
Abstract
The genome of Oceanusvirus kaneohense isolate, Tetraselmis virus 1 (TetV-1), was previously sequenced, but little was known about its infection cycle. We determined the eclipse period (4–8 h), latent period (16 h), and burst size (~1000), and observed ultrastructural changes in the host during infection. Putative viral factories and inclusion bodies appear in the cytoplasm by 8 and 16 h post-infection, respectively. Of 830 viral transcripts detected, those related to transcription, DNA synthesis, and host immune repression appeared earlier (0.25 and 4 h), and virus structural genes appeared later (8–16 h). Host lipid metabolism and endocytosis-related transcripts were upregulated during the early phase, while protein modification genes were downregulated. In the later stages, host transcripts associated with basic cellular processes were relatively more common, while developmental genes became less so. Many highly expressed host and virus genes were of unknown function, highlighting a need for additional functional studies.
Similar content being viewed by others


Introduction
Viruses in the phylum Nucleocytoviricota, the new taxonomic designation for the Nucleocytoplasmic Large DNA Viruses (NCLDVs), are characterized by their large double-stranded DNA genomes (up to 2.5 Mbp) and large virion sizes (up to 1500 nm)1,2,3. The largest members in this phylum, often referred to colloquially as “giant viruses”4, contain hundreds of genes that afford them a higher degree of autonomy compared to “smaller” viruses5, the smallest of which encode as few as two genes. In addition to a typical suite of DNA replication and transcription machinery, giant virus genomes may encode a diverse suite of products involved in metabolic functions previously only associated with cells. For example, some encode dozens of tRNAs and hundreds of components of the translation machinery6,7. Others may encode rhodopsins8,9, enzymes involved in the TCA cycle and glycolysis10,11, histones12,13, cytoskeletal proteins (actin, kinesin, myosin)14,15, or, as was found for the virus studied here, TetV-1, enzymes involved in fermentation16. In addition to the genes with diverse predicted functions, giant viruses also encode many ORFans17 or genes with predicted amino acid sequences that are so divergent from any characterized protein that their functions remain a mystery. Even for the genes for which confident functions are assigned, how and when those genes are used by the virus is not always clear.
Transcriptional analysis complements genome sequencing by providing detailed insights into the operational program of a virus18. Transcriptome sequences can reveal errors in genome sequencing and assembly17 and improve genome annotations. Genes not predicted ab initio, for example, may be revealed by their presence in the transcript pool19,20,21. Conversely, initially predicted genes may be absent from the transcriptome22. Transcriptional analysis can also shed light on how gene expression is regulated, revealing, for example, changes in the promoter motifs used at different stages of infection19,23, alternative splicing24,25, transcription start and termination sites19, and transcription termination mechanisms18,26. Transcriptomics also allows for inferences about the interplay between virus and host metabolism23,27,28,29. In some cases, host genes are upregulated that carry out functions not encoded by the virus30,31. In other cases, the virus and host share homologous genes, but the version expressed (host vs virus) changes during infection27,32.
Inferences about the timing of replication based on transcriptional patterns can be corroborated and augmented by observations of ultrastructural changes in infected cells. Electron microscopy of thin sections provides information on the mechanisms of virion entry33,34, the ___location of replication and the formation of viral factories (VFs)35, the timing of the eclipse period, and other virus-induced changes to cellular ultrastructure7,32. In this study, we investigated the infection cycle of the green alga-infecting Tetraselmis virus 1 (TetV-1) using both electron microscopy and transcriptomics to (i) identify ultrastructural changes accompanying the infection progression, (ii) temporal expression of viral genes, and (iii) host responses to infection.
Results
TetV-1 infection cycle and ultrastructure
In an initial experiment tracking the infection cycle of TetV-1, free virions remained consistently low for the first 15.5 hours post-infection (hpi), began a rapid increase by 16 hpi, and plateaued at 17–18 hpi (Fig. 1). Based on the number of new viruses produced and the host counts at the beginning of the infection, and assuming all cells are lysed, the burst size was estimated to be around 800–1000.

Virions were counted by flow cytometry. Subsequently, estimates of the latent period and burst size were determined. The eclipse period was determined using electron micrographs. Samples for electron microscopy and RNA sequencing were obtained throughout the time series except at 20 hpi (solid lines on the arrow below the graph).
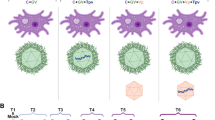
Examination of thin sections of uninfected algal cells by electron microscopy revealed the intact organelles expected in a healthy cell such as the nucleus, nucleolus, chloroplasts, mitochondria, vacuoles, pyrenoids, and an eyespot (Fig. 2a). Four basal bodies (base of flagella) are also evident (Supplementary Fig. 1). For infected cells, the initial attachment and entry process of the virus was not observed in any of the examined cells, likely because of the low temporal resolution of sampling. Cells at 15 min and 4 hpi were similar to the uninfected cells, with no apparent changes to subcellular structures or evidence of virion assembly (Fig. 2b, c). Intracellular virions were first observed in samples taken at 8 hpi, which puts the eclipse period somewhere between 4 and 8 hpi (Fig. 2d). By 8 hpi, there was also a change in the fine-scale structure of the cytoplasm. At this point, organelles appeared to be compressed around the periphery of the cell, the expanded area of cytoplasm was more granular, and putative VFs were present. The VFs are localized areas within the cytoplasm with incomplete capsids near the center and complete capsids further out. VFs were present for the remainder of the latent period (Fig. 2e, f) but multiple, well-defined, darker staining inclusion bodies (IBs) also appeared in the cytoplasm by 16 hpi (Fig. 2f). At this point most cells were filled with virions, anticipating the imminent lysis, which is evident from the rapid increase in free virions over the next 2 h (Fig. 1). Some intact cells at the late stages of infection with both VF and IB were still observed at 20 hpi (Fig. 2g–i), either because of a non-synchronous start of infection, or variation in latent period length. Higher magnification views of one such cell illustrate the incomplete capsid structure at the core of the VF and a darker staining edge of the IB, suggesting a possible membrane-bound compartment (Fig. 2g–i).

a Apparent in the uninfected algae are typical organelles such as the nucleus (NC), nucleolus (NCL), chloroplast (CHL), vacuoles (VAC), mitochondrion (MITO), as well as algae-specific photoreceptive organelles (eyespot; EYSP) and CO2-fixation center (pyrenoid; PYR). b, c No changes were observed in the early stages of infection (15 min and 4 hpi). c, d Further inspection of electron micrographs showed the first appearance of intracellular virions at 8 hpi but not 4 hpi, indicating an eclipse period between 4 and 8 hpi. e–g Representative images of the virocell close to the latent period show the presence of (g, h) a putative virus factory (VF) and (i) inclusion bodies (IB). Scale bar = 0.8 µm (a–g) or 0.2 µm (h, i).
TetV-1 transcriptome
The TetV-1 genome was sequenced previously and predicted to contain 663 genes using Prodigal16. The current RNA-seq pipeline detected 830 transcripts, of which 167 (20%) are multi-exon. Included in these are 292 transcripts that are longer than the Prodigal-predicted genes, 27 that contain overlapping exons with Prodigal-predicted genes, one transcript that is within an intron, and 10 transcripts contain exons that overlap with Prodigal-predicted genes but are on the opposite strand.
