
Abstract
Inhibition of Ras trafficking was the first approach to target Ras in the clinic. With the advent of direct Ras inhibitors, trafficking inhibition may appear obsolete. However, targeting certain trafficking hubs may still offer unexpected opportunities. To exploit these, we need to learn more about the functioning of Ras in specific organelles, which are associated with e.g., cell differentiation. We here discuss future opportunities in that regard.
Similar content being viewed by others
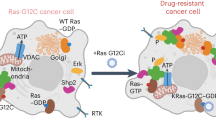

Introduction
The small GTPase Ras operates as a membrane-anchored switchable recruitment site for downstream effectors. Given that membrane anchorage is indispensable, the first Ras drugs evaluated in the clinic were farnesyltransferase inhibitors, which block the attachment of the first obligate lipid modification on the C-terminus of Ras (Fig. 1). In the early 2000s, both inhibitors tipifarnib and lonafarnib failed and Ras was deemed undruggable. At that time, it was underappreciated that the proteins encoded by the most frequently mutated RAS gene KRAS, K-Ras4A and K-Ras4B, and N-Ras can be alternatively prenylated with a geranylgeranyl-moiety1.

Example compounds that inhibit or modulate indicated targets are shown with their names. Several are approved or clinical-stage drugs (all compounds in the bottom row). The left-hand side depicts lipid modification pathways of Ras, while the right side shows the action of trafficking chaperones of Ras. On the endoplasmic reticulum, all Ras proteins are initially farnesylated on the C-terminal cysteine of the CAAX-box (C: cysteine, A: aliphatic residue, X: any residue) followed by subsequent RCE1-mediated AAX-tripeptide cleavage and carboxymethylation of the cysteine by ICMT (enzymes shown in turquoise). Subsequently, K-Ras4B becomes trapped on the recycling endosome (not shown), while the other Ras isoforms are trapped on the Golgi-apparatus by palmitoylation. Enzymes mediating re- and depalmitoylation are shown in gray hues. All Ras are subsequently vesicularly transported to the plasma membrane, where they laterally segregate into distinct nanoclusters. Phosphatidylserine (PS) has emerged as an important lipid to mediate this process by directly interacting with the most C-terminal residues of Ras proteins. Extraction or dissociation from the plasma membrane is enhanced after depalmitoylation. In addition, endocytosis may return Ras into the cycle for vesicular transport. Trafficking chaperones (purple hues) solubilize Ras whenever it has to move between membranes by binding to the farnesyl moiety thus allowing its long-range diffusion. This is necessary to fuel the transport cycle to the plasma membrane and other organelles. Given the distinct client spectrum and their potential involvement in specific cell- and developmental pathways, trafficking chaperones may offer more discrete drug targeting opportunities than broad inhibition of the lipidation machinery.
Fortunately, the inhibitors that were developed during this period experienced a late renaissance. Lonafarnib was found to be a well-tolerated, FDA-approved treatment for the accelerated aging disease Hutchinson-Gilford progeria syndrome, and tipifarnib was FDA-approved in 2021 for the treatment of head and neck cancers with mutated HRAS2,3.
In this perspective, we mostly focus on K-Ras, due to its high mutation frequency in cancer and the possibility to target it directly4. There are now two approved direct K-Ras-G12C inhibitors in the clinic5, but also a deluge of direct Ras inhibitors under development, which promise even greater success. As compared to approved OFF-state inhibitors, Ras (ON)-state inhibitors, target the predominant, GTP-loaded active conformation of oncogenic Ras6 (Table S1). The reversible pan-K-Ras (OFF) inhibitor BI-2865 can block wild-type and several oncogenic KRAS alleles7. An even broader activity is expected with the reversible tri-complex Ras (ON)-multi-selective inhibitor RMC-6236, which targets multiple wildtype and oncogenic Ras isoforms and remains potent in K-Ras-G12C inhibitor-resistant preclinical models8. All these new inhibitors have the potential to overcome some or several limitations associated with the first generation of K-Ras-G12C inhibitors. Excitingly, recent work further suggests that the cryptic switch II pocket is also targetable in other Ras- and Rho-family members, suggesting an enormous potential to broadly target small GTPases in the future by capitalizing on the chemical scaffolds established with covalent K-Ras-G12C inhibitors9. This opportunity conversely indicates a liability, as inhibitors with this scaffold may spuriously target multiple GTPases or non-mutant Ras isoforms leading to side effects.
