.e5 (2020).
Zabelskii, D. et al. Viral rhodopsins 1 are an unique family of light-gated cation channels. Nat. Commun. 11, 5707 (2020).
Gann, E. R., Gainer, P. J., Reynolds, T. B. & Wilhelm, S. W. Influence of light on the infection of Aureococcus anophagefferens CCMP 1984 by a ‘giant virus. PLOS ONE 15, e0226758 (2020).
Thamatrakoln, K. et al. Light regulation of coccolithophore host–virus interactions. N. Phytol. 221, 1289–1302 (2019).
Juneau, P., Lawrence, J., Suttle, C. & Harrison, P. Effects of viral infection on photosynthetic processes in the bloom-forming alga Heterosigma akashiwo. Aquat. Microb. Ecol. 31, 9–17 (2003).
Seaton, G. G. R., Lee, K. & Rohozinski, J. Photosynthetic shutdown in Chlorella NC64A associated with the infection cycle of Paramecium bursaria Chlorella Virus-1. Plant Physiol. 108, 1431–1438 (1995).
Philosof, A., Battchikova, N., Aro, E. M. & Béja, O. Marine cyanophages: tinkering with the electron transport chain. ISME J. 5, 1568–1570 (2011).
Sharon, I. et al. Photosystem I gene cassettes are present in marine virus genomes. Nature 461, 258–262 (2009).
Finke, J. F., Hunt, B. P. V., Winter, C., Carmack, E. C. & Suttle, C. A. Nutrients and other environmental factors influence virus abundances across oxic and hypoxic marine environments. Viruses 9, 152 (2017).
Martin M., “Cutadapt removes adapter sequences from high-throughput sequencing reads,” EMBnet.journal, 17, 1, https://doi.org/10.14806/ej.17.1.200 2011.
Li, D., Liu, C.-M., Luo, R., Sadakane, K. & Lam, T.-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Kang, D. D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359 (2019).
Aylward, F. O. & Moniruzzaman, M. ViralRecall—a flexible command-line tool for the detection of giant virus signatures in ‘omic data. Viruses 13, 150 (2021).
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11, 2864–2868 (2017).
Shen, W., Le, S., Li, Y. & Hu, F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLOS ONE 11, e0163962 (2016).
Grzymski, J. J. & Dussaq, A. M. The significance of nitrogen cost minimization in proteomes of marine microorganisms. ISME J. 6, 71–80 (2012).
Ha, A. D. & Aylward, F. O. Automated classification of giant virus genomes using a random forest model built on trademark protein families. Npj Viruses 2, 1–9 (2024).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Camargo, A. P. et al. Identification of mobile genetic elements with geNomad. Nat. Biotechnol. 42, 1303–1312 (2024).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419 (2021).
Grazziotin, A. L., Koonin, E. V. & Kristensen, D. M. Prokaryotic Virus Orthologous Groups (pVOGs): a resource for comparative genomics and protein family annotation. Nucleic Acids Res. 45, D491–D498 (2017).
Tatusov, R. L., Galperin, M. Y., Natale, D. A. & Koonin, E. V. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36 (2000).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Eren, A. M. et al. Community-led, integrated, reproducible multi-omics with anvi’o. Nat. Microbiol. 6, 3–6 (2021).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Acknowledgements
This study leveraged a large number of publicly available metagenomic datasets, and we thank the researchers who have made their data available. We also thank the researchers who made their bioinformatic tools and pipelines available for public use. We also gratefully acknowledge the computational resources provided by the Frost Institute for Data Science and Computing (IDSC), University of Miami.
Author information
Authors and Affiliations
Contributions
B.M. and M.M. jointly developed the research idea. B.M. performed the data analysis, bioinformatic pipeline development, and wrote the manuscript. M.M. supervised the research, contributed writing and editing of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Minch, B., Moniruzzaman, M. Expansion of the genomic and functional diversity of global ocean giant viruses. npj Viruses 3, 32 (2025). https://doi.org/10.1038/s44298-025-00122-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s44298-025-00122-z
This article is cited by
-
A deep dive into giant viruses
npj Viruses (2025)