The relative abundance of reads that map to TetV-1 steadily increased early in the infection and peaked at 8 hpi, at which point they accounted for ~58% of the total reads (Supplementary Fig. 2A). The proportion of viral reads subsequently decreased, presumably when transcription slows down and other processes, such as packaging, and assembly predominate until lysis at around 16 hpi (Supplementary Fig. 2A). The average RNA yield remained constant in the control sample, while it seems to decrease in the infected samples (Supplementary Fig. 2B). Global clustering of TetV-1 gene expression profiles resulted in the earliest time points (15 mpi and 4 hpi) forming one cluster and the three later time points (8–16 hpi) forming another (Supplementary Fig. 3A). Clustering based on standardized TPM per transcript further refined the temporal groups, in which the early stage was differentiated into immediate early (15 mpi) and late early (4 hpi) while the late stage (8–16 hpi) remained grouped (Supplementary Fig. 3B). In some analyses, we distinguished between the two early groups, but in other cases we retain the simpler designations of “early” or “late,” as these categories are equivalent to eclipse and post-eclipse periods.
After a filtering step to remove transcripts that were relatively constant (those with a variance less than one across samples), 94% of the viral genes (782 out of 830) were retained. Nine of the top ten highly expressed genes are of unknown function (TetV_630, 071, 278, 511, 015, 023, 024, 013, and 405) (Fig. 3). A search for motifs across all transcripts revealed one potential promoter motif (ATCCGGA) that was “universal,” i.e., was observed in at least some portion of all the temporal expression groups but was most prevalent among immediate early transcripts (92%) compared to the other groups (29–39%) (Fig. 3A; Supplementary Fig. 4A). A search for motifs specific to each temporal cluster resulted in additional potential promoters with varying prevalence in each group: immediate early (52%), late early (17%), late (77%) (Fig. 3B).

(A) Universal and (B) enriched motifs were identified across viral temporal classes. (C) Temporal expression of select TetV-1 genes. Genes are grouped based on their function, as described above. HP hypothetical protein (percentage shows the relative abundance in proteome with respect to capsid), DCP ___domain-containing protein, vMiSTRA TetV-1 viral mitochondria stress response algae, Ub Ubiquitin, M1DPH mannitol 1-phosphate dehydrogenase, 5-CAI 5-(carboxyamino)imidazole, rNDP ribonucleoside-diphosphate reductase, IF initiation factor, EF elongation factor, LCMC large-conductance mechanosensitive channel, FP family protein, PFL pyruvate formate lyase, PFL AE pyruvate formate lyase 1 activating enzyme, TPM Transcript per Million.
Early infection stage
Expression of viral transcripts was detected at least as early as 15 min after infection (Fig. 3, Supplementary Fig. 4, Supplementary Table 1). Four highly expressed transcripts at the 15 min post-infection have no known or predicted function (TetV_026, 357, 104, and 431).
Several enzymes involved in carbohydrate metabolism are expressed early such as alpha-galactosidase (TetV_601), glycosyl hydrolase (TetV_615), arabinose 5-phosphate isomerase (TetV_403) and mannitol 1-phosphate dehydrogenase (M1DPH, TetV_320) (Fig. 3). A putative host immune suppression gene, vMISTRA (TetV_113), and various RING ___domain-containing proteins (TetV_277, 420, 434) that are implicated in apoptosis inhibition are also expressed early as is the virus-encoded ribonuclease-3 gene (TetV_554) (Fig. 3). Most ubiquitination genes are expressed throughout the infection (TetV_035, 358, 621) except one that seems to be expressed early, E3 ubiquitin-protein ligase MIEL-1 like protein (TetV_138). The virus-encoded fermentation genes, pyruvate formate lyase (vPFL; TetV_428) and pyruvate formate lyase activating enzyme (vPFLA; TetV_456), are somewhat higher in the early phase than in late stages but were essentially constitutively expressed (Fig. 3).
Expression of genes involved in transcription began in the early stage and expression of some was sustained throughout the infection (Fig. 3, Supplementary Fig. 4B). Transcription factors such as transcription initiation factor TFIIIB Brf1 (TetV_036), transcription activator SWIB/MDM2 (TetV_204), and TATA-box binding proteins (TetV_094 and 195) were among those expressed early. In addition, transcription elongation factor TFIIS (TetV_526) and late transcription factor VLTF3-like (TetV_254) were expressed early and were sustained throughout the infection (Fig. 3). Similarly, DNA-directed RNA polymerase subunits were expressed early and sustained in the late stage. These included Rpb2 (TetV_196, 616), Rpb3/Rpb11 (TetV_250), Rpb5 (TetV_078), Rpb6 (TetV_177), Rpb7 (TetV_286), and Rpb10/RpoN (TetV_156) (Supplementary Fig. 4). Expression of mRNA capping enzymes (TetV_090 and 219) also began in the early stage (Supplementary Fig. 4).
Early genes involved in nucleotide synthesis included ribonucleoside-diphosphate reductase subunit alpha (TetV_458) and beta (TetV_533), thymidylate kinase (TetV_225), thymidylate synthase complementing protein (TetV_240), 5-(carboxyamino) imidazole ribonucleotide mutase (TetV_495), and dUTP pyrophosphatase (TetV_595) (Fig. 3). Early genes involved in DNA replication, ATP synthesis and repair genes included DNA polymerase B (TetV_567), DNA topoisomerase 2 (TetV_404), and DNA clamp, proliferating cell nuclear antigen (TetV_607) (Supplementary Fig. 4). Genes involved in DNA repair (DNA repair exonuclease SbcCD, TetV_472; DNA repair ATPase SbcC, TetV_662) appeared early, as did others that remained relatively high through the late stages such as DNA mismatch repair ATPase MutS (TetV_243) and deoxyribodipyrimidine photolyases (TetV_298 and 522) (Supplementary Fig. 4). Finally, expression of eukaryotic initiation factor 4E (TetV_117) and translation initiation factor 1A (TetV_059) are first detected in the early stages of infection (Fig. 3).
Late infection stage
Genes encoding proteins related to virion structure and packaging, such as the major capsid protein (TetV_001) and VV A32-like virion packaging ATPase (TetV_186) (Fig. 3) were highly expressed in the late stage. Genes encoding a functional potassium ion channel (TetV_029)36, a large-conductance mechanosensitive channel (TetV_044), and a transmembrane protein/ion channel (TetV_199) were also expressed in late stages (Fig. 3). Genes encoding some enzymes were also expressed in the late stage such as glycosyltransferases (TetV_241 and 318), sulfatases (TetV_008 and 050), lipid catabolic enzymes, lipase (TetV_056) and phosphatase (TetV_413), proteases (TetV_461 and 556), and redox enzymes (TetV_019, 191, 236, and 626) (Supplementary Fig. 4). Different methylation genes appear to be expressed at distinct stages of the infection. There is one methylation gene expressed early, the SET ___domain-containing protein (TetV_181), whereas four methyltransferases are expressed in the late stage (TetV_211, 484, 501, 608) (Supplementary Fig. 4).
Tetraselmis sp. (host) response
Before the host transcriptome assembly, TetV-1 reads were removed, leaving 308 million paired-end reads. Trinity de novo assembly yielded 159,006 transcripts corresponding to 69,817 genes (Supplementary Table 2). The assembly is of high quality based on metrics such as N50 (3244 bp), read alignment (99%), and the presence of 96% of Chlorophyta single-copy genes (Supplementary Table 2). Two additional filtering steps were implemented: First, only transcripts with enough read support according to TransRate were retained, leaving 91% of the genes. TransRate calculates contig scores based on read mapping evidence, identifying contigs that are chimeric or misassembled, among others. Second, only the highest expressed isoform per gene was retained for downstream analysis (Supplementary Table 3). Of the remaining 63,860 genes, 83.7% are expressed at >1 TPM (transcripts per million transcripts), while only 37% are expressed at >5 TPM. Around 14–15% of the genes have BLAST/HMMER matches to SwissProt Uniprot and Pfam databases (Supplementary Fig. 5). Out of the few annotated genes (10,000), 46.4% are closely matching eukaryotic genes (the majority being Viridiplantae homologs), while the rest are closely matching Bacteria (51.3%), Archaea (1.6%), and viral (0.6%) genes (Supplementary Fig. 5). This translates to 5290 genes (8.3% of the total) that are prokaryotic homologs.