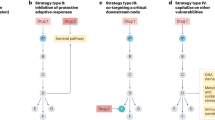
An obvious way to increase clinical efficacy and circumvent the occurrence of resistance is to apply drug combination regimes10. However, ideally, one would want to target an activity of oncogenic Ras that drives the emergence of resistant clones. Our current understanding of Ras is that it relays mitogenic stimuli into the cell and drives cell cycle progression and proliferation. Thus, it would seem plausible that oncogenic Ras-transformed clones have a proliferative advantage to outcompete other clones. This concept is implied in countless depictions of the Ras pathway topography, where the signal input at the top (mitogen) has a matched output at the bottom (proliferation). This simplifying pathway thinking, which predominates our current “systems-level” view, has its roots in genetic experiments performed in model organisms. Genetic dependency experiments could tell if a gene was up- or downstream of another. However, typically, these statements were limited to a certain developmental and/or tissue context.
How can we get back to the tissue and developmental stage-specific context of signaling events in Ras and cancer research? The role of Ras isoforms during development and cell differentiation is only poorly understood. Scant evidence suggests that the differentiation of cells and self-renewal of progenitors depend on specific Ras isoforms H-Ras, N-Ras, K-Ras4A, or K-Ras4B11,12. This is remarkable, as it points to Ras being involved in a fundamental process of metazoan life.
Given that several mouse models have already investigated the overall developmental impact of these Ras isoforms, their specific role in cell differentiation in simple cellular models may appear less noteworthy. However, the impressions obtained with Ras mouse models are not exactly coherent. On the one hand, knockouts of HRAS and/or NRAS are viable and were regarded as largely normal13,14, while on the other hand, recent data show that surviving double-knockouts of these two genes display RASopathy phenotypes15. It is surprising that single knockouts appear normal, while the double knockout shows a disease phenotype. This may suggest that RASopathies can also be associated with the loss of certain Ras isoforms, such as in the double knockout. Indeed, several RASopathy KRAS mutants carry also loss-of-function features, such as reduced effector binding16. Thus, MAPK-pathway activity is only slightly, if at all, elevated as compared to oncogenic hotspot RAS mutants. These are typically not seen in RASopathies, with the exception of HRAS-G12S-associated Costello syndrome. Its transgenic mouse model is driven by an HRAS-G12V mutation and displays grossly abnormal developmental defects similar to what is seen in human patients17. Given that HRAS mutations cause Costello syndrome in humans, while mutations in KRAS or NRAS are associated with the RASopathy Noonan syndrome, it appears that the type of developmental syndrome is also associated with specific Ras isoforms18.
Understanding the developmental activities of Ras requires the longitudinal examination of Ras activity and, probably, the study of signaling events localized to subcellular structures as compared to averaged signals in the bulk of cell lines or tissues11,19. On the flip side, targeting Ras trafficking to these subcellular structures may become again more relevant in drug development.
In this perspective, we will keep these big questions of the role of Ras isoforms during normal and abnormal development in mind when reflecting on untapped drug targeting opportunities in Ras trafficking pathways. For comprehensive recent reviews of Ras trafficking and membrane organization and its drug targeting potential en route to the plasma membrane, we refer to recent reviews20,21.
Ras lipid domains of the Golgi apparatus and the plasma membrane may offer new targeting opportunities
The cancer-associated Ras isoforms differ mostly in their last ~20 C-terminal residues and their engagement of the membrane via their allosteric lobe (residues 87-171), which largely determine their subcellular trafficking and lateral organization in plasma membrane lipid domains20,22. Therefore, trafficking and membrane localization must carry a significant amount of biological information that explains isoform differences. It is undisputed that Ras executes its major activity at the plasma membrane, where mitogen-sensing receptors are embedded. Ras can subsequently redistribute to many membranous organelles of the cell, whether it is internalized by endocytosis or detached by dissociation23,24. Targeting of Ras endocytosis may still hold drug development potential and deserves further investigation in the future.
Interference with both the de- and repalmitoylation cycles of Ras was shown to delocalize H-Ras, N-Ras, and K-Ras4A in cells24,25. Both H-Ras and N-Ras are trapped on the Golgi apparatus for S-palmitoylation and subsequent vesicular transport to the plasma membrane26. K-Ras4A is found on mitochondrial membranes when depalmitoylated, where it directly binds and activates hexokinase I as a novel effector27.
