
Abstract
Modern approaches to copper-mediated radiolabeling have proven an important addition to the radiochemical toolbox. Radiopharmaceuticals prepared using this methodology have been translated from preclinical PET studies into clinical trials, and it has been adapted for radionuclides beyond fluorine-18, enabling theranostic applications. The methodology is also beginning to benefit from AI-assisted radiochemistry development. This perspective discusses the history, state-of-the-art, and potential future impact of copper-mediated radiochemistry on radiopharmaceutical development.
Similar content being viewed by others

Introduction
The significant potential of targeted radiopharmaceuticals to enhance patient diagnosis and treatment has played an essential role in advancing nuclear medicine into mainstream clinical practice1. As such, there has been a rapid rise in demand for new and established radiopharmaceuticals for the clinical diagnosis, stratification, and treatment of cancer, neurodegenerative diseases, and numerous other conditions. Furthermore, nuclear molecular imaging techniques such as positron emission tomography (PET) and single photon emission computed tomography (SPECT) are increasingly acknowledged as powerful tools for preclinical research and drug discovery in both academic and industrial settings2,3,4,5. The Food and Drug Administration’s (FDA) approvals of blockbuster theranostic products like Pluvicto6 (Novartis, [177Lu]Lu-PSMA-617; for metastatic castration-resistant prostate cancer), Lutathera7 (Novartis, [177Lu]Lu-DOTATATE, for somatostatin-positive gastroenteropancreatic neuroendocrine tumors), and Xofigo8 (Bayer, [223Ra]RaCl2, for bone metastases) have also sparked significant commercial interest and investment in radiopharmaceutical development, transforming what was once a niche academic subfield into a powerful standard of care and a multibillion-dollar industry1,9. All evidence suggests that nuclear medicine and the application of targeted theranostic radiopharmaceutical pairs will continue to grow as a central component of an increasingly personalized approach to patient diagnosis and treatment. However, developing and producing novel radiopharmaceuticals is a non-trivial task. All of the usual challenges of drug development are further complicated by the use of short-lived, highly radioactive materials that require specialized infrastructure, equipment, and expertise for their safe manufacture, handling, and distribution1. The rapidly expanding demand for novel and established radiopharmaceuticals is already exerting pressure on the global radiopharmaceutical production infrastructure and skilled workforce10, and this has put radiochemistry front and center as a field ripe for innovation9. One of the most significant radiochemical advancements of the last decade has been the introduction of a new class of copper-mediated radiolabeling reaction, and this work has gone a long way to improving the chemical space that can be radiolabeled. However, there remains a need for more efficient and reliable radiopharmaceutical production technologies capable of reducing the time it takes to optimize and translate a copper-mediated (or any other) radiolabeling method for routine production. Both aspects of the radiopharmaceutical manufacturing paradigm will be discussed in the context of copper-mediated radiolabeling throughout this perspective.
The new family of copper-mediated radiolabeling reactions introduced in the mid-2010s were originally pioneered for [18F]fluoride ([18F]F-) but, given their widespread utility, has also subsequently been adapted for numerous other radionuclides (vide infra). Cu-mediated radiofluorination (CMRF) represents an efficient and general method for the direct late-stage oxidative radiofluorination of aromatic rings, with a mechanism analogous to that of the classical Chan-Lam cross-coupling11,12,13. Before the development of this chemistry, accessible radiofluorinations of aromatic systems using nucleophilic cyclotron-produced [18F]fluoride were largely achieved using nucleophilic aromatic substitution reactions (SNAr) on highly electron-deficient aromatic or heteroaromatic ring systems. Electron-rich aromatic rings were typically labeled using electrophilic [18F]F2 gas; however, this approach was far from desirable as it used F2 gas and produced products of low molar activity14,15. In most laboratories, these constraints significantly limited the types of molecular scaffolds that could be labeled with 18F. Radiochemists were forced to adopt complex multistep radiosynthesis procedures incompatible with standard cGMP-compliant automation equipment used for routine production or to compromise on molecular design by either choosing alternative (often non-optimal) radiolabeling sites or using auxiliary prosthetic group strategies12.
To address these limitations, several groups began exploring the development of new radiofluorination methodologies compatible with diverse heteroaromatic precursors, including electron-rich rings. Two main approaches have been investigated: (i) generating new precursors to expand the substrate scope compatible with SNAr and (ii) transition-metal-mediated processes. A significant development in SNAr chemistry was the report by Pike’s group that enabled arenes to be labeled in a single step via SNAr via radiofluorination of diaryliodonium salts16. Reactions utilized no-carrier-added 18F- and were heated at 85–110 oC for 35–40 min. Although yields were somewhat low and regioselectivity modest, particularly in the case of electron-rich aromatics (e.g., anisole), this was an important development for radiochemists. Subsequent improvements (for recent reviews, see refs. 17,18) have expanded the scope of this transformation (e.g., treatment of (2-thienyl)(aryl)iodonium salts with [18F]KF selectively affords [18F]fluoroarenes because the highly electron-rich 2-thienyl group directs radiofluorination to the other aryl group on iodine with reasonable selectivity), but the starting materials can be challenging to access and can also suffer from low stability. Related to this approach, the radiofluorination of iodonium ylides was also first published in 2014 by Vasdev and Liang. This was another significant advancement towards a broadly applicable method for the radiofluorination of electron-rich aromatic rings19, however, issues around the accessibility and shelf-stability of the iodonium ylide precursors significantly constrain its application. Besides iodonium precursors, there have also been several reports of sulfonium-type precursors for 18F-labeling of arenes. Pike and colleagues reported the production of [18F]fluoroarenes via the radiofluorination of diaryl sulfoxides, while the Årstad, Ametamey and Ritter groups independently described 18F-labeling of aryl sulfonium salts20,21,22. These various operationally simple, robust, and metal-free SNAr protocols mean there is no need to check clinical GMP products for trace metal contaminants (cf. transition-metal-mediated protocols).
The second approach to radiofluorination methodology development involves the use of transition-metal mediators to label diverse precursor molecules. There are historical examples of transition-metal-mediated radiofluorination protocols, such as Buchwald’s PdII-mediated radiofluorination of aryl triflates23, adapted for radiochemistry by Ermert and Coenen24, that demonstrated proof-of-concept but which suffered from low yields and the need to add carrier fluoride. A paradigm shift came with the discovery that high oxidation state transition metals (e.g., PdIV, CuIII) can promote radiofluorination reactions, and seminal work by Ritter and Hooker showed both Pd- and Ni-mediated radiofluorinations of diverse heteroaromatics25,26,27. Although these transition-metal-mediated methods for the radiofluorination of electron-rich and neutral aromatic rings have not been widely adopted, likely because of the toxicity of palladium and that they require the synthesis of air-sensitive transition-metal precursor complexes that are difficult to implement into routine productions, they ushered in a new hunt for metal-mediated reactions for 18F-labeling that ultimately resulted in the introduction of CMRF. In contrast to the complex organometallic chemistry of the early methods, the robustness, operational simplicity, and exceptional functional group tolerance of CMRF, as well as the lower toxicity of copper, make this transformation broadly applicable to numerous previously “difficult-to-access” pharmaceutically relevant scaffolds. This has enabled the exploration of both novel and established in vivo imaging targets.
The basis for the development of CMRF was the Cu-mediated “cold” fluorination of iodonium salts and organoboron reagents reported in 2013 by the Sanford lab28,29. Building on this precedent, the first example of a CMRF reaction was disclosed in 2014 by the Scott and Sanford groups involving the copper-catalyzed radiofluorination of electrophilic (mesityl)(aryl)iodonium salts (Fig. 1A)30. This work demonstrated that copper-mediators in combination with stable and synthetically accessible mesityl iodonium salts directed oxidative addition (and thus radiofluorination) to the smaller aryl group on iodine, regardless of its electronic properties, under mild conditions (85 oC, 20 min). The approach overcomes both: (i) the need for high temperatures when using SNAr to radiofluorinate iodonium salt precursors, which likely exacerbates the limited stability and shelf-life of precursors like (2-thienyl)(aryl)iodonium salts (resulting in more variable yields from SNAr compared to CMRF), and (ii) the modest regioselectivity of the original SNAr process.

A–D Early examples of the copper-mediated radiofluorination reactions with various aryl precursors. E The proposed general mechanism of the copper-mediated radiofluorination of organoboron and tin reagents is analogous to the Chan-Lam coupling reaction.