We found seven host transcripts related to fermentation with homology to pyruvate formate lyase activating enzyme (PFLA), but no transcripts related to pyruvate formate lyase (PFL) (Supplementary Fig. 6). Four host transcripts matched to formate acetyltransferase (PFLB) (not shown). Only one of the seven host PFLA genes showed significant expression, and, like the viral-encoded PFLA, the host PFLA seems to be expressed at high levels early in the infection.
To determine the host response to infection, differentially expressed genes were determined at early (15 mpi, 4 hpi) and late (8 hpi, 12 hpi) stages of infection (Fig. 4). The control sample corresponding to the 16 hpi sample was potentially cross-contaminated with viral reads so, this time point was excluded from analysis (Supplementary Fig. 2). A gene ontology (GO) enrichment test was performed to identify overrepresented functions in the differentially expressed genes after removal of prokaryotic homologs.

Figure generated through DESeq2 output. The number of down- or upregulated genes shown in parentheses are DESeq2 DEGs cross-validated by edgeR. Colored points represent significant genes at FDR (false discovery rate) ≤ 0.05. FC fold change. 0.25 and 4 hpi represent early-stage samples, while 8 and 12 hpi represent late-stage samples.
A few differentially expressed genes were observed during the early stage (30 upregulated and 38 downregulated out of 63,860 genes). The upregulated gene set shows enriched GO terms for carbohydrate and lipid metabolism and endocytosis (mainly attributable to adapter protein complex 4; AP4E gene) (Figs. 4 and 5a, Supplementary Table 4). The downregulated gene set includes GO terms related to protein modification/transport/turnover (e.g., UPL6, E3 ubiquitin-protein ligase; SC61A, transport protein Sec61 subunit), cytoskeleton (e.g., Dynein heavy chain 7; DYH7) and chlorophyll catabolic process (e.g., NOL, Chlorophyll(ide) b reductase) (Fig. 4, Supplementary Fig. 5B, Supplementary Table 5).

Enriched GO (gene ontology) terms illustrating the functions that were either upregulated (a and c) or downregulated (b and d) during the early and late stages of the infection.
A much greater proportion of genes were differentially expressed in the late stage (875 upregulated and 256 downregulated) (Fig. 4). Among the genes upregulated were those with GO terms indicating involvement in immune and stress response, nucleotide, and lipid metabolism, methylation, RNA processing and transcription, ion transport, and protein modification (Fig. 5c, Supplementary Table 6). Interestingly, putative endogenous major capsid proteins were also upregulated in the Tetraselmis host (Figs. 4 and 5, Supplementary Fig 7). In the downregulated gene set, enriched GO terms included stress/defense response, plant-related morphogenesis, and cytoskeleton (Fig. 5d, Supplementary Table 7).
Discussion
With this study, we provide details of the infection cycle of TetV-1 when infecting a marine coastal strain of the green alga Tetraselmis sp. (UHM1315). Latent period and burst size are among the most important parameters to parameterize when modeling the spread of viral infections through phytoplankton populations37. However, they are not fixed traits. A virus that infects a cell under sub-optimal conditions (e.g., growth-limiting temperature, irradiance, or nutrient supply) may present a significantly longer latent period and reduced burst size38. Despite the context-dependence of these traits, our estimates for TetV-1 were very similar to those reported for its closest cultivated relative, Pyramimonas orientalis virus, or PoV-01B. Like TetV-1, PoV-01B is a green alga-infecting virus in the family Allomimiviridae. The latent period (14–19 h) and burst size (800–1000) reported for PoV-01B39 were indistinguishable from our findings for TetV-1, despite the former being assayed at lower temperature (15 °C) and irradiance (40 µmol photons per m−2 s−1). Undoubtedly, these values would change for either virus under more stringent growth limitations, but our objective in determining the latent period in this study was primarily to determine the endpoint of the virus life cycle as context for interpreting changes in ultrastructure and gene expression under the same conditions. Under our controlled conditions, the eclipse period accounted for more than a quarter, but less than half of the latent period, and provided a convenient visual break point for early and late stages of infection.
The appearance of VF in the cytoplasm and the persistent integrity of the nucleus during infection are similar to observations for some members of the families Mimiviridae and Poxviridae35,40. Moreover, the reduction in Tetraselmis chloroplast size during TetV-1 infection is similar to observations in Emiliania huxleyi when infected with EhV, but in the latter case, the nucleus was degraded32. VFs are also observed in other giant virus infections, including Acanthamoeba polyphaga mimivirus (APMV)41 and Tupanvirus42. The vesicular or angular shapes of apparent capsid components seen in TetV-1 virus factories are also evident in PBCV-1 infection43. However, the TetV-1 putative VF appears as an area of aggregation, but not as a well-defined electron-dense zone with clear boundaries as observed in APMV41. In APMV, the VF is the source of viral DNA and is associated with empty virus capsids44. The putative VF produced by TetV-1 is similar to structures that have also been referred to variously as viroplasm, replication organelles, assembly sites, or IBs in other virus-host systems45,46,47,48. VFs concentrate viral replication machinery49, which is proposed to protect viruses from host defenses45,48.
In addition to the VFs, we observed other structures we refer to generically as “inclusion bodies” (IBs), which is a broad term for protein aggregates found in various cells50,51. Here, we differentiate the TetV-1 IBs from putative VFs. It remains to be seen if the distinct IBs play a specific role in TetV-1 replication or if they are simply a cytopathic byproduct of the significant metabolic disruption caused by infections.
Transcription is the first major biosynthetic step after infection with a dsDNA virus. Immediate transcription is not unusual for giant viruses, with transcription happening within minutes in some cases28,52. For poxviruses, transcription initiates within virion core particles that maintain structural integrity upon entry18. Rapid transcription is possible because transcriptional proteins are packaged within the virion18,53,54. Like other NCLDVs such as AaV, APMV, PBCV-1, and CroV, all known TetV-1 genes, as well as the 167 multi-exon transcripts discovered here, are expressed at least once in the infection cycle.
Multi-exon transcripts have been described in other giant viruses: 26 splice junctions in Faustovirus25 and 133 junctions (in ~65 genes) in PBCV-126. The modest number of splice junctions reported for PBCV-1 compared to TetV-1 may simply reflect the limited window within which PBCV-1 transcription was surveyed (only 1 hpi). Similar to other Imitervirales, the TetV-1 MCP transcripts consist of 3 exons24. The number of multi-exon transcripts detected in TetV-1 is not so unusual when compared to some other dsDNA viruses, even those with considerably fewer genes. For example, Human adenovirus type 5 can reach up to 11,000 splicing patterns in ~38 genes55. It has been hypothesized that the presence of exons and introns in viruses can disrupt nuclease cleavage sites56 thereby providing protection from host nucleases, and this may be the case for giant viruses. Another possibility is that splicing does not benefit the virus but is a remnant of horizontally transferred genes (HGTs) that host spliceosomes recognize. Regardless of the selective forces that lead to their presence, the existence of multi-exon transcripts and alternatively spliced genes in TetV-1, and previous studies highlighting the rich alternative splicing repertoire of NCLDVs20,24,25, emphasize that gene annotations based solely on genome sequences miss much of the complexity that underlies viral replication.