Inhibition of protein palmitoyltransferase activity has so far proven difficult28. The most commonly used compound, 2-bromopalmitate, is overall non-selective, blocking the formation of acylase intermediates and fatty acid biosynthesis. Interestingly, inhibition of depalmitoylating enzymes acyl protein thioesterases APT1 and α/β hydrolase ___domain 17 (ABHD17) by palmostatin B led to Ras being redistributed to internal membranes29,30. Remarkably, the APT1- and APT2-specific inhibitors ML348 and ML349, respectively, or knockdown of their targets does not inhibit the growth of NRAS mutant melanoma cells31. By contrast, palmostatin B does, suggesting that inhibition of ABHD17 is more relevant in this context (Fig. 1, left).
The dually palmitoylated Golgi-resident protein golgin subfamily A number 7 (GOLGA7) stabilizes the N-Ras and H-Ras protein acyltransferase ZDHHC9. Its knockout however led to the mislocalization specifically of N-Ras from the plasma membrane32. This might point to N-Ras and H-Ras being palmitoylated in distinct compartments of the Golgi. Targeting of such compartments may open new isoform-specific drug targeting opportunities.
Targeting the lateral segregation of Ras proteins into plasma membrane nanoclusters represents another untapped opportunity to block specific Ras isoforms. Ras nanoclusters are proteo-lipid assemblies of di-/oligomeric Ras, which serve as platforms for effector recruitment33. The recent years have seen great progress in our understanding of the lipid specificity that is encoded in the C-terminus of Ras. The polybasic lysine stretch of K-Ras4B does not electrostatically anchor the protein to the membrane, but instead adopts an ensemble of distinct conformations that mediate lipid recognition, in particular of phosphatidylserine (PS)34,35. PS is thereby important to maintain the proper lateral segregation between H-Ras and K-Ras4B and maintain nanoclusters of K-Ras4B20. Interference with PS trafficking by fendiline treatment reduced K-Ras4B nanoclustering, cancer cell proliferation, and tumor growth in vivo36. Likewise, ablation of PS transport proteins ORP5 and ORP8 significantly blocked growth of pancreatic cancer cell xenografts37. The Hancock group furthermore discovered that metabolic reprogramming by oncogenic KRAS alters a subset of glycosphingolipids on the outer leaflet. One of these, GM3, couples through the lipid bilayer to maintain PS on the inner leaflet and thus K-Ras4B nanoclustering38. Targeting of this intricate feedforward loop blocked pancreatic cancer cell growth in vitro and in vivo. However, with the inhibition of enzymes responsible for Ras lipidation or of lipids of specific membrane domains, a broad effect on the trafficking and membrane organization of many other proteins can be expected and may contribute to undesired biological effects.
Different oncogenic mutants of K-Ras4B display distinct conformational dynamics, leading to lipid headgroup and acyl chain-specific engagements of the G-___domain with the plasma membrane39. By using a covalent switch II pocket inhibitor that was modified with a long, lipophilic acyl chain, it could be demonstrated that restraining the plasma membrane orientation of K-Ras4B-G12C with such a compound disrupts its nanoclustering more potently than the parental MRTX84940. Therefore, taking the conformational dynamics of oncogenic Ras in its native membrane environment into account may present novel, more specific inhibitor design opportunities.
Targeting Ras prenylation and prenyl-binding chaperones
As mentioned at the outset, the historically first approach to target Ras was to inhibit farnesyltransferase, which culminated in the development of tipifarnib and lonafarnib. Once it was recognized that K-Ras and N-Ras become geranylgeranylated upon inhibition of farnesyltransferase, CAAX-mimetic inhibitors of geranylgeranyltransferase I and dual inhibitors targeting both prenyltransferases were developed. While GGTI-2418 progressed into the clinic, it was soon recognized that dual inhibition of farnesyltransferase and geranylgeranyltransferase was too toxic41.
Likewise, inhibition of other CAAX-processing enzymes, isoprenylcysteine carboxylmethyltransferase (ICMT) and Ras converting enzyme 1 (RCE1), may be too toxic due to their broad impact1. Relatively little progress was made in advancing inhibitors of these enzymes to the clinic, with the exception of salirasib, which inhibits ICMT with micromolar (Ki = 2.6 μM) potency42,43.