This initial CMRF of iodonium salts was followed shortly thereafter by the Gouverneur group's publication of the copper-mediated radiofluorination of nucleophilic aryl boronic acid pinacol ester substrates (Fig. 1B)31. These two reactions provided robust methods to synthesize previously inaccessible 18F-labeled electron-rich and -neutral aromatic rings from readily accessible precursors using nucleophilic [18F]fluoride. The CMRF reaction of organoboron reagents is believed to proceed via a mechanism akin to the Chan-Lam cross-coupling, whereby an aryl nucleophile undergoes transmetalation with a solvated copper(II)-ligand-[18F]fluoride) complex. Oxidation to form an organoCu(III) is followed by C(sp2)–18F bond-forming reductive elimination to release the radiolabeled product (Fig. 1E). The Scott and Sanford groups quickly expanded this methodology to include aryl boronic acid and aryl stannane substrates, making CMRF a versatile synthetic platform for the 18F-fluorination of a diverse array of aromatic precursors (Fig. 1C, D)12,32,33. The CMRF reaction was revolutionary in providing an operationally simple and automatable method utilizing relatively accessible, shelf-stable, and non-toxic reagents and precursors. In certain instances, CMRF has enabled access to imaging agents where numerous other labeling methods, including SNAr, failed to yield any product34. In this perspective, we highlight recent developments in: (i) CMRF; (ii) Cu-mediated introduction of other radionuclides, (iii) cutting-edge preclinical and clinical applications of Cu-mediated radiolabeling; and (iv) applications of these reactions in theranostics. We also discuss how CMRF is being used to evaluate emerging technologies such as high-throughput radiochemistry, statistical design-of-experiments (DoE), artificial intelligence (AI), and machine learning (ML) to reimagine the radiochemistry workflow and accelerate radiopharmaceutical discovery.
Highlights of new Cu-mediated radiofluorination developments
In the decade since its introduction, considerable work has been undertaken to improve the original Cu-mediated labeling protocols. For example, the Neumaier and Zlatopolskiy groups have reported alcohol-enhanced CMRF conditions and next-generation copper-mediators for the efficient production of 18F-labeled aromatics (Fig. 2A). These include Cu(4-PhPy)4(ClO4)2, Cu(3,4-Me2Py)4(OTf)2 and Cu(3,4-Me2Py)4(ClO4)2, all of which, when used in conjunction with DMI or nBuOH/DMI as reaction media, enable efficient 18F-labeling of BPin and stannane precursors35,36. Alternative sources of [18F]fluoride have also been investigated. For instance, Haveman and Vugts have explored [18F]triflyl fluoride as a [18F]fluoride source for several base-sensitive reactions, including CMRF, while the Zhou lab utilized [18F]TsF in combination with an amide directing group for the preparation of [18F]olaparib37,38. Meanwhile, the Schirrmacher and Hall groups have addressed the long-standing challenge of competing protodeborylation in CMRF reactions (i.e., concurrent formulation of ArH that can be difficult to separate from Ar18F). By utilizing a new Cu-mediator (Cu(ONf)2) and replacing pyridine/DMA with 18-crown-6/tBuOH, they were able to maintain RCY of the desired 18F-labeled product while reducing the formation of protonated side-products by 10-20-fold (Fig. 2C)39. Lastly, variations of the original reactions have also been described by Scott and Sanford that allow for the directed radiofluorination (and radiocyanation) of both aryl C-H bonds (Fig. 2D–F)40,41,42,43,44 and aryl halides (Fig. 2B)45,46 These stable, readily accessible precursors are particularly advantageous when the requisite iodonium salt or organoboron precursor is challenging to synthesize or isolate.

A Improved copper-mediators and solvents for CMRF35. B The directed (DG: Directing Group) radiofluorination of aryl halides with a Cu-N-Heterocycle-carbene (Cu-NHC)45. C Improved CMRF conditions for the suppression of protodeborylated byproduct formation39. D Aminoquinoline-directed CMRF of aromatic C-H bonds43. E The CMRF of aryl C-H bonds via the in-situ formation of aryl iodonium salts42. F Selective Sequential Ir/Cu-mediated radiofluorination of Ar C-H bonds41.
Cu-mediated radiochemistry in preclinical research and clinical production
Some of the first applications of CMRF were to improve the syntheses of established yet “difficult-to-access” radiotracers, with [18F]FDOPA as an obvious example. [18F]FDOPA is a well-established amino acid radiotracer of presynaptic dopamine synthesis and is thus used to study the presynaptic dopaminergic system in a variety of neurophysiological disorders, including schizophrenia and Parkinson’s disease, for which it received FDA approval in 201947,48. [18F]FDOPA has also shown great promise for the imaging of glioma and other primary brain malignancies as well as neuroendocrine tumors and disorders including, phaeochromocytoma, insulinoma, and congenital hyperinsulinism49,50,51. Despite its immense promise as a diagnostic and research tool, the routine use of [18F]FDOPA was hampered by its relative inaccessibility through traditional radiosynthetic approaches. These required harsh radiolabeling conditions using low molar activity electrophilic [18F]F2 or long multistep synthetic sequences that were complex and difficult to automate reliably due to the need to radiofluorinate a highly electron-rich catechol ring14,52. The synthesis of [18F]FDOPA was so challenging that it was commonly used as the primary performance benchmark for emerging aromatic radiofluorination methodologies. CMRF chemistry involving aryl iodonium, boronate, and stannane precursors has afforded the radiochemical community relatively simple and automatable one-pot procedures to access [18F]FDOPA using standard radiochemistry equipment validated for cGMP clinical production53,54,55,56. Using CMRF enabled direct fluorination of FDOPA precursors protected with acid labile (e.g. MOM, Boc) groups, allowing a straightforward 1 pot, 2 step radiofluorination/HCl-mediated deprotection using a standard fluorine-18 radiochemistry module45,46. While work continues to further improve the CMRF synthesis yields and overall production reliability (Fig. 3A)14, this work highlights two important aspects of CMRF that have led to its widespread adoption by the radiochemistry community. First, it enables the facile radiofluorination of complex, electronically diverse aromatic rings from common precursors. Second, the workflow has been designed for ease of use by non-experts and compatibility with existing radiochemistry infrastructure (e.g., synthesis modules, HPLC purification, sep-pak reformulation). As such, the barrier to entry is low for sites wanting to use CMRF themselves. Reflecting this, the radiosyntheses of other clinically and preclinically important radiotracers, such as [18F]flumazenil (Fig. 3B), have also received CMRF overhauls to their cGMP production procedures from different groups, increasing synthesis reliability and yield, and hence, the number of patient doses that can be administered for imaging from a single production57,58.

A Clinically Validated CMRF synthesis of [18F]FDOPA54. B Clinically Validated CMRF synthesis of [18F]Flumazinil57. C Initial human imaging experience with [18F]TRACK – brain PET (left, reproduced from ref. 61 with permission©. American Chemical Society) and whole-body dosimetry (right, reproduced from ref. 55 under a CC BY 4.0 license). D Human imaging with [18F]-SynVesT-1 (Left, coregistered parametric VT images of representative baseline scan, 120-min PET data) and [18F]-SynVesT-2 (Right, summed SUV PET images of representative baseline scan, 40 to 60 min post-injection). Reproduced from refs. 69,70 with permission©. SNMMI. E Clinical imaging with [11C]LY2795050 prepared via Cu-mediated radiocyanation (representative bolus + infusion equilibrium ratio (EQR) images on two planes in a normal control (peak EQR = 2.8; EQR = distribution volume ratio, under equilibrium conditions). Parametric EQR images are calculated from the average of frames from 40 to 80 min post-injection, normalized by the value in cerebellar gray matter). Images reproduced from ref. 63 with permission©. American Chemical Society. F Nonhuman primate imaging with [11C]CN-Nociceptin prepared via Cu-mediated radiocyanation (summed SUV PET images 20–60 min post-injection). Images reproduced from ref. 46 with permission©. American Chemical Society.