Viral “early” genes are typically those that are involved in DNA replication, transcription, or translation, putative immune evasion, or nucleotide metabolism genes, as was the case for TetV-1. Also apparent at the early stages was the expression of enzymes that are likely involved in the destruction/degradation of host cell wall (alpha-galactosidase, TetV_601; glycosyl hydrolase, TetV_615), virion-cell recognition (arabinose 5-phosphate isomerase, TetV_403) or controlling intracellular osmolarity (mannitol 1-phosphate dehydrogenase M1PHD, TetV_320)57 (Fig. 3). Mannitol has also been shown to suppress ROS-based defense (reactive oxygen species) against pathogens58, thus it is possible that the virus is increasing the mannitol pool to counter host defenses. The early expression of nucleotide metabolism genes such as ribonucleotide reductase, rNDP (TetV_458, 533), thymidylate kinase (TetV_225), and thymidylate synthase complementing protein (TetV_240) is likely a response to the increased nucleotide demands of the virocell. The expression of viral RNA polymerase subunits suggests that, like other NCLDVs such as ASFV and poxvirus18,59, viral transcription likely relies on these viral-encoded enzymes. However, the host RNA polymerase is also constitutively expressed (Supplementary Table 8). Whether the virus relies exclusively on its own RNA polymerase or takes advantage of host polymerase activity is unknown.
Other “early” genes may be involved in evading the host’s innate immune response. The putative immune suppression gene, vMISTRA (viral homolog of MItochondria STress Response Algae, TetV_113), is hypothesized to counteract viral-induced apoptosis60. In addition, RNAse III (ribonuclease III) is known for its role in gene silencing with the potential to shift virocell global gene expression. Ubiquitination proteins (TetV_035, 358, 621) and other nucleases may play a role in keeping host proteins and DNA at bay (Fig. 3). Various RING ___domain-containing proteins implicated in apoptosis inhibition, are also expressed early (TetV_27, 420, 434) (Fig. 3). This is the case for the Ascovirus IAPs (Inhibitors of Apoptosis) gene which encodes an E3 ubiquitin ligase with RING ___domain61. TetV-1 encodes multiple copies of RING ___domain-containing genes that might represent gene duplications. Expansions of viral genetic content through gene duplication (i.e., a “gene accordion”) were suggested to be a mechanism by which poxviruses adapt to host antiviral defenses, as new, duplicated genes can freely acquire mutations that facilitate virus evasion of the cellular immune system62. RNAse III and ubiquitin-related proteins have non-immune evasion functions. RNAse III plays a role in gene regulation and ribosomal RNA processing63 while ubiquitin is utilized for general protein homeostasis and signaling64,65, so their roles in immune evasion is entirely speculative at this point.
The classification of viral gene temporal classes allows examination of mechanisms that govern coordinated expression. Indeed, a motif search identified potential promoters enriched within temporal groups (Fig. 3A). These motifs are different from those discovered in EhV23 or ASFV19. Whether these putative promoters are functional requires experimental evidence beyond an in-silico survey. Furthermore, spatial viral genome regulation is observed in EhV wherein a specific subregion (EhV218-366) is highly expressed at 1 hpi32, and in PBCV-1, the top 50 expressed genes at 7 min post-infection are also co-located. However, spatial clustering and temporally coordinated expression are not observed in other NCLDVs, such as the Marseillevirus29.
While the early phase of infection can be characterized as the preparation stage for viral production, the late stage is characterized by transcripts suggesting sustained biosynthesis of materials needed for viral assembly and egress. The late-stage transcripts are structural proteins, DNA packaging materials, transcripts/proteins used for egress, or transcripts/proteins that may be packaged in virus particles, among others.
Strikingly, most of the highly expressed late genes have unknown functions. These hypothetical genes/proteins that are co-expressed with the major capsid protein in high relative abundance in both transcriptome and virion proteome (TetV_511 and 630) are likely structural and excellent candidates for further functional characterization (Fig. 3, Supplementary Table 1)66. It is also apparent that there are genes with unknown functions that display low to moderate gene expression (Supplementary Table 1) but are found in TetV-1 virion proteome, such as TetV_155, 247, 258, 362, 378, 412, 463, 481, and 59766. This could also be subject to further functional screening. Various transmembrane proteins and ion channels are also expressed late (TetV_029, 044, 199) (Fig. 3). These proteins are likely involved in controlling cellular ion concentration or pH and could be involved in either virus entry or exit, depending on whether they are used in late-stage infection or packaged inside the virions. If a given transmembrane protein is used in the late stage, its function might be to alter cell osmotic pressure, facilitating exit. If packaged inside the virions, an ion channel may act at the early stage of infection in a manner similar to that observed for PBCV-1, where the potassium ion channel triggers depolarization and loss of ions, resulting in lower cellular pressure and triggering injection of viral DNA into the host67,68. Here, among the three transmembrane proteins/ion channels, only the large-conductance mechanosensitive channel (TetV_044) seems to be packaged inside the virions, based on the virion proteome66. It is also possible that ion channels are crucial in controlling a balance of intracellular ions favorable for viral protein activity or mediating calcium/potassium-dependent signaling36,69.
The expression of thioredoxin in the late stage (Supplementary Fig. 4) can be attributed to its role in regulating the cell redox state associated with cell death70,71. In addition, late-stage methyltransferases (TetV_211, 484, 501, 608) might be responsible for the methylation of newly synthesized viral DNA (Supplementary Fig. 4). The sulfatase (TetV_008) that is highly expressed as transcripts in the late stage and has a high signal in the virion proteome66 suggests the packaging of this specific enzyme (Supplementary Fig. 4). This enzyme can play a role in host surface or cytoplasm remodeling, as it catabolizes sulfated algal polysaccharides and glycolipids72.
Here, we used conventional bulk RNA-seq, which can be considered as the average state of the virocell transcriptome. Because not all cells are infected at precisely the same time, there is some loss of resolution in the timing of expression when bulk sampling, as illustrated by single-cell transcriptomics of E. huxleyi-EhV23. Furthermore, other RNA sequencing approaches could offer additional insights. For instance, CAGE-seq and 3′ RNA-seq will be required to ascertain transcriptional start and termination sites19. Despite these limitations, our transcriptome analysis allowed the delineation of gene boundaries, the identification of spliced transcripts, and the determination of TetV-1 genes and their temporal profiles.
This study demonstrated host responses to infection by analyzing differential gene expression and GO enrichment analysis. These analyses paint a broad picture of host metabolic reprogramming in response to the virus attack. Host reprogramming can be accomplished through various mechanisms, such as viral-encoded nucleases that preferentially degrade host mRNAs, viral methylases and epigenetic modification systems, and virus-encoded transcriptional and post-transcriptional regulatory controls that affect host gene expression, among others. Here, we cannot ascertain the mechanism of host reprogramming, but nonetheless observed relative up- or downregulation of host genes as a response to giant virus infection. GO enrichment test was done after removal of prokaryotic homologs, which could represent HGTs or simply contaminating prokaryotic sequences included from the non-axenic cultures despite poly-A-enrichment during RNA-seq library preparation. Green algae have been shown to contain a higher proportion of prokaryotic HGTs accounting for 7847 genes (2.73%) for Bathycoccus prasinos73 and 9851 genes (2.97%) for Cococmyxa subellipsoidea74 (reviewed in ref. 75). However, we cannot ascertain that these are indeed prokaryotic HGTs; thus, we only used the eukaryotic homologs for downstream analysis.