Three prenyl-binding trafficking chaperones have been identified, which solubilize the hydrophobic prenyl-moiety of several Ras proteins to allow for their long-range diffusion through the cytoplasm, thus feeding the cycle that transports Ras proteins to the plasma membrane44. These chaperones, SmgGDS-558, calmodulin (CaM), and PDE6D, promiscuously facilitate solubilization also of other small GTPases45,46,47. CaM in addition has the ability to extract K-Ras4B from the membrane47,48. Furthermore, the retromer coat protein VPS35 emerged from a proteomics screen as a specific trafficking chaperone of farnesylated N-Ras49. All these chaperones bind primarily the prenyl moiety of small GTPases, with some contribution of polybasic residues at the C-terminus of the cargo to CaM and SmgGDS-558 binding. Therefore, these chaperones generally do not discriminate between the active or inactive conformation of Ras. However, specific contacts between cargo and chaperone define distinct cargo specificities45,46,50,51,52. In general, palmitoylation close to the prenylated cysteine blocks binding to these chaperones. By contrast, carboxymethylation of the farnesylated cysteine increases the affinity of cargo proteins to the trafficking chaperone PDE6D50. Therefore, inhibitors of ICMT can be predicted to act synergistically with PDE6D inhibitors. Such an activity may be combined with the ICMT inhibitor salirasib, which, as a farnesyl-derivative, also seems to inhibit PDE6D (DA unpublished observations). This combination opportunity may kindle novel interest in the development of ICMT inhibitors.
Phosphorylation of Ser181 by PKC or PKG2 at the C-terminus of K-Ras4B reduces its affinity to both CaM and PDE6D50,53,54. This illustrates an interesting selectivity opportunity to inhibit the binding preferentially of only those few small GTPases to these chaperones, which can be phosphorylated at their C-terminus in a similar manner as K-Ras4B55. In combination with incomplete chaperone inhibition, this could limit the expected broad impact of full chaperone inhibition to potentially only a few small GTPases. Moreover, it is plausible to assume that the three trafficking chaperones have significantly overlapping functions. However, there may be (patho)biological conditions that are still insufficiently understood, where the role of one or the other chaperone predominates. Therefore, targeting of trafficking chaperones offers a more focused opportunity for the inhibition of prenylated proteins, as compared to the inhibition of prenyltransferases (Fig. 1, right).
Since the 1980s, several CaM inhibitors have been developed, which naturally target multiple activities of CaM beyond those of chaperoning Ras. CaM inhibiting neuroleptic phenothiazines at some stage even advanced into the clinic, given the involvement of CaM in cell proliferation56. The most potent non-covalent CaM inhibitor is calmidazolium57. The natural product ophiobolin A is a potent covalent inhibitor of CaM, but also reacts with several other proteins, explaining its broad toxicity. Recently a less toxic and more selective synthetic analog, calmirasone1, was identified, which more selectively reduces K-Ras4B membrane association58. Interestingly, these inhibitors also target the interaction of K-Ras4B with centrin1, a protein highly related to CaM59. Surprisingly, oncogenic K-Ras4B mutants show a higher engagement with both proteins, suggesting complexation in concert with effectors58,59,60,61. At the same time, centrin1 depletion has a minor effect on K-Ras4B membrane anchorage. Thus, CaM more so than centrin1 may act as a trafficking chaperone in cells, while in addition being in complex with active K-Ras4B and effectors59. It can be speculated that such complexes are formed during mitosis and cytokinesis when CaM localizes to the plasma membrane and both CaM and centrin1 are found on centrosomes59,62,63. At both locations, these chaperones may thus likewise serve as transient docking sites of K-Ras4B. However, a function of centrosomal K-Ras4B is not described.
The other major Ras trafficking chaperone, PDE6D was nominated as a surrogate target of K-Ras4B. This led to the development of multiple PDE6D inhibitors, notably by the Waldmann group64,65 (Table S2). Shortcomings of these inhibitors included off-target effects and poor solubility. The latter may seem inevitable when raising inhibitors against a highly hydrophobic pocket. While the recently developed inhibitor Deltaflexin3 overcomes these issues, it shares the relatively poor inhibitory activity on MAPK-signaling with the other compounds55. By combining it with the approved drug Sildenafil, which facilitates the phosphorylation of K-Ras4B Ser181 and thus lowers its PDE6D affinity, the inhibitory effect could be increased but remained modest. This is most easily rationalized by accepting that this trafficking chaperone contributes only to an estimated 25-50% of K-Ras4B membrane trafficking55. A way forward in PDE6D inhibitor development may be inspired by the fact that natural cargo modulates its affinity to the chaperone at the entrance of the prenyl-binding pocket51.