In addition to its use as a synthetic tool to simplify routine GMP radiotracer productions, CMRF chemistry has been widely adopted by radiopharmaceutical research groups worldwide to access a chemically diverse and rapidly expanding list of novel radiotracers for preclinical and clinical evaluation. In 2020, Scott and Sanford extensively reviewed the many examples where CMRF chemistry has been applied to synthesize novel radiopharmaceuticals for use in (pre)clinical imaging studies11. Since then, the labeling methodology has provided many more novel radiopharmaceuticals of preclinical interest, while several older examples have continued to be further evaluated in human subjects. The uptake and dosimetry profile of [18F]TRACK (Fig. 3C), a radiotracer for the tropomyosin receptor kinase (TrkA/B/C) family prepared by CMRF of the BPin precursor and discussed in our previous review, has since been evaluated in humans by the Schirrmacher group. Initial human studies established brain uptake and favorable dosimetry, and the tracer is now being evaluated to assess Trk expression in various CNS pathologies, including Alzheimer’s disease and certain cancers59,60,61,62,63. CMRF chemistry also played a pivotal role in the development of validated GMP-certified syntheses for [18F]-SynVesT-1 and [18F]-SynVesT-218,F-labeled derivatives of the synaptic vesicle glycoprotein (SV2A) radiotracer, [11C]UCB-J, being explored by the pioneers of SV2A imaging at the Yale PET Center (Cai, Huang, Carson) and elsewhere (Fig. 3D)64,65,66,67. Both [18F]-SynVesT-1 and [18F]-SynVesT-2, prepared via CMRF of the corresponding stannane precursors, show promising imaging characteristics in healthy controls and are under clinical evaluation as radiotracers to monitor changes in synaptic density and function in a variety of neuropsychiatric disorders, including addiction, schizophrenia, depression, multiple sclerosis, autism, epilepsy, and post-traumatic stress disorder as well as neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease68,69,70.
Cu-mediated radiochemistry has become an essential synthetic tool for incorporating [18F]fluoride into small aromatic molecules but, as noted above, the CMRF mechanistic manifold has also been expanded to other medically important radiosynthons and radionuclides beyond 18F. In particular, there is considerable interest in the complementary reaction with [11C]cyanide. Building on the historical precedent of reactions with [11C]cyanide promoted by metals like copper71, the Buchwald and Hooker groups reported a Pd-mediated radiocyanation72, while we and others have undertaken significant work developing Cu-mediated radiocyanation reactions. In 2017, the Vasdev lab reported the copper-mediated radiocyanation of boronic acids73 while, in 2018, we demonstrated the copper-mediated radiocyanation ([11C]CN-) of aryl boronate esters and aryl stannane precursors, subsequently demonstrating how 11C-aryl nitriles could be readily diversified into a variety of versatile radiolabeled products. In one example, we have used Cu-mediated radiocyanation for routine production of [11C]LY2795050 (Fig. 3E)74,75, a kappa opioid receptor radioligand based upon an Eli Lilly asset and developed by the Yale PET Center76,77, that is currently under clinical evaluation at our site.
We have also reported methods for the Cu-mediated radiocyanation of aryl iodides with [11C]KCN. The N,N’-dimethylethylene diamine (DMEDA) ligand accelerates oxidative addition, enabling the radiocyanation of diverse substrates, including unprotected amino acids46. In the event precursors are difficult to access for a given substrate and/or require a complex synthesis of the requisite iodide precursor (since natural amino acids do not contain aryl iodides), a telescoped C–H iodination/CMRF sequence has also been developed and applied to the 11CN-radiolabeling of tyrosine and tryptophan residues in native peptides44. This methodology is suitable for the radiolabeling of unprotected amino acids and peptides, and as a proof-of-concept, we applied it to the labeling of nociceptin (11C-CN-NOP, Fig. 3F), a 17-amino acid neuropeptide. Nonhuman primate imaging demonstrated unexpected CNS penetration of the peptide, which, until this result, we had assumed did not cross the blood-brain barrier.
Copper-mediated radiochemistry for theranostics
In addition to radiotracers for in vivo diagnostic imaging, copper-mediated radiochemistry is also poised to play an important role in the rapidly developing area of radio-halogenated theranostics. Theranostics are the same (or very similar) molecules labeled with both diagnostic (e.g., PET) and therapeutic (e.g. α, β–) radionuclides that are the state-of-the-art when it comes to precision cancer care78. Many radiotheranostic agents currently under evaluation are large bioconjugates (e.g., peptides, antibodies) labeled with chelated radioactive metal ions (e.g., 68Ga or 89Zr for imaging177, Lu or 225Ac for therapy). However, there is also interest in labeling the imaging agents with fluorine-18. In some instances (e.g., commercialization)18,F-labeled peptide-based theranostics offer certain advantages over their 68Ga counterparts in terms of costs, yield, transport/distribution (due to the longer half-life of 18F (110 min) compared to 68Ga (68 min)), and image resolution79. Radiolabeling can be accomplished by direct radiofluorination or by preparation of an 18F-labeled building block or prosthetic group. CMRF, for example, has been used to prepare [18F]fluorobenzyl alcohols and activated esters for downstream labeling80,81.
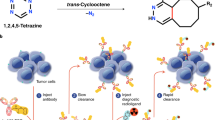
Due to the long circulation time of antibodies (hours to days), these are frequently labeled using 89Zr (half-life = 78 h) so that imaging can occur days after administration. However, there is also interest in labeling them with fluorine-18 due to its ready availability compared to 89Zr, and its more favorable dosimetry profile82. A key challenge is that the long circulation time of antibodies is not readily compatible with the 110-min half-life of fluorine-18, meaning direct or prosthetic group late-stage labeling is not possible. To circumvent this issue, considerable work has been undertaken to develop the concept of pretargeting (Fig. 4A)83. Antibodies tagged with an appropriate labeling handle are injected and allowed to accumulate at the target site over several days. Subsequently, an 18F-labeled small molecule with fast kinetics and bearing the reaction partner to the labeling handle is administered and undergoes a bioorthogonal click reaction with the antibody. Any unreacted radioactivity rapidly clears, allowing imaging of any sites where the antibody has accumulated.

A Schematic for in vivo pretargeting. Recreated from ref. 83 (using https://BioRender.com) with permission©, SNMMI. B The rapid biorthogonal inverse-electron-demand Diels–Alder (click) reaction between trans-cyclooctene and an aryl tetrazine serves as the chemical basis for some in vivo pretargeting approaches. C Copper-mediated Radiofluorination was successfully employed to radiolabel methyl-phenyl-tetrazine. Adapted from ref. 85 under a Creative Commons Attribution 3.0 Unported Licence.
A powerful way to achieve pretargeting involves the tetrazene (Tz) ligation, an inverse-electron-demand Diels–Alder cycloaddition between a radiolabeled electron-deficient Tz and an antibody functionalized with a strained trans-cyclooctene (TCO) (Fig. 4B). The Herth lab has been at the forefront of developing this technology for routine use, synthesizing and screening libraries of 18F-labeled tetrazines to determine optimal parameters for use in pretargeting84,85,86. Notably, while the high reactivity of tetrazines is ideal for bioorthogonal click chemistry, it makes their preparation and radiolabeling somewhat challenging. In a recent example, Herth’s group was investigating direct radiofluorination of aryl-tetrazines on the aromatic ring, but their efforts to label via SNAr were unsuccessful because nucleophilic basic conditions and high temperatures proved incompatible with the Tz moieties. Gratifyingly, CMRF requires less base and lower temperatures and could be used to prepare libraries of 18F-labeled tetrazines in >30% RCC. Stannanes were found to be the optimal precursors, although, in the case of some less reactive tetrazines, BPin esters were also suitable (Fig. 4C). The Herth lab has used similar stannane precursors to also prepare 211At-labeled tetrazines via electrophilic astatination to click with TCO-functionalized antibodies for the preparation of the corresponding alpha-emitting therapeutics87.
While radiometal chelated bioconjugates are often relatively easy to produce and are highly selective for their targets, they are also often sterically large and have long in vivo half-lives, slow pharmacokinetics, and, in most cases, are unable to cross the blood-brain barrier (BBB). On the other hand, small-lipophilic molecules generally have faster in vivo pharmacokinetics and significant work has been undertaken to optimize BBB penetration (and cell membrane permeability) of radiopharmaceuticals to obtain accurate imaging and deliver radiotherapeutic payloads to radiosensitive intracellular targets88,89,90. Indeed, one of the earliest theranostics, metaiodobenzylguanidine (MIBG), is a small molecule developed at the University of Michigan in the 1970s, that has been widely used for the diagnosis (123I-MIBG) and treatment (131I-MIBG) of neural crest tumors such as pheochromocytomas and neuroblastomas91. Given the enormous current interest in the development of new theranostics92, lipophilic small-molecule radiopharmaceuticals labeled with theranostic radionuclide pairs are important targets. With ready access, the diversity of radionuclides available (PET, SPECT, therapy), and the ease of incorporation into small molecules, radiohalogens such as 76Br (β+) and 77Br (auger e-), the various diagnostic and therapeutic isotopes of radioiodine (123/124/125/131I), and the promising combination of 18F (β+) and 211At (α), offer particularly attractive opportunities for the development of small-molecule theranostics. Reflecting this, there is growing interest in methods that allow for the mild incorporation of a variety of radiohalogens into diverse theranostic constructs and, given the low toxicity and ready accessibility of the precursors, mild labeling conditions, and high substrate tolerance, Cu-mediated radiolabeling holds promise in this regard.