Few host genes showed significant changes in expression during the early stage (68 genes out of 63,860 Tetraselmis genes). A host gene related to endocytosis, adapter protein complex 4 subunit epsilon (AP4E), was upregulated. Adapter protein complexes are host factors that interact with viruses to facilitate entry [reviewed in ref. 76]. A related NCLDV, Vaccinia virus, for example, was shown to interact with its host’s adapter protein complex 2 (AP2)77. This circumstantial evidence suggests that AP4 may act as a critical host factor for TetV-1 infection, although confirmatory tests are needed. A relatively higher number of host genes (1131) are differentially expressed in the late stage. During this stage, we observed extensive changes in the ultrastructure, so, unsurprisingly, many cellular processes are disrupted relative to the control. Putative endogenous MCPs are upregulated at the late stage, which agrees with a recent survey that shows that protists, including various Tetraselmis species, express endogenous MCPs78. We are unsure if these are functional capsids or are degraded or repurposed genes that retained their transcriptional signals and are thus expressed upon infection. The lipid metabolism genes upregulated in both early and late stages are presumably associated with cytoplasmic rearrangement, creating lipid-rich virus factories46. Downregulated host genes across the infection include those associated with the cytoskeleton, morphogenesis, and development, which seems consistent with the disruption of cellular structure in favor of creating virus factories. It is worth noting, the constitutive expression of both viral and host-encoded fermentation genes (PFL and PLFA). A host version of the pyruvate formate lyase (PFL) was not detected in the transcriptome; however, Tetraselmis striata, the only Tetraselmis species with a published genome, contains a PFL (Protein ID: 421160; TSEL_012203.t1)79. Hence, it is possible that PFL is also present in Tetraselmis sp. KB-FL45, but is below the detection limit of our sequencing approach.
In conclusion, by tracking changes in the transcriptional activity and ultrastructure of a green alga infected with an allomimivirus, several broad patterns emerged consistent with responses to infection seen in other NCLDVs and viruses in general. Specifically, viral genes expressed early in infection were those involved with carbohydrate degradation, putative immune evasion, transcription initiation, and nucleotide metabolism, among others. In contrast, viral genes expressed later were structural proteins and DNA packaging materials. Transcriptional changes in host genes in response to infection suggested metabolic reprogramming to accommodate the production of virus factories and disruption of host metabolism and development. The transcriptional data from this study expands and improves the TetV-1 genome annotation while highlighting that many of the highly expressed viral genes have unknown functions. These would be good candidates for functional characterization to further improve our understanding of how giant viruses commandeer the host metabolic machinery and facilitate their replication.
Methods
Virus and algal culture
The virus Tetraselmis virus 1, or TetV-1 for short16, was isolated from Kāneʻohe Bay (21.42973° N, 157.7919° W), a coastal embayment on the northeast shore of Oʻahu, Hawaiʻi that is protected by a barrier reef. A unicellular green alga Tetraselmis sp. AL-FL03 (UHM1300) isolated from open ocean waters of Station ALOHA (22.75° N, 158.00° W) in the North Pacific Subtropical Gyre was the initial host used to isolate and propagate the virus, but other strains of Tetraselmis isolated from Station ALOHA and Kāneʻohe Bay are susceptible and permissive to the virus16. In this study, a strain from Kāneʻohe Bay, Tetraselmis sp. KB-FL45 (UHM1315) was used for the experiments. The uni-algal (but not axenic) culture was maintained in an f/2 medium80,81 at 24 °C on a cycle of 12 h light:12 h dark with a photon flux of 250–280 µE m−2 s−1 during the light period (07:30–19:30 h). The virus was recently assigned to a new genus and species, Oceanusvirus kaneohense, within the family Allomimiviridae82.
Flow cytometry counts
Algal cells and virions were counted by flow cytometry (AttuneTM NxT; Thermo Fisher Scientific) using side scatter (SSC) and either chlorophyll autofluorescence (algae) or DNA fluorescence after staining with SYBR Green I (Thermo Fisher Scientific) for ~30 min (virions) to discriminate populations. Autoclaved, 0.2 µm-filtered seawater was used as a blank to verify that background events in the algae and virion windows were minimal for the flow cytometer analyses. TetV-1 counting followed the previously published protocol using green fluorescence and side-scatter83.
Latent period and burst size estimation
For latent period determination, triplicate algal cultures in the exponential growth phase (238 mL each at 1.2 × 105 cells mL−1) were each inoculated 4.5 h into the light period with fresh, filtered (0.45 µm, Sterivex, Millipore) lysate (~8 mL at ~1.8 × 108 virions mL−1) resulting in a virion-to-host cell ratio of ~50:1. After inoculation, cultures were subsampled periodically up to 30 hpi. The cultures, both infected and control, were mixed by swirling gently before sampling. Subsamples (1 mL) were preserved with glutaraldehyde (0.5% v:v final conc.), flash frozen in liquid nitrogen, then stored at −80 °C. Samples were thawed immediately prior to flow cytometric analysis, as described above. Burst size estimates (viruses produced per cell) were calculated by dividing the change in viral counts before (15 h) and after (30 h) the latent period by the host counts at the beginning of infection. We made the limiting assumption that all cells contributed to viral production because cell debris and clumping at the end of the latent period precluded accurate quantification of any residual unlysed cells. The burst size estimate may, therefore, be somewhat underestimated.
Experiment for ultrastructural and transcriptional analysis
Guided by data from the first experiment, a second infection experiment was conducted to analyze changes in ultrastructure and transcriptional activity through the latent period. Four independent algal cultures (1350 mL each) were initiated (three experimental and one uninfected control) and grown to exponential phase (~1.48 × 105 cells mL−1) under the routine conditions noted above. Approximately 100 mL of filtered virus lysate at ~1.0 × 108 virions mL−1 was added to the three experimental cultures at noon (designated as t = 0 h) for a virion-to-host ratio of ~50 as above, and incubations continued for another 20 h. Subsamples were collected from each culture at t = −1, 0.25, 4, 8, 12, 16, and 20 hpi for analyses as follows. One milliliter of the subsample was fixed with glutaraldehyde, frozen, and stored as described above for flow cytometry. Fifty milliliters (50 mL) of the subsample were preserved and processed for electron microscopy, as described below. Another 50 mL was immediately filtered through a 2.0 µm polycarbonate filter by vacuum filtration to collect the cells. The filters were transferred to a 2 mL cryovial, flash frozen, and stored at −80 °C for subsequent RNA extraction, as described below.
Transmission electron microscopy (TEM)
Samples for electron microscopy were preserved with a fixative (final concentration: 0.1 M sodium cacodylate, 2% glutaraldehyde, 0.005 M CaCl2, 0.06 g glucose/mL). Cells were pelleted at 2400 × g and embedded in ~3.3% agarose gel. The embedded samples were washed twice with 0.1 M cacodylate buffer with 0.44 M sucrose for 20–30 min. The samples were then post-fixed with 1% osmium tetroxide in 0.1 M cacodylate buffer for 1 h. Post-fixed samples were dehydrated twice in a graded ethanol series (30%, 50%, 70%, 85%, 95%) for each dilution and thrice in 100% ethanol for 10 min each. Samples were then substituted with propylene oxide thrice for 10 min each and then infiltrated with 1:1 propylene oxide:epoxy resin (LX112) overnight. They were then immersed in a freshly made 100% epoxy resin for 2 h, followed by another exchange for 3 h. Finally, the samples were placed in molds with epoxy resin and polymerized at 60 °C for 2–3 days. Ultrathin (60–80 nm) sections were generated on an RMC MT6000-XL ultramicrotome, viewed unstained on a Hitachi HT7700 TEM at 100 kV, and photographed with an AMT XR-41B 2k × 2k CCD camera. The EM image contrast was adjusted for better clarity.
RNA extraction and sequencing
Total RNA was extracted from cells collected on filters using a Zymo Direct-zol RNA miniprep kit following the manufacturer’s protocol with the addition of bead beating with ZR bashing beads at the TRIzol lysis step. RNA integrity was checked with gel electrophoresis. RNA concentration was determined by fluorescence using a Qubit RNA High Sensitivity kit (Thermo Fisher Scientific). Quality control using RNA ScreenTape, library preparations, poly-A selection, and Illumina sequencing (HiSeq 2 × 150 bp) was performed by a commercial facility (Azenta Life Sciences, NJ, USA). The triplicate infected samples at each time point were pooled equally into one sequencing library. This translates to 12 sequencing libraries with one experimental and one control library for each of six time points (−1, 0.25, 4, 8, 12, and 16 hpi).