A new perspective on how to target Ras trafficking in diseases
The natural cargos of PDE6D include many proteins destined for the primary cilium, a crucial antenna-like organelle on the surface of stem- and progenitor-cells, which senses developmental signaling cues51. The cilium emerges from the basal body, which originates from the mother centrosome in resting cells. K-Ras4B has been detected inside the cilium, in line with the fact that PDE6D-cargo release factor Arl3 is active in the cilium and can facilitate ejection of both high- and low-affinity cargo, such as K-Ras4B into the cilium66,67. However, in another instance only a mutant with increased PDE6D affinity was detected in the cilium, while the wild-type K-Ras4B was not68.
It is interesting to note that both PDE6D and CaM are associated with the two centriolar organelles, the primary cilium and centrosome. Both organelles are not only involved in cell division but have distinctive roles during cell differentiation, where inheritance of the mother centrosome and the remnant of the primary cilium protect stemness in the inheriting daughter cell69. Importantly, this affords a general mechanism for asymmetric cell divisions, a process that is critical as tissues need to regenerate in the adult organism.
The profound importance of PDE6D during development is highlighted by the fact that its germline loss-of-function mutations are associated with the ciliopathy Joubert-Syndrome, which affects multiple organ systems70. By contrast, CaM is not directly associated with a developmental disease, which can be explained by the fact that humans have three gene copies of it, CALM1-3. Instead, CaM is associated with the “ADORA2B mediated anti-inflammatory cytokine production” SuperPath (https://tinyurl.com/2d2wapxc), which incidentally is associated with several RASopathies (https://tinyurl.com/3s9dx3a5). ADORA2B is an adenosine-activated G-protein coupled receptor that can influence the Ras-pathway via its second messenger cAMP and is negatively associated with cancer survival, while its activation is positively associated with axon regeneration in mouse retinal ganglion cells71,72. Indeed, ciliopathies and RASopathies share several of their multi-organ phenotypic defects, including musculoskeletal and neurodevelopmental defects, and some RASopathies exhibit loss of cilia in animal models73.
Could this mean that Ras proteins have a role in the functioning of the primary cilium? It would place Ras at the center of a stem- and progenitor-cell resident organelle that regulates major developmental decisions, which are not only important during organismal development but also during repair in the adult74.
Cancer has often been described as ‘the wound that never heals’, i.e. an incomplete repair process. In this context, it is interesting to note that oncogenic Ras transformation alone can significantly block terminal cell differentiation11,75,76. This process would not only keep cells in a less differentiated state but also allow them to continue proliferating slowly, thus imparting two major hallmarks observed in cancer. Implicitly, this would mean that transformation by Ras alone sustains clones that continue to proliferate and are not fully differentiated and immaturity traits that could appear as “stem-like” such as seen in cancer stem cells19. Moreover, Ras-transformed cells and cancer cells typically do not form a primary cilium, thus transformation also subtracts from the pool of tissue-resident stem-/progenitor cells that would promote normal repair.
Given our currently still insufficient knowledge about the details of the underlying cellular and molecular processes, it remains difficult to predict if inhibition, in particular of K-Ras4B trafficking to the cilium or the centrosome, would be beneficial in Ras-driven diseases. However, it is clear that targeting K-Ras4B trafficking may not only affect processes associated with plasma membrane-bound K-Ras4B but also those (unknown) processes tied to other organelles. Hence, the microscopic and mesoscopic spatio-temporal context, i.e. inside the cell during the cell cycle, or in tissues during development, should probably become part of the consideration of how to target K-Ras4B (trafficking) in diseases. By combining Ras trafficking inhibitors with currently developed direct Ras-inhibitors, one may not only identify novel synergistic activities but possibly also enable fine tuning of the total inhibitory activity.