Radioiodine has long been established as an important radionuclide for diagnosis and therapy. [125/131I]NaI was the first theranostic agent used to image and treat thyroid cancer and thyrotoxicosis, and radio-iodinated radiopharmaceuticals are routinely used in nuclear medicine departments worldwide93. Building on historical copper-mediated radioiodination chemistry94,95, Several groups employing different radioiodine isotopes have studied the copper-mediated radioiodination of aryl boronate and stannane substrates, demonstrating improved production of several clinically relevant radiopharmaceuticals, including [125I]iodouracil, [123/125I]MIBG, [123I]DPA-713, [123I]KX-1, [123I]IMPY, [123I]IPEB and others96,97,98,99,100,101,102. [123/131I]MIBG is a well-established theranostic radiotherapeutic pair for the localization and treatment of neuroblastoma103. To improve the therapeutic outcomes, [211At]MABG is being considered as a possible alternative alpha-emitting theranostic partner to [125I]MIBG and is currently in clinical trials104,105. To date, most site-specific heavy radiohalogen radiochemistry still relies on the use of electrophilic halodestannylation to incorporate the respective radionuclides into aromatic compounds. However, the low toxicity of the organoboron reagents used for copper-mediated radiohalogenation is more conducive to clinical production and translation, and several efforts have been made to adapt CMRF chemistry for use with these radionuclides.
In 2018, the Mach lab demonstrated this approach by introducing the copper-mediated [211At]astatination and [125I]iodination of boronic acid esters106. The reaction proceeded at room temperature in methanol/acetonitrile within 10 min, resulting in high radiochemical conversion for a wide array of radio-astatinated and -iodinated aromatic products, including several difficult-to-label heteroaromatic scaffolds. The mild conditions and high radionuclide incorporations of the Cu-mediated radioiodination and astatination of aryl boronic acid esters compared to the original CMRF reactions may be rationalized by the comparably greater relative nucleophilicity of both [125I]I- and [211A]At- relative to [18F]F-. The authors used the methodology for the single-step radioastatination and iodination of several (pre-)clinically interesting scaffolds, noting the method to be generally higher yielding than the standard electrophilic astatatodestannylation approach. Theranostic pairing with the CMRF reaction manifold was demonstrated by synthesizing radioiodinated and astatinated derivatives of two well-established 18F-labeled PARP expression imaging agents [18F]PARPi (based on olaparib) and [18F]fluorthanatrace ([18F]FTT, based on rucaparib) (Fig. 5A). In contrast to many of the large radiotheranostic bioconjugates, small-molecule radiotheranostics like these PARP inhibitors possess the cell membrane permeability to deliver their therapeutic payloads to the radiosensitive PARP DNA repair complexes within the nuclei of tumor cells with upregulated PARP expression.

In 2018, the Katzenellenbogen group demonstrated the use of copper-mediated radiobromination chemistry label N-succinimidyl-4-[77Br]bromobenzoate ([77Br]SBB), which was then used to synthesize [77Br]PARPi107. More recently, the Ellison lab has reported a variation on these conditions that efficiently radio-brominates various aromatic and heteroaromatic rings under mild conditions and at room temperature108. This approach was applied to the synthesis of 77Br-labeled analogs of the PSMA targeting radiopharmaceutical [18F]DCFPyL (Pylarify) as well as [77Br]PARPi (Fig. 5B). These Cu-mediated labeling conditions were also effective for labeling a rucaparib derivative with bromine-76 and -77 to produce a true theranostic pair from the same radiopharmaceutical precursor. With demand for new theranostics steadily increasing, copper-mediated radiochemistry is expected to become an important and versatile synthetic tool for developing small-molecule radiohalogen-based theranostic agents against intracellular targets.
Disruptive technologies with the potential to enhance copper-mediated radiolabeling
CMRF chemistry has profoundly impacted the radiopharmaceutical sciences, enabling access to new regions of small-molecule radiopharmaceutical space for research and development. However, as the power of CMRF chemistry as a radiosynthetic tool became apparent to the community, its adoption into routine use faced some significant hurdles. For example, preliminary attempts to automate CMRF radiosyntheses at production scales often showed significantly reduced reaction performance and were prone to unexpected failures. Work by the Neumeier and Gouverneur groups, Scott and Sanford labs, and numerous other labs has further studied the reaction’s critical parameters35,109,110,111,112, identified the methodology’s limitations110,113, and offered practical and reliable production strategies that have since been applied to multiple real-world examples36,114,115,116,117. While we now have a much better understanding of the factors affecting the reaction’s probability for success, its multicomponent nature often makes optimizing the conditions for new untested precursors a time and labor-intensive endeavor. The reaction’s performance is affected by a complex interplay between multiple discrete and continuous reaction variables. Furthermore, the optimal conditions are highly sensitive to the nature and structure of the substrate118. Unfortunately, many radiopharmaceutical development efforts that use CMRF chemistry still fail to find conditions capable of reliably affording the desired product with sufficient yield or molar activity. Therefore, the radiolabeling of each new scaffold still presents a unique set of optimization challenges that must be overcome before a reliable and high-yielding CMRF protocol can enter routine (pre)clinical production.
In their 2017 commentary, “Bridging the Gaps in 18F Tracer Development," Campbell and colleagues, including Gouverneur, Hooker, and Ritter, called for a reevaluation of traditional radiopharmaceutical development pipelines, emphasizing the need for improved process development and optimization workflows to accelerate the adoption of new radiochemical methodologies for routine radiopharmaceutical production119. One of the most significant impacts of CMRF chemistry is that it has prompted the radiochemical community to reassess its conventional approaches to radiopharmaceutical development. The challenges associated with implementing CMRF and other emerging methodologies have sparked a wave of innovation that aims to integrate advanced automation technologies120,121, internet-connected laboratories122, high-throughput radiochemistry workflows120,123, statistically optimal experimental design strategies118, and advanced data analysis techniques based on computational and machine learning methods88,124,125.
Preliminary application of these methods to the CMRF labeling of challenging substrates has shown enormous promise. For example, the Maurer group applied a statistical design-of-experiments (DoE) approach for reaction optimization to establish reliable and robust one-pot radiosyntheses of several novel radiotracers, including the CLIP-Tag reporter probe [18F]pFBC126 and [18F]talazoparib127, (Fig. 6), a PARP1-targeted imaging probe that was previously synthesized by a complex 4-step, multi-pot manual procedure. This work laid the foundation for a DoE-based radiotracer development pipeline using CMRF chemistry118. Using DoE to systematically study these process optimizations allowed reliable synthesis conditions to be found within weeks, a significant improvement over the multiple months such optimizations can take using traditional “one-variable-at-a-time” approaches. Extending the concept further, Scott and Sanford collaborated with Merck to develop high-throughput radiochemistry that allows radiochemistry reactions to be run in 24- and 96-well plate format123. Combining this high-throughput workflow with the systematic and statistical advantages of DoE facilitated the identification of CMRF conditions for the labeling [18F]crizotinib within two 3-h experimental sessions using just 27.8 µmol of the precursor (Fig. 6)128. We expect these advancements in radiochemical technologies and techniques will continue to expedite the discovery and delivery of innovative radiopharmaceuticals for preclinical evaluation and clinical application, maximizing their future impact. With the rising demand for new radiopharmaceuticals and ongoing challenges with limited production infrastructure capacity and skilled labor availability, embracing new, more efficient radiochemical technologies will aid in advancing nuclear medicine's development and growth in the future.

Systematic approaches to radiosynthesis optimization that combine designed data collection, high-throughput experimentation, and data science techniques will drastically accelerate radiopharmaceutical development timelines.