Virus transcriptome assembly and analysis
Low-quality sequencing reads (average quality scores < Q15) were removed. Adapters and low-quality bases were trimmed from the ends of remaining sequences with Trimommatic84 using the following parameters (IlluminaClip:TruSeq3-PE-2.fa:2:30:10:1:true Leading:30 Trailing:30 SlidingWindow:4:15 MinLen:30) and checked with FastQC85. Genome-guided transcriptome assembly was implemented using the Tuxedo protocol (HISAT2, StringTie, and Ballgown)86,87,88. Briefly, the RNA-seq paired-end reads were aligned to the TetV-1 genome using HISAT2. StringTie assembled and quantified expressed genes and transcripts and merged all transcripts from all samples to create a reference set. These Tuxedo-based transcripts were compared to the TetV-1 reference annotation based on Prodigal16. The transcript abundance was reported as TPM (Transcripts per Million). We report normalized TPM to further examine viral gene clusters as opposed to log scale visualization that might ignore relatively small but significant changes. The normalized TPM for a transcript is calculated by dividing its TPM by the total TPM of that transcript across all time points. To identify potential promoters driving gene expression, we extracted a 200 bp region upstream of the transcription start site using BEDTools89. Transcripts were then grouped based on their expression patterns. Promoter motifs enriched within the different groups were identified using MEME90. In addition, enriched motifs per group relative to the whole set were performed using STREME91.
Host transcriptome assembly and analysis
The genome of Tetraselmis sp. KB-FL45 has not been sequenced, so de novo transcriptome assembly was implemented using Trinity92 after removal of TetV-1 reads using Bowtie293. To assess the completeness of the transcriptome, BUSCO was used to detect Chlorophyta single-copy genes94. To ensure quality of transcripts, only those that passed TransRate metrics were retained for further analysis95. TransRate evaluates de novo assemblies based on read mapping evidence to detect chimeras, structural and base errors, and incomplete assemblies. Transcript and gene quantification were done using RSEM (RNA-seq by Expectation-Maximization)96, whereas differential expression (DE) analysis was performed using DESeq2 in Trinity97. Another DE analysis was performed per time point using edgeR98 to cross-validate the DESeq2 output (Supplementary Fig 8). Coding sequence identification and functional annotation were performed using TransDecoder and Trinotate99, respectively, using the following databases: UniProt SwissProt100 (Feb. 2021 release; accessed 04/09/21), Pfam101 (v34.0; accessed 04/09/21), GO102,103 (Feb. 2021 release; accessed 04/09/21), Eggnog terms104 (v4.5; accessed 04/09/21). Before performing the enrichment analysis, we removed prokaryotic homologs from our DEGs to eliminate potential bacterial contaminants that may have escaped poly-A selection. Furthermore, we retained DESeq2-identified DEGs only if they were statistically significant (FDR ≤ 0.05, using an edgeR dispersion value of 0.4 for increased stringency) and if they were also identified as common DEGs by edgeR for the corresponding infection stage (early stage: DEGs common to 15 mpi and 4 hpi; late stage: DEGs common to 8 and 12 hpi). Finally, to assess the enriched functional categories in DEGs, GO enrichment analysis was performed using topGO.
Data availability
The raw sequencing reads used in this paper have been deposited in the NCBI Short Read Archive as BioProject PRJNA1102674. The host transcriptome assembly, annotation, and differential expression data are available at FigShare (https://figshare.com/s/98d2742f5f1c1f3bf6fb, and https://figshare.com/s/aad55ae781891e083b09). All other data are available as supplementary material.
Code availability
Codes used in this study are deposited in GitHub https://github.com/andriangajigan/TetV_RNA/.
References
Koonin, E. V. & Yutin, N. Evolution of the Large Nucleocytoplasmic DNA Viruses Of Eukaryotes And Convergent Origins Of Viral Gigantism. Adv. Virus Res. 103, 167–202 (2019).
Legendre, M. et al. Thirty-thousand-year-old distant relative of giant icosahedral DNA viruses with a pandoravirus morphology. Proc. Natl. Acad. Sci. USA 111, 4274 (2014).
Philippe, N. et al. Pandoraviruses: Amoeba viruses with genomes up to 2.5 Mb reaching that of parasitic eukaryotes. Science 341, 281–286 (2013).
Aylward, F. O. & Moniruzzaman, M. Viral complexity. Biomolecules 12, 1061 (2022).
Moniruzzaman, M. et al. Virologs, viral mimicry, and virocell metabolism: the expanding scale of cellular functions encoded in the complex genomes of giant viruses. FEMS Microbiol. Rev. 47, fuad053 (2023).
Abrahão, J. et al. Tailed giant Tupanvirus possesses the most complete translational apparatus of the known virosphere. Nat. Commun. 9, 749 (2018).
Andreani, J. et al. Morphological and genomic features of the new Klosneuvirinae isolate Fadolivirus IHUMI-VV54. Front. Microbiol. 12, 719703 (2021).
Needham, D. M. et al. A distinct lineage of giant viruses brings a rhodopsin photosystem to unicellular marine predators. Proc. Natl. Acad. Sci. USA 116, 20574–20583 (2019).
Santini, S. et al. Genome of Phaeocystis globosa virus PgV-16T highlights the common ancestry of the largest known DNA viruses infecting eukaryotes. Proc. Natl. Acad. Sci. USA 110, 10800–10805 (2013).
Blanc-Mathieu, R. et al. A persistent giant algal virus, with a unique morphology, encodes an unprecedented number of genes involved in energy metabolism. J. Virol. 95, 10–1128 (2021).
Moniruzzaman, M., Martinez-Gutierrez, C. A., Weinheimer, A. R. & Aylward, F. O. Dynamic genome evolution and complex virocell metabolism of globally-distributed giant viruses. Nat. Commun. 11, 1–11 (2020).
Boyer, M. et al. Giant Marseillevirus highlights the role of amoebae as a melting pot in emergence of chimeric microorganisms. Proc. Natl. Acad. Sci. USA 106, 21848 (2009).
Yoshikawa, G. et al. Medusavirus, a novel large DNA virus discovered from hot spring water. J. Virol. 93, 25 (2019).
Da Cunha, V. et al. Giant viruses encode actin-related proteins. Mol. Biol. Evol. 39, msac022 (2022).
Ha, A. D., Moniruzzaman, M. & Aylward, F. O. High transcriptional activity and diverse functional repertoires of hundreds of giant viruses in a coastal marine system. mSystems 6, 15 (2021).
Schvarcz, C. R. & Steward, G. F. A giant virus infecting green algae encodes key fermentation genes. Virology 518, 423–433 (2018).
Claverie, J.-M. & Abergel, C. Mimivirus and its virophage. Annu. Rev. Genet. 43, 49–66 (2009).
Broyles, S. S. Vaccinia virus transcription. J. Gen. Virol. 84, 2293–2303 (2003).
Cackett, G. et al. The African swine fever virus transcriptome. J. Virol. 94, e00119–e00120 (2020).
Legendre, M. et al. mRNA deep sequencing reveals 75 new genes and a complex transcriptional landscape in Mimivirus. Genome Res. 20, 664–674 (2010).
Legendre, M., Santini, S., Rico, A., Abergel, C. & Claverie, J.-M. Breaking the 1000-gene barrier for Mimivirus using ultra-deep genome and transcriptome sequencing. Virol. J. 8, 99 (2011).