We conclude that inhibition of specific K-Ras4B trafficking routes still holds some potential. To enable this, we advocate to reexamine the trafficking-dependent mechanisms underlying the disease-associated differentiation activities of Ras. In the foreseeable future, however, direct Ras inhibitors will dominate the development of therapies against aberrant Ras activities.
Data availability
No datasets were generated or analyzed during the current study.
References
Berndt, N., Hamilton, A. D. & Sebti, S. M. Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 11, 775–791 (2011).
Suzuki, M. et al. FDA approval summary for lonafarnib (Zokinvy) for the treatment of Hutchinson-Gilford progeria syndrome and processing-deficient progeroid laminopathies. Genet Med. 25, 100335 (2023).
Ho, A. L. et al. Tipifarnib in head and neck squamous cell carcinoma With HRAS mutations. J. Clin. Oncol. 39, 1856–1864 (2021).
Punekar, S. R., Velcheti, V., Neel, B. G. & Wong, K. K. The current state of the art and future trends in RAS-targeted cancer therapies. Nat. Rev. Clin. Oncol. 19, 637–655 (2022).
Steffen, C. L., Kaya, P., Schaffner-Reckinger, E. & Abankwa, D. Eliminating oncogenic RAS: back to the future at the drawing board. Biochem. Soc. Trans. 51, 447–456 (2023).
Sharma, A. K. et al. Revealing the mechanism of action of a first-in-class covalent inhibitor of KRASG12C (ON) and other functional properties of oncogenic KRAS by (31)P NMR. J. Biol. Chem. 300, 105650 (2024).
Kim, D. et al. Pan-KRAS inhibitor disables oncogenic signalling and tumour growth. Nature 619, 160–166 (2023).
Holderfield, M. et al. Concurrent inhibition of oncogenic and wild-type RAS-GTP for cancer therapy. Nature 629, 919–926 (2024).
Morstein, J. et al. Targeting Ras-, Rho-, and Rab-family GTPases via a conserved cryptic pocket. Cell, https://doi.org/10.1016/j.cell.2024.08.017 (2024).
Sattler, M., Mohanty, A., Kulkarni, P. & Salgia, R. Precision oncology provides opportunities for targeting KRAS-inhibitor resistance. Trends Cancer 9, 42–54 (2023).
Chippalkatti, R. et al. RAS isoform-specific activities are disrupted by disease-associated mutations during cell differentiation. Eur. J. Cell Biol. 103, 151425 (2024).
Quinlan, M. P., Quatela, S. E., Philips, M. R. & Settleman, J. Activated Kras, but not Hras or Nras, may initiate tumors of endodermal origin via stem cell expansion. Mol. Cell Biol. 28, 2659–2674 (2008).
Malumbres, M. & Barbacid, M. RAS oncogenes: the first 30 years. Nat. Rev. Cancer 3, 459–465 (2003).
Esteban, L. M. et al. Targeted genomic disruption of H-ras and N-ras, individually or in combination, reveals the dispensability of both loci for mouse growth and development. Mol. Cell Biol. 21, 1444–1452 (2001).
Fuentes-Mateos, R. et al. Combined HRAS and NRAS ablation induces a RASopathy phenotype in mice. Cell Commun. Signal 22, 332 (2024).
Gremer, L. et al. Germline KRAS mutations cause aberrant biochemical and physical properties leading to developmental disorders. Hum. Mutat. 32, 33–43 (2011).
Schuhmacher, A. J. et al. A mouse model for Costello syndrome reveals an Ang II-mediated hypertensive condition. J. Clin. Investig. 118, 2169–2179 (2008).
Rauen, K. A. The RASopathies. Annu Rev. Genom. Hum. Genet 14, 355–369 (2013).
Chippalkatti, R. & Abankwa, D. Promotion of cancer cell stemness by Ras. Biochem. Soc. Trans. 49, 467–476 (2021).
Zhou, Y., Gorfe, A. A. & Hancock, J. F. RAS nanoclusters selectively sort distinct lipid headgroups and acyl chains. Front. Mol. Biosci. 8, 686338 (2021).
Pavic, K., Chippalkatti, R. & Abankwa, D. Drug targeting opportunities en route to Ras nanoclusters. Adv. Cancer Res. 153, 63–99 (2022).
Abankwa, D., Gorfe, A. A., Inder, K. & Hancock, J. F. Ras membrane orientation and nanodomain localization generate isoform diversity. Proc. Natl. Acad. Sci. USA 107, 1130–1135 (2010).