Challenges and Limitations
Copper-mediated radiochemistry has benefited from more than a decade of research investment from many radiochemistry groups following its introduction in 2014. The result is a powerful synthetic technique that, as described in this perspective, has had a pronounced impact on the synthesis of radiopharmaceuticals for diagnostic imaging and theranostic applications. With that said, there remain several challenges and limitations that users should be aware of when working with the methodology in their own radiochemistry facilities. The most significant is perhaps the issue of competing protodeborylation, giving rise to the protonated byproduct (ArH) that can be difficult to separate from the desired radiopharmaceutical product (e.g. Ar18F, Ar11CN). While there are preliminary solutions aimed at reducing the formation of the protonated byproduct as noted above (Schirrmacher and Hall reduced the formation of ArH by 10-20-fold using Cu(ONf)2 in conjunction with 18-crown-6/tBuOH39), or separating it from the desired radiopharmaceutical product if formed (we have separated ArH from Ar18F using a perfluorophenyl-capped HPLC columns115), currently how much ArH is formed varies between syntheses and needs to be accounted for on a case-by-case basis until a general solution is developed.
A second challenge is that certain substrates are incompatible with copper-mediated radiolabeling, and despite efforts at derisking the process from the Gouverneur lab113, it is still not straightforward to predict when this will be the case or not. For substrates that can be labeled with these methods, optimizing the conditions for new untested precursors remains time, equipment and labor-intensive. This is becoming ever more challenging, given the rapid growth in nuclear medicine and burgeoning demand for radiopharmaceuticals worldwide. To predict whether a given substrate can be labeled using a copper-mediated method and, if so, what the optimal precursor and/or labeling conditions might be is not straightforward, but preliminary results with machine learning techniques are encouraging and worthy of further investigation. However, several challenges need to be overcome to capitalize on this technology. The first is a data access challenge, involving both how to generate big data for copper-mediated radiolabeling and how to store and curate it into a format appropriate for machine learning, while the second challenge stems from ongoing issues with shortages in the workforce pipeline.
High-throughput experimentation and DoE are the first approaches to generating the large datasets needed to effectively apply machine learning techniques to radiochemistry, although there is still work to be done on automation and ease of use before radiochemistry facilities around the world can readily deploy these techniques. There is also a need to expand the infrastructure for conducting this type of work. Much is standard and commercially available (HTE/DoE equipment and software) or available on college campuses (supercomputers for machine learning), but is not standard fare in most radiochemistry labs today. Meanwhile, initiatives to organize data collection are underway, but will also take time. Efforts to standardize radiochemistry nomenclature a couple of years ago were a critical first step in this process129,130, defining how data should be reported, and we recommend that similar guideline paper(s) are conceptualized for the type and format that data should be collected in to enable, for example, machine learning.
Workforce pipeline issues also need to be urgently addressed. Recent papers have drawn attention to a critical shortage of radiochemists, and we strongly support the recommendations by the authors of these articles on how to train more10,131. However, it is important to also note that the number of radiochemists with the necessary lab skills to conduct both copper-mediated radiolabeling and computer science savvy to deploy the sophisticated data processing and machine learning techniques being developed is even smaller. To address this shortage, we encourage groups with interest in this type of work to be intentional in their efforts to train students both in radiochemistry and advanced data analysis techniques. This will ensure that the field can truly capitalize on the benefits that data science and machine learning offer when it comes to accelerating radiopharmaceutical R&D.
Conclusions and future outlook
Copper-mediated radiolabeling, introduced over a decade ago, has proven a transformative methodology for radiochemists. Immediately following its introduction for radiofluorination, the approach was adopted by numerous labs around the world to both improve the synthesis of underutilized, historically difficult-to-access radiopharmaceuticals as well as to synthesize new ones that were previously inaccessible. These early successes spurred the refinement of the first-generation methods to include more precursor options, new copper-mediators, reduction of side-products, and inclusion of additives such as n-BuOH to enhance radiochemical yields. Furthermore, the platform technology has been adapted for use with other diagnostic and therapeutic radionuclides, including 11C123/125, I211, At, and 76/77Br. This shows an important future role for copper-mediated radiolabeling in the emerging field of theranostics. Indeed, copper-mediated radiofluorination and astatination have already been used to label tetrazines for pretargeted theranostics. The result of these successes means that copper-mediated radiolabeling is being applied to radiolabel increasingly diverse chemical space. As such, over the next decade, we anticipate that new optimization paradigms will be needed because of the growth that nuclear medicine and theranostics are experiencing. Research groups are beginning to pioneer such approaches, showing the early promise that high-throughput radiochemistry workflows, statistically optimal experimental design strategies, and advanced data analysis techniques based on computational and machine learning methods have for accelerating the optimization of copper-mediated radiolabeling for any given substrate. As these methods mature, they are expected to dramatically improve the efficiency of the radiochemistry workplace, ultimately accelerating the design, evaluation, and clinical translation of new theranostic radiopharmaceuticals.
Data availability
No datasets were generated or analyzed during the current study.
References
Lapi, S. E. et al. Recent advances and impending challenges for the radiopharmaceutical sciences in oncology. Lancet Oncol. 25, e236–e249 (2024).
Piel, M., Vernaleken, I. & Rösch, F. Positron emission tomography in CNS drug discovery and drug monitoring. J. Med. Chem. 57, 9232–9258 (2014).
Matthews, P. M., Rabiner, E. A., Passchier, J. & Gunn, R. N. Positron emission tomography molecular imaging for drug development. Br. J. Clin. Pharmacol. 73, 175–186 (2012).
Donnelly, D. J. in Handbook of Radiopharmaceuticals: Methodology and Applications: Second Edition. 703–725 (John Wiley and Sons, 2021).
Ghosh, K. K. et al. Positron emission tomographic imaging in drug discovery. Drug Discov. Today 27, 280–291 (2022).
Novartis Pharmaceuticals Corporation. Pluvicto® ((177Lu)Lutetiumvipivotidtetraxetan) Package Insert [Internet]. Available from: https://www.novartis.com/us-en/sites/novartis_us/files/pluvicto.pdf (2022). Accessed 4-Mar-2024.
Novartis Pharmaceuticals Corporation. LUTATHERA® (Lutetium Lu 177 Dotatate) Package insert. [Internet]. Available from: https://www.novartis.com/us-en/sites/novartis_us/files/lutathera.pdf (2018). Accessed 4-Mar-2024.
Bayer Inc. XOFIGO® (Radium Ra 223 Dichloride) Package Insert [Internet]. Available from: https://labeling.bayerhealthcare.com/html/products/pi/Xofigo_PI.pdf (2013). Accessed 4-Mar-2024.
Bowden, G. D., Scott, P. J. H. & Boros, E. Radiochemistry: a hot field with opportunities for cool chemistry. ACS Cent. Sci. 9, 2183–2195 (2023).
Scott, A. M. et al. Trends in nuclear medicine and the radiopharmaceutical sciences in oncology: workforce challenges and training in the age of theranostics. Lancet Oncol. 25, e250–e259 (2024).
Wright, J. S. et al. Copper-mediated late-stage radiofluorination: five years of impact on preclinical and clinical PET imaging. Clin. Transl. Imaging 8, 167–206 (2020).
Brooks, A. F., Topczewski, J. J., Ichiishi, N., Sanford, M. S. & Scott, P. J. H. Late-stage [18F]fluorination: new solutions to old problems. Chem. Sci. 5, 4545–4553 (2014).
Preshlock, S., Tredwell, M. & Gouverneur, V. 18F-Labeling of arenes and heteroarenes for applications in positron emission tomography. Chem. Rev. 116, 719–766 (2016).
Krasikova, R. N. Nucleophilic synthesis of 6-l-[18F]FDOPA. Is copper-mediated radiofluorination the answer?. Molecules 25, 4365 (2020).
Forsback, S., Eskola, O., Haaparanta, M., Bergman, J. & Solin, O. Electrophilic synthesis of 6-[18F]fluoro-L-DOPA using post-target produced [18F]F2. Radiochim. Acta 96, 845–848 (2008).
Pike, V. W. & Aigbirhio, F. I. Reactions of cyclotron-produced [18F]fluoride with diaryliodonium salts—a novel single-step route to no-carrier-added [18]fluoroarenes. J. Chem. Soc. Chem. Commun. 1995, 2215–2216 (1995).