Aherfi, S. et al. A large open pangenome and a small core genome for giant pandoraviruses. Front. Microbiol. 9, 1486 (2018).
Ku, C. et al. A single-cell view on alga-virus interactions reveals sequential transcriptional programs and infection states. Sci. Adv. 6, eaba4137 (2020).
Boratto, P. V. M. et al. Analyses of the Kroon virus major capsid gene and its transcript highlight a distinct pattern of gene evolution and splicing among Mimiviruses. J. Virol. 92, e01782–17 (2018).
Cherif Louazani, A., Baptiste, E., Levasseur, A., Colson, P. & La Scola, B. Faustovirus E12 transcriptome analysis reveals complex splicing in capsid gene. Front. Microbiol. 9, 2534 (2018).
Blanc, G. et al. Deep RNA sequencing reveals hidden features and dynamics of early gene transcription in Paramecium bursaria Chlorella Virus 1. PLoS ONE 9, e90989 (2014).
Derelle, E., Yau, S., Moreau, H. & Grimsley, N. H. Prasinovirus attack of Ostreococcus is furtive by day but savage by night. J. Virol. 92, 16 (2018).
Moniruzzaman, M., Gann, E. R. & Wilhelm, S. W. Infection by a giant virus (AaV) induces widespread physiological reprogramming in Aureococcus anophagefferens CCMP1984—a harmful bloom algae. Front. Microbiol. 9, 752 (2018).
Rodrigues, R. A. L. et al. Analysis of a Marseillevirus transcriptome reveals temporal gene expression profile and host transcriptional shift. Front. Microbiol. 11, 651 (2020).
Bachy, C. et al. Transcriptional responses of the marine green alga Micromonas pusilla and an infecting prasinovirus under different phosphate conditions: host-virus interactions under varied nutrients. Environ. Microbiol. 20, 2898–2912 (2018).
Rowe, J. M. et al. Global analysis of chlorella variabilis NC64A mRNA profiles during the early phase of Paramecium bursaria Chlorella Virus-1 Infection. PLoS ONE 9, e90988 (2014).
Rosenwasser, S. et al. Rewiring host lipid metabolism by large viruses determines the fate of Emiliania huxleyi, a bloom-forming alga in the ocean. Plant Cell 26, 2689–2707 (2014).
Ghigo, E. et al. Ameobal pathogen Mimivirus infects macrophages through phagocytosis. PLoS Pathog. 4, e1000087 (2008).
Rodrigues, R. A. L., Abrahão, J. S., Drumond, B. P. & Kroon, E. G. Giants among larges: how gigantism impacts giant virus entry into amoebae. Curr. Opin. Microbiol. 31, 88–93 (2016).
Mutsafi, Y., Zauberman, N., Sabanay, I. & Minsky, A. Vaccinia-like cytoplasmic replication of the giant Mimivirus. Proc. Natl. Acad. Sci. USA 107, 5978–5982 (2010).
Kukovetz, K. et al. A functional K+ channel from Tetraselmis Virus 1, a member of the Mimiviridae. Viruses 12, 1107 (2020).
Flynn, K. J. et al. Modelling the effects of traits and abiotic factors on viral lysis in phytoplankton. Front. Mar. Sci. 8, 667184 (2021).
Zimmerman, A. E. et al. Metabolic and biogeochemical consequences of viral infection in aquatic ecosystems. Nat. Rev. Microbiol. 18, 21–34 (2020).
Sandaa, R.-A., Heldal, M., Castberg, T., Thyrhaug, R. & Bratbak, G. Isolation and characterization of two viruses with large genome size infecting Chrysochromulina ericina (Prymnesiophyceae) and Pyramimonas orientalis (Prasinophyceae). Virology 290, 272–280 (2001).
Mutsafi, Y., Fridmann-Sirkis, Y., Milrot, E., Hevroni, L. & Minsky, A. Infection cycles of large DNA viruses: emerging themes and underlying questions. Virology 466–467, 3–14 (2014).
Suzan-Monti, M., Scola, B. L., Barrassi, L., Espinosa, L. & Raoult, D. Ultrastructural characterization of the giant volcano-like virus factory of Acanthamoeba polyphaga Mimivirus. PLoS ONE 2, e328 (2007).
Silva, L. C. F. et al. Microscopic Analysis Of The Tupanvirus Cycle in Vermamoeba vermiformis. Front. Microbiol. 10, 671 (2019).
Meints, R. H., Lee, K. & Van Etten, J. L. Assembly site of the virus PBCV-1 in a Chlorella-like green alga: ultrastructural studies. Virology 154, 240–245 (1986).
Zauberman, N. et al. Distinct DNA exit and packaging portals in the virus Acanthamoeba polyphaga mimivirus. PLoS Biol. 6, e114 (2008).
de Castro, I. F., Volonté, L. & Risco, C. Virus factories: biogenesis and structural design: Biogenesis and architecture of virus factories. Cell. Microbiol. 15, 24–34 (2013).
Netherton, C. L. & Wileman, T. Virus factories, double membrane vesicles and viroplasm generated in animal cells. Curr. Opin. Virol. 1, 381–387 (2011).
Novoa, R. R. et al. Virus factories: associations of cell organelles for viral replication and morphogenesis. Biol. Cell 97, 147–172 (2005).
Sagan, S. M. & Weber, S. C. Let’s phase it: viruses are master architects of biomolecular condensates. Trends Biochem. Sci. 48, 229–243 (2023).
Katsafanas, G. C. & Moss, B. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe 2, 221–228 (2007).
García-Fruitós, E., Sabate, R., De Groot, N. S., Villaverde, A. & Ventura, S. Biological role of bacterial inclusion bodies: a model for amyloid aggregation. FEBS J. 278, 2419–2427 (2011).
Kopito, R. R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10, 524–530 (2000).
Kawasaki, T., Tanaka, M., Fujie, M., Usami, S. & Yamada, T. Immediate early genes expressed in chlorovirus infections. Virology 318, 214–223 (2004).
Fischer, M. G., Kelly, I., Foster, L. J. & Suttle, C. A. The virion of Cafeteria roenbergensis virus (CroV) contains a complex suite of proteins for transcription and DNA repair. Virology 466–467, 82–94 (2014).
Renesto, P. et al. Mimivirus giant particles incorporate a large fraction of anonymous and unique gene products. J. Virol. 80, 11678–11685 (2006).
Donovan-Banfield, I., Turnell, A. S., Hiscox, J. A., Leppard, K. N. & Matthews, D. A. Deep splicing plasticity of the human adenovirus type 5 transcriptome drives virus evolution. Commun. Biol. 3, 124 (2020).
Chelkha, N., Levasseur, A., La Scola, B. & Colson, P. Host–virus interactions and defense mechanisms for giant viruses. Ann. N. Y. Acad. Sci. 1486, 39–57 (2021).
Sand, M. et al. Mannitol-1-phosphate dehydrogenases/phosphatases: a family of novel bifunctional enzymes for bacterial adaptation to osmotic stress. Environ. Microbiol. 17, 711–719 (2015).
Meena, M., Prasad, V., Zehra, A., Gupta, V. K. & Upadhyay, R. S. Mannitol metabolism during pathogenic fungal–host interactions under stressed conditions. Front. Microbiol. 6, 1019 (2015).
Rodríguez, J. M. & Salas, M. L. African swine fever virus transcription. Virus Res. 173, 15–28 (2013).
Sorouri, M., Chang, T., Jesudhasan, P., Pinkham, C., Elde, N. C. & Hancks, D. C. Signatures of host–pathogen evolutionary conflict reveal MISTR—A conserved MItochondrial STress Response network. PLOS Biology, 18, e3001045 (2020).