Ross, T. L., Ermert, J., Hocke, C. & Coenen, H. H. Nucleophilic 18F-fluorination of heteroaromatic iodonium salts with no-carrier-added [18F]fluoride. J. Am. Chem. Soc. 129, 8018–8025 (2007).
Yusubov, M. S., Svitich, D. Y., Larkina, M. S. & Zhdankin, V. V. Applications of iodonium salts and iodonium ylides as precursors for nucleophilic fluorination in positron emission tomography. Arkivoc 2013, 364–395 (2013).
Rotstein, B. H., Stephenson, N. A., Vasdev, N. & Liang, S. H. Spirocyclic hypervalent iodine(III)-mediated radiofluorination of non-activated and hindered aromatics. Nat. Commun. 5, 4365 (2014).
Sander, K. et al. Sulfonium salts as leaving groups for aromatic labelling of drug-like small molecules with fluorine-18. Sci. Rep. 5, 9941 (2015).
Mu, L. et al. 18F-radiolabeling of aromatic compounds using triarylsulfonium salts. Eur. J. Org. Chem. 2012, 889–892 (2012).
Xu, P. et al. Site-selective late-stage aromatic [18F]fluorination via aryl sulfonium salts. Angew. Chem. 132, 1972–1976 (2020).
Watson, D. A. et al. Formation of ArF from LPdAr(F): catalytic conversion of aryl triflates to aryl fluorides. Science 325, 1661–1664 (2009).
Cardinale, J. et al. Carrier-effect on palladium-catalyzed, nucleophilic 18F- fluorination of aryl triflates. J. Label. Comp. Radiopharm. 55, 450–453 (2012).
Lee, E., Hooker, J. M. & Ritter, T. Nickel-mediated oxidative fluorination for PET with aqueous [18F]fluoride. J. Am. Chem. Soc. 134, 17456–17458 (2012).
Ren, H. et al. Synthesis and imaging validation of [18F]MDL100907 enabled by Ni-mediated fluorination. ACS Chem. Neurosci. 5, 611–615 (2014).
Lee, E. et al. A fluoride-derived electrophilic late-stage fluorination reagent for PET imaging. Science 334, 639–642 (2011).
Ye, Y., Schimler, S. D., Hanley, P. S. & Sanford, M. S. Cu(OTf)2-mediated fluorination of aryltrifluoroborates with potassium fluoride. J. Am. Chem. Soc. 135, 16292–16295 (2013).
Ichiishi, N., Canty, A. J., Yates, B. F. & Sanford, M. S. Cu-catalyzed fluorination of diaryliodonium salts with KF. Org. Lett. 15, 5134–5137 (2013).
Ichiishi, N. et al. Copper-catalyzed [18F]fluorination of (mesityl)(aryl)iodonium salts. Org. Lett. 16, 3224–3227 (2014).
Tredwell, M. et al. A general copper-mediated nucleophilic 18F fluorination of arenes. Angew. Chem. Int Ed. 53, 7751–7755 (2014).
Mossine, A. V. et al. Synthesis of [18F]arenes via the copper-mediated [18F]fluorination of boronic acids. Org. Lett. 17, 5780–5783 (2015).
Makaravage, K. J., Brooks, A. F., Mossine, A. V., Sanford, M. S. & Scott, P. J. H. H. Copper-mediated radiofluorination of arylstannanes with [18F]KF. Org. Lett. 18, 5440–5443 (2016).
Cole, E. et al. Radiochemistry challenges and progression for incorporation of 18F into a complex substituted 6-18F-fluoroquinoline BMS-986205 for IDO imaging. J. Nucl. Med. 59, 605 (2018). (Suppl 1).
Hoffmann, C. et al. Next generation copper mediators for the efficient production of 18F-labeled aromatics. Chem. Eur. J. 29, e202202965 (2023).
Zischler, J., Kolks, N., Modemann, D. & Neumaier, B. & Zlatopolskiy, B. D. Alcohol-enhanced Cu-mediated radiofluorination. Chem. Eur. J. 23, 3251–3256 (2017).
Haveman, L. Y. F., de Kruijff, A. M. T., van Eeden, S. P. P., Windhorst, A. D. & Vugts, D. J. Triflyl [18F]fluoride as a solution for base-sensitive late-stage nucleophilic aromatic 18F-fluorination reactions. Chem. Eur. J. 21, e202403127 (2025).
Zhou, D., Chu, W., Chen, H. & Xu, J. Exploration of directing-group-assisted, copper-mediated radiofluorination and radiosynthesis of [18F]Olaparib. ACS Med. Chem. Lett. 15, 116–122 (2024).
Sun, J., Jaworski, C., Schirrmacher, R. & Hall, D. G. Suppressing protodeboronation in Cu-mediated 19F/18F-fluorination of arylboronic acids: a mechanistically guided approach towards optimized PET probe development. Chem. Eur. J. 30, e202400906 (2024).
Wright, J. S., Sharninghausen, L. S., Lapsys, A., Sanford, M. S. & Scott, P. J. H. C–H labeling with [18F]fluoride: an emerging methodology in radiochemistry. ACS Cent. Sci. 10, 1674–1688 (2024).
Wright, J. S. et al. Sequential Ir/Cu-mediated method for the Meta -selective C–H radiofluorination of (hetero)arenes. J. Am. Chem. Soc. 143, 6915–6921 (2021).
McCammant, M. S. et al. Cu-mediated C-H 18F-fluorination of electron-rich (hetero)arenes. Org. Lett. 19, 3939–3942 (2017).
Lee, S. J., Makaravage, K. J., Brooks, A. F., Scott, P. J. H. & Sanford, M. S. Copper-mediated aminoquinoline-directed radiofluorination of aromatic C−H bonds with K18F. Angew. Chem. Int. Ed. 58, 3119–3122 (2019).
Horikawa, M. et al. C-H radiocyanation of bioactive molecules via sequential iodination/copper-mediated cross-coupling. Chem. Sci. 14, 12068–12072 (2023).
Sharninghausen, L. S. et al. NHC-copper mediated ligand-directed radiofluorination of aryl halides. J. Am. Chem. Soc. 142, 7362–7367 (2020).
Sharninghausen, L. S. et al. Copper-mediated radiocyanation of unprotected amino acids and peptides. J. Am. Chem. Soc. 144, 7422–7429 (2022).
Pretze, M., Wängler, C. & Wängler, B. 6-[18F]fluoro-L-DOPA: a well-established neurotracer with expanding application spectrum and strongly improved radiosyntheses. Biomed. Res. Int. 2014, 674063 (2014).
Dhawan, V. et al. Prospective F-18 FDOPA PET Imaging Study in Human PD. Nucl. Med. Mol. Imaging 56, 147–157 (2022).
Somme, F., Bender, L., Namer, I. J., Noël, G. & Bund, C. Usefulness of 18F-FDOPA PET for the management of primary brain tumors: a systematic review of the literature. Cancer Imaging 20, 70 (2020).
Minn, H., Kauhanen, S., Seppänen, M. & Nuutila, P. 18.F-FDOPA: a multiple-target molecule. J. Nucl. Med. 50, 1915–1918 (2009).
Santhanam, P. & Taïeb, D. Role of 18F-FDOPA PET/CT imaging in endocrinology. Clin. Endocrinol. 81, 789–798 (2014).
Neves, ÂC. B. et al. Advances in the automated synthesis of 6-[18F]Fluoro-L-DOPA. EJNMMI Radiopharmy Chem. 6, 11 (2021).
Mossine, A. V. et al. One-pot synthesis of high molar activity 6-[18F]fluoro-l-DOPA by Cu-mediated fluorination of a BPin precursor. Org. Biomol. Chem. 17, 8701–8705 (2019).
Mossine, A. V. et al. Synthesis of high-molar-activity [18F]6-fluoro-l-DOPA suitable for human use via Cu-mediated fluorination of a BPin precursor. Nat. Protoc. 15, 1742–1759 (2020).
Orlovskaya, V. V. et al. Alcohol-supported Cu-mediated 18F-fluorination of iodonium salts under “minimalist” conditions. Molecules 24, 3197 (2019).
Zarrad, F., Zlatopolskiy, B. D., Krapf, P., Zischler, J. & Neumaier, B. A practical method for the preparation of 18F-labeled aromatic amino acids from nucleophilic [18F]fluoride and stannyl precursors for electrophilic radiohalogenation. Molecules 22, 2231 (2017).
Gendron, T. et al. Multi-patient dose synthesis of [18F]Flumazenil via a copper-mediated 18F-fluorination. EJNMMI Radiopharm. Chem. 7, 5 (2022).