Zaghloul, H. A. H., Hice, R., Arensburger, P. & Federici, B. A. Transcriptome analysis of the Spodoptera frugiperda Ascovirus in vivo provides insights into how its apoptosis inhibitors and caspase promote increased synthesis of viral vesicles and virion progeny. J. Virol. 91, 10–1128 (2017).
Elde, N. C. et al. Poxviruses deploy genomic accordions to adapt rapidly against host antiviral defenses. Cell 150, 831–841 (2012).
Lejars, M., Kobayashi, A. & Hajnsdorf, E. RNase III, ribosome biogenesis and beyond. Microorganisms 9, 2608 (2021).
Damgaard, R. B. The ubiquitin system: from cell signalling to disease biology and new therapeutic opportunities. Cell Death Differ. 28, 423–426 (2021).
Komander, D. & Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 81, 203–229 (2012).
Schvarcz, C. R. Cultivation and characterization of viruses infecting eukaryotic phytoplankyon from the Tropical North Pacific Ocean. (Doctoral dissertation, University of Hawai‘i at Mānoa, 2018).
Lee, S.-W., Lee, E.-H., Thiel, G., Van Etten, J. L. & Saraf, R. F. Noninvasive measurement of electrical events associated with a single chlorovirus infection of a microalgal cell. ACS Nano 10, 5123–5130 (2016).
Thiel, G., Moroni, A., Dunigan, D. & Etten, J. L. Initial events associated with virus PBCV-1 infection of Chlorella NC64A. in Progress in Botany Vol. 71 (eds Lüttge, U. E., Beyschlag, W., Büdel, B. & Francis, D.) 169–183 (Springer Berlin Heidelberg, 2010).
Zhou, Y., Frey, T. K. & Yang, J. J. Viral calciomics: interplays between Ca2+ and virus. Cell Calcium 46, 1–17 (2009).
Ueda, S. et al. Redox control of cell death. Antioxid. Redox Signal. 4, 405–414 (2002).
Masutani, H., Ueda, S. & Yodoi, J. The thioredoxin system in retroviral infection and apoptosis. Cell Death Differ. 12, 991–998 (2005).
Hettle, A. G., Vickers, C. J. & Boraston, A. B. Sulfatases: critical enzymes for algal polysaccharide processing. Front. Plant Sci. 13, 837636 (2022).
Moreau, H. et al. Gene functionalities and genome structure in Bathycoccus prasinos reflect cellular specializations at the base of the green lineage. Genome Biol. 13, R74 (2012).
Blanc, G. et al. The genome of the polar eukaryotic microalga Coccomyxa subellipsoidea reveals traits of cold adaptation. Genome Biol. 13, R39 (2012).
Van Etten, J. & Bhattacharya, D. Horizontal gene transfer in eukaryotes: Not if, but how much?. Trends Genet. 36, 915–925 (2020).
Geljic, I. S. et al. Viral interactions with adaptor-protein complexes: a ubiquitous trait among viral species. Int. J. Mol. Sci. 22, 5274 (2021).
Husain, M. & Moss, B. Role of receptor-mediated endocytosis in the formation of vaccinia virus extracellular enveloped particles. J. Virol. 79, 4080–4089 (2005).
Bellas, C. et al. Large-scale invasion of unicellular eukaryotic genomes by integrating DNA viruses. Proc. Natl. Acad. Sci. USA 120, e2300465120 (2023).
Steadman Tyler, C. R. et al. High-quality draft genome sequence of the green alga Tetraselmis striata (Chlorophyta) generated from PacBio sequencing. Microbiol. Resour. Announc. 8, e00780-19 (2019).
Guillard, R. R. L. Culture of Phytoplankton for Feeding Marine Invertebrates. in Culture of Marine Invertebrate Animals: Proceedings—1st Conference on Culture of Marine Invertebrate Animals Greenport (eds Smith, W. L. & Chanley, M. H.) 29–60. https://doi.org/10.1007/978-1-4615-8714-9_3 (Springer US, 1975).
Guillard, R. R. L. & Ryther, J. H. Studies of marine planktonic diatoms: I. Cytotella nana Hustedt, and Detonula confervacea (Cleve) Gran. Can. J. Microbiol. 8, 229–239 (1962).
Aylward, F. O. et al. Taxonomic update for giant viruses in the order Imitervirales (phylum Nucleocytoviricota). Arch. Virol. 168, 283 (2023).
Brussaard, C. P. D., Payet, J. P., Winter, C. & Weinbauer, M. G. Quantification of aquatic viruses by flow cytometry. In Manual of Aquatic Viral Ecology (eds Wilhelm, S. W., Weinbauer, M. G. & Suttle, C. A.) 102–109 (ASLO, 2010).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina Sequence Data. Bioinformatics https://doi.org/10.1093/bioinformatics/btu170 (2014).
Andrew, S. FastQC: a quality control tool for high throughput sequence data. (2010).
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667 (2016).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Frazee, A. C. et al. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat. Biotechnol. 33, 243–246 (2015).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Bailey, T. L., Williams, N., Misleh, C. & Li, W. W. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 34, W369–W373 (2006).
Bailey, T. L. STREME: accurate and versatile sequence motif discovery. Bioinformatics 37, 2834–2840 (2021).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: Assessing genome assembly and annotation completeness. in Gene Prediction Vol. 1962 (ed. Kollmar, M.) 227–245 (Springer New York, 2019).
Smith-Unna, R., Boursnell, C., Patro, R., Hibberd, J. M. & Kelly, S. TransRate: reference-free quality assessment of de novo transcriptome assemblies. Genome Res. 26, 1134–1144 (2016).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. 16 (2011).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Bryant, D. M. et al. A tissue-mapped axolotl de novo transcriptome enables identification of limb regeneration factors. Cell Rep. 18, 762–776 (2017).
The UniProt Consortium UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169 (2017).
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230 (2014).
The Gene Ontology Consortium et al. The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 49, D325–D334 (2021).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
Huerta-Cepas, J. et al. EGGNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293 (2016).
Acknowledgements
This project is funded by the US National Science Foundation to K.F.E. and G.F.S. (OCE #1559356 and OIA #1736030), and a Simons Foundation Early Career Award to K.F.E. We are indebted to Tina Carvalho of the Biological Electron Microscope Facility, Pacific Bioscience Research Center, for her assistance with the transmission electron microscopy. The computational work in this paper was implemented at the University of Hawai‘i High Performance Computer. The technical support and advanced computing resources from the University of Hawaii Information Technology Services—Cyberinfrastructure, funded partly by the National Science Foundation MRI award # 1920304, are gratefully acknowledged. We thank Feresa Cabrera for assisting with one of the time series. We are also grateful to Rosie Alegado for valuable suggestions on improving this manuscript. This work is supported by the Hawai‘i Institute of Geophysics and Planetology Denise B. Evans Fellowship, CMAIKI (Center for Microbiome Analysis and Investigation Graduate Research Competition Fund, UH Mānoa), and Uehiro Center for the Advancement of Oceanography Graduate Student Fellowship to A.P.G. (UCAO contribution number 19).
Author information
Authors and Affiliations
Contributions
A.P.G., C.R.S. and G.F.S. conceived of the paper. A.P.G. and C.R.S. performed the experiments. All authors (A.P.G., C.R.S., C.C., K.F.E., G.F.S.) contributed to the data analysis, writing, and approving the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gajigan, A.P., Schvarcz, C.R., Conaco, C. et al. Ultrastructural and transcriptional changes during a giant virus infection of a green alga. npj Viruses 3, 47 (2025). https://doi.org/10.1038/s44298-025-00128-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s44298-025-00128-7
This article is cited by
-
A deep dive into giant viruses
npj Viruses (2025)