Haskali, M. B. et al. Effective preparation of [18F]flumazenil using copper-mediated late-stage radiofluorination of a stannyl precursor. Molecules 27, 5931 (2022).
Schirrmacher, R. et al. Radioligands for tropomyosin receptor kinase (Trk) positron emission tomography imaging. Pharmaceuticals 12, 7 (2019).
Thiel, A. et al. Dosimetry of [18F]TRACK, the first PET tracer for imaging of TrkB/C receptors in humans. EJNMMI Radiopharm. Chem. 8, 33 (2023).
Bailey, J. J. et al. First-in-Human Brain Imaging of [18F]TRACK, a PET tracer for Tropomyosin Receptor Kinases. ACS Chem. Neurosci. 10, 2697–2702 (2019).
Bernard-Gauthier, V. et al. Identification of [18F]TRACK, a Fluorine-18-Labeled Tropomyosin Receptor Kinase (Trk) Inhibitor for PET Imaging. J. Med. Chem. 61, 1737–1743 (2018).
Kang, M. S. et al. [18F]TRACK, a PET tracer for imaging of tropomyosin receptor kinases (Trk): efficient radiosynthesis and preliminary evaluation in the TgF344-AD rat model of Alzheimer’s disease. Alzheimer’s Dement. 19, e065138 (2023). (Suppl 2).
Dahl, K. et al. Good manufacturing procedure production of [18F]SynVesT-1, a radioligand for in vivo positron emission tomography imaging of synaptic vesicle glycoprotein 2A. J. Label. Comp. Radiopharm. 65, 315–322 (2022).
Cai, Z. et al. Synthesis and preclinical evaluation of an 18F-labeled synaptic vesicle glycoprotein 2A PET imaging probe: [18F]SynVesT-2. ACS Chem. Neurosci. 11, 592–603 (2020).
Li, S. et al. Synthesis and in vivo evaluation of a novel PET radiotracer for imaging of synaptic vesicle glycoprotein 2A (SV2A) in nonhuman primates. ACS Chem. Neurosci. 10, 1544–1554 (2019).
Chen, L. et al. GMP-compliant automated radiosynthesis of [18F]SynVesT-1 for PET imaging of synaptic vesicle glycoprotein 2 A (SV2A). EJNMMI Radiopharm. Chem. 9, 66 (2024).
Howes, O., Marcinkowska, J., Turkheimer, F. E. & Carr, R. Synaptic changes in psychiatric and neurological disorders: state-of-the art of in vivo imaging. Neuropsychopharmacol 50, 164–183 (2024).
Naganawa, M. et al. First-in-human evaluation of 18F-SynVesT-1, a radioligand for PET imaging of synaptic vesicle glycoprotein 2a. J. Nucl. Med. 62, 561–567 (2021).
Drake, L. R. et al. First-in-Human Study of 18F-SynVesT-2: An SV2A PET Imaging Probe with Fast Brain Kinetics and High Specific Binding. J. Nucl. Med. 65, 462–469 (2024).
Ponchant, M., Hinnen, F., Demphel, S. & Crouzel, C. [11C]copper(I) cyanide: a new radioactive precursor for 11C-cyanation and functionalization of haloarenes. Appl Radiat. Isot. 48, 755–762 (1997).
Lee, H. G., Milner, P. J., Placzek, M. S., Buchwald, S. L. & Hooker, J. M. Virtually instantaneous, room-temperature [11C]-cyanation using biaryl phosphine Pd(0) complexes. J. Am. Chem. Soc. 137, 648–651 (2015).
Ma, L., Placzek, M. S., Hooker, J. M., Vasdev, N. & Liang, S. H. [11C]Cyanation of arylboronic acids in aqueous solutions. Cheml. Commun. 53, 6597–6600 (2017).
Kaur, T. et al. Strategies for the production of [11C]LY2795050 for clinical use. Org. Process. Res. Dev. 27, 373–381 (2023).
Makaravage, K. J. et al. Copper(II)-mediated [11C]cyanation of arylboronic acids and arylstannanes. Org. Lett. 20, 1530–1533 (2018).
Naganawa, M. et al. Kinetic modeling of 11C-LY2795050, a novel antagonist radiotracer for PET imaging of the kappa opioid receptor in humans. J. Cereb. Blood Flow. Metab. 34, 1818–1825 (2014).
Zheng, M. Q. et al. Synthesis and evaluation of 11C-LY2795050 as a k-opioid receptor antagonist radiotracer for PET imaging. J. Nucl. Med. 54, 455–463 (2013).
Herrmann, K. et al. Radiotheranostics: a roadmap for future development. Lancet Oncol. 21, e146–e156 (2020).
De Man, K. et al. 18F-PSMA-11 Versus 68Ga-PSMA-11 positron emission tomography/computed tomography for staging and biochemical recurrence of prostate cancer: a prospective double-blind randomised cross-over trial. Eur. Urol. 82, 501–509 (2022).
Nagachinta, S. et al. Fully automated 18F-fluorination of N-succinimidyl-4-[18F]fluorobenzoate ([18F]SFB) for indirect labelling of nanobodies. Sci. Rep. 12, 18655 (2022).
Stotz, S., Bowden, G. D., Cotton, J. M., Pichler, B. J. & Maurer, A. Covalent 18F-radiotracers for SNAPTag: a new toolbox for reporter gene imaging. Pharmaceuticals 14, 897 (2021).
Duvenhage, J., Kahts, M., Summers, B., Zeevaart, J. R. & Ebenhan, T. Highlighting new research trends on zirconium-89 radiopharmaceuticals beyond antibodies. Semin. Nucl. Med. 54, 801–811 (2024).
Altai, M., Membreno, R., Cook, B., Tolmachev, V. & Zeglis, B. M. Pretargeted imaging and therapy. J. Nucl. Med. 58, 1553–1559 (2017).
Battisti, U. M. et al. Development of the first aliphatic 18F-labeled tetrazine suitable for pretargeted PET imaging - expanding the bioorthogonal tool box. J. Med. Chem. 64, 15297–15312 (2021).
García-Vázquez, R. et al. Direct Cu-mediated aromatic 18F-labeling of highly reactive tetrazines for pretargeted bioorthogonal PET imaging. Chem. Sci. 12, 11668–11675 (2021).
Shalgunov, V. et al. Pretargeted imaging beyond the blood-brain barrier. RSC Med. Chem. 14, 444–453 (2022).
Poulie, C. B. M. et al. Comparison of different organometallics towards electrophilic aromatic 211At-astatinations of highly reactive tetrazines. Chem. Eur. J. 30, e202403446 (2024).
Jackson, I. M., Webb, E. W., Scott, P. J. H. & James, M. L. In silico approaches for addressing challenges in CNS radiopharmaceutical design. ACS Chem. Neurosci. 13, 1675–1683 (2022).
Tolboom, N. et al. Theranostics in neurooncology: heading toward new horizons. J. Nucl. Med. 65, 167–173 (2024).
Albert, N. L. et al. Policy review translating the theranostic concept to neuro-oncology: disrupting barriers. Lancet Oncol. 25, e441–e451 (2024).
Agrawal, A., Rangarajan, V., Shah, S., Puranik, A. & Purandare, N. MIBG (metaiodobenzylguanidine) theranostics in pediatric and adult malignancies. Br. J. Radiol. 91, 20180103 (2018).
Fischer, J. R. Theranostics, other trends drive radiopharma VC investments from $63 million to $408 million. [Internet] Available from: https://www.dotmed.com/news/story/61964 (2023). Accessed 4-Mar-2025.
Silberstein, E. B. Radioiodine. The classic theranostic agent. Semin. Nucl. Med. 42, 164–170 (2012).
Moerlein, S. M. Regiospecific aromatic radioiodination via no-carrier-added copper(I) chloride-assisted iododebromination. Radiochim. Acta 50, 55–62 (1990).
El-Wetery, A. S. et al. Catalytic effect of copper(II) chloride on the radioiodination of L-P-Iodo phenylalanine. Arab J. Nucl. Sci. Appl 30, 117 (1997).
McErlain, H., Andrews, M. J., Watson, A. J. B., Pimlott, S. L. & Sutherland, A. Ligand-enabled copper-mediated radioiodination of arenes. Org. Lett. 26, 1528–1532 (2024).
Wilson, T. C. et al. Radiosynthesis of SPECT tracers: via a copper mediated 123I iodination of (hetero)aryl boron reagents. Chem. Commun. 52, 13277–13280 (2016).
Zhou, D., Chu, W. & Xu, J. A practical protocol for large-scale copper-mediated radioiodination of organoboronic precursors: radiosynthesis of [123I]KX-1 for Auger radiotherapy: Copper-mediated radioiodination of [123I]KX-1 for Auger radiotherapy. J. Label. Comp. Radiopharm. 66, 435–439 (2023).
Kondo, Y. et al. One-pot two-step radioiodination based on copper-mediated iododeboronation and azide–alkyne cycloaddition reaction. Chem. Commun. 60, 714–717 (2024).
Zhang, P. et al. A highly efficient copper-mediated radioiodination approach using aryl boronic acids. Chem. Eur. J. 22, 16783–16786 (2016).
Kondo, Y. et al. Effect of water on direct radioiodination of small molecules/peptides using copper-mediated iododeboronation in water-alcohol solvent. ACS Omega 8, 24418–24425 (2023).
Kondo, Y. et al. Copper-mediated radioiodination reaction through aryl boronic acid or ester precursor and its application to direct radiolabeling of a cyclic peptide. J. Label. Comp. Radiopharm. 64, 336–345 (2021).
Wiseman, G. A. et al. Usefulness of123I-MIBG scintigraphy in the evaluation of patients with known or suspected primary or metastatic pheochromocytoma or paraganglioma: results from a prospective multicenter trial. J. Nucl. Med. 50, 1448–1454 (2009).
Batra, V. et al. Preclinical development of [211At]metaastatobenzylguanidine ([211At]MABG) as an alpha particle radiopharmaceutical therapy for neuroblastoma. Clin. Cancer Res. 28, 4146–4157 (2022).
Albertsson, P. et al. Astatine-211 based radionuclide therapy: Current clinical trial landscape. Front. Med. 9, 1076210 (2023).
Reilly, S. W., Makvandi, M., Xu, K. & Mach, R. H. Rapid Cu-catalyzed [211At]astatination and [125I]iodination of boronic esters at room temperature. Org. Lett. 20, 1752–1755 (2018).
Zhou, D., Chu, W., Voller, T. & Katzenellenbogen, J. A. Copper-mediated nucleophilic radiobromination of aryl boron precursors: Convenient preparation of a radiobrominated PARP-1 inhibitor. Tetrahedron Lett. 59, 1963–1967 (2018).
Mixdorf, J. C. et al. Copper-mediated radiobromination of (hetero)aryl boronic pinacol esters. J. Org. Chem. 88, 2089–2094 (2023).
Preshlock, S. et al. Enhanced copper-mediated 18F-fluorination of aryl boronic esters provides eight radiotracers for PET applications. Chem. Commun. 52, 8361–8364 (2016).
Zlatopolskiy, B. D. et al. Copper-mediated aromatic radiofluorination revisited: Efficient production of PET tracers on a preparative scale. Chem. Eur. J. 21, 5972–5979 (2015).
Zhou, D., Chu, W. & Katzenellenbogen, J. A. Exploration of alcohol-enhanced Cu-mediated radiofluorination toward practical labeling. J. Label. Comp. Radiopharm. 65, 13–20 (2022).
Antuganov, D. et al. Copper-mediated radiofluorination of aryl pinacolboronate esters: a straightforward protocol by using pyridinium sulfonates. Eur. J. Org. Chem. 2019, 918–922 (2019).
Taylor, N. J. et al. Derisking the Cu-mediated 18 F-fluorination of heterocyclic positron emission tomography radioligands. J. Am. Chem. Soc. 139, 8267–8276 (2017).
Bowden, G. D., Chailanggar, N., Pichler, B. J. & Maurer, A. Scalable 18F processing conditions for copper-mediated radiofluorination chemistry facilitate DoE optimization studies and afford an improved synthesis of [18F]olaparib. Org. Biomol. Chem. 19, 6995–7000 (2021).
Mossine, A. V. et al. Automated synthesis of PET radiotracers by copper-mediated 18F-fluorination of organoborons: Importance of the order of addition and competing protodeborylation. J. Label. Comp. Radiopharm. 61, 228–236 (2018).
Mossine, A. V. et al. Development of customized [18F]fluoride elution techniques for the enhancement of copper-mediated late-stage radiofluorination. Sci. Rep. 7, 233 (2017).
Lahdenpohja, S. O., Rajala, N. A., Rajander, J. & Kirjavainen, A. K. Fast and efficient copper-mediated 18F-fluorination of arylstannanes, aryl boronic acids, and aryl boronic esters without azeotropic drying. EJNMMI Radiopharm. Chem. 4, 28 (2019).
Bowden, G. D., Pichler, B. J. & Maurer, A. A design of experiments (DoE) approach accelerates the optimization of copper-mediated 18F-fluorination reactions of arylstannanes. Sci. Rep. 9, 11370 (2019).
Campbell, M. G. et al. Bridging the gaps in 18F PET tracer development. Nat. Chem. 9, 1–3 (2017).
Jones, J., Do, V., Lu, Y. & van Dam, R. M. Accelerating radiochemistry development: Automated robotic platform for performing up to 64 droplet radiochemical reactions in a morning. Chem. Eng. J. 468, 143524 (2023).
Rios, A. et al. Microliter-scale reaction arrays for economical high-throughput experimentation in radiochemistry. Sci. Rep. 12, 10263 (2022).
Thompson, S., Kilbourn, M. R. & Scott, P. J. H. Radiochemistry, PET imaging, and the internet of chemical things. ACS Cent. Sci. 2, 497–505 (2016).
Webb, E. W. et al. Development of high-throughput experimentation approaches for rapid radiochemical exploration. J. Am. Chem. Soc. 146, 10581–10590 (2024).
Webb, E. W. & Scott, P. J. H. Potential applications of artificial intelligence and machine learning in radiochemistry and radiochemical engineering. PET Clin. 16, 525–532 (2021).
McGale, J. P. et al. Integrating artificial intelligence and PET imaging for drug discovery: a paradigm shift in immunotherapy. Pharmaceuticals 17, 210 (2024).
Bowden, G. D. et al. [18F]pFBC, a covalent CLIP-tag radiotracer for detection of viral reporter gene transfer in the murine brain. Bioconjug Chem. 35, 254–264 (2024).
Bowden, G. D. et al. DoE optimization empowers the automated preparation of enantiomerically pure [18F]talazoparib and its in vivo evaluation as a PARP radiotracer. J. Med Chem. 64, 15690–15701 (2021).
Bowden, G. et al. Leveraging statistical ‘Design of Experiments’ (DoE) with high-throughput experimentation workflows to accelerate the development of novel radiopharmaceuticals. J. Nucl. Med. 65, 241788 (2024).
Coenen, H. H. et al. Consensus nomenclature rules for radiopharmaceutical chemistry—Setting the record straight. Nucl. Med Biol. 55, v–xi (2017).
Herth, M. M. et al. On the consensus nomenclature rules for radiopharmaceutical chemistry—reconsideration of radiochemical conversion. Nucl. Med. Biol. 93, 19–21 (2021).
Gee, A. D. et al. Training the next generation of radiopharmaceutical scientists. Nucl. Med. Biol. 88–89, 10–13 (2020).
Acknowledgements
Financial support of this work by the NIH (R01EB021155), the Danmarks Frie Forskningsfond (1032-00177B) and the Mistletoe Research Foundation (MRF) is gratefully acknowledged. We also thank all the staff and trainees from the Scott and Sanford labs involved in the development of CMRF over the last 10+ years, as well as colleagues at Vanderbilt University (Bhuminder Singh, Gary Sulikowski, Kwangho KimKyuok Jeon, Adam Rosenberg) and Merck (Stefan Verhoog, Michael Wismer, Dipannita Kalyani, Shane Krska) for their collaboration on high-throughput radiochemistry.
Author information
Authors and Affiliations
Contributions
G.D.B., M.M., and P.J.H.S. conceptualized and wrote the manuscript. M.M.H., M.S.S., and P.J.H.S. provided funding and supervision and edited the manuscript. All authors approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bowden, G.D., Müller, M., Herth, M.M. et al. Copper-mediated radiochemistry: historical impact, current trends, and future possibilities. npj Imaging 3, 25 (2025). https://doi.org/10.1038/s44303-025-00087-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s44303-025-00087-x