Schmick, M. et al. KRas localizes to the plasma membrane by spatial cycles of solubilization, trapping and vesicular transport. Cell 157, 459–471 (2014).
Rocks, O. et al. The palmitoylation machinery is a spatially organizing system for peripheral membrane proteins. Cell 141, 458–471 (2010).
Tsai, F. D. et al. K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc. Natl. Acad. Sci. USA 112, 779–784 (2015).
Rocks, O. et al. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science 307, 1746–1752 (2005).
Amendola, C. R. et al. KRAS4A directly regulates hexokinase 1. Nature 576, 482–486 (2019).
Hu, X. et al. A mini review of small-molecule inhibitors targeting palmitoyltransferases. Eur. J. Med. Chem. Rep. 5, 100041 (2022).
Dekker, F. J. et al. Small-molecule inhibition of APT1 affects Ras localization and signaling. Nat. Chem. Biol. 6, 449–456 (2010).
Lin, D. T. & Conibear, E. ABHD17 proteins are novel protein depalmitoylases that regulate N-Ras palmitate turnover and subcellular localization. Elife 4, e11306 (2015).
Vujic, I. et al. Acyl protein thioesterase 1 and 2 (APT-1, APT-2) inhibitors palmostatin B, ML348 and ML349 have different effects on NRAS mutant melanoma cells. Oncotarget 7, 7297–7306 (2016).
Liu, C. et al. GOLGA7 is essential for NRAS trafficking from the Golgi to the plasma membrane but not for its palmitoylation. Cell Commun. Signal 22, 98 (2024).
Abankwa, D. & Gorfe, A. A. Mechanisms of Ras membrane organization and signaling: Ras rocks again. Biomolecules 10, https://doi.org/10.3390/biom10111522 (2020).
Zhou, Y., Prakash, P. S., Liang, H., Gorfe, A. A. & Hancock, J. F. The KRAS and other prenylated polybasic ___domain membrane anchors recognize phosphatidylserine acyl chain structure. Proc. Natl. Acad. Sci. USA 118, https://doi.org/10.1073/pnas.2014605118 (2021).
Zhou, Y. et al. Lipid-sorting specificity encoded in K-Ras membrane anchor regulates signal output. Cell 168, 239–251 e216 (2017).
van der Hoeven, D. et al. Sphingomyelin metabolism is a regulator of K-Ras function. Mol. Cell Biol. 38, https://doi.org/10.1128/MCB.00373-17 (2018).
Kattan, W. E. et al. Components of the phosphatidylserine endoplasmic reticulum to plasma membrane transport mechanism as targets for KRAS inhibition in pancreatic cancer. Proc. Natl. Acad. Sci. USA 118, https://doi.org/10.1073/pnas.2114126118 (2021).
Liu, J. et al. Glycolysis regulates KRAS plasma membrane localization and function through defined glycosphingolipids. Nat. Commun. 14, 465 (2023).
Arora, N., Mu, H., Liang, H., Zhao, W. & Zhou, Y. RAS G-domains allosterically contribute to the recognition of lipid headgroups and acyl chains. J. Cell Biol. 223, https://doi.org/10.1083/jcb.202307121 (2024).
Morstein, J. et al. Direct modulators of K-Ras-membrane interactions. ACS Chem. Biol. 18, 2082–2093 (2023).
Lobell, R. B. et al. Evaluation of farnesyl:protein transferase and geranylgeranyl:protein transferase inhibitor combinations in preclinical models. Cancer Res. 61, 8758–8768 (2001).
Marom, M. et al. Selective inhibition of Ras-dependent cell growth by farnesylthiosalisylic acid. J. Biol. Chem. 270, 22263–22270 (1995).
Riely, G. J. et al. A phase II trial of Salirasib in patients with lung adenocarcinomas with KRAS mutations. J. Thorac. Oncol. 6, 1435–1437 (2011).
Schmick, M., Kraemer, A. & Bastiaens, P. I. Ras moves to stay in place. Trends Cell Biol. 25, 190–197 (2015).
Brandt, A. C., Koehn, O. J. & Williams, C. L. SmgGDS: an emerging master regulator of prenylation and trafficking by small GTPases in the Ras and Rho families. Front. Mol. Biosci. 8, 685135 (2021).
Chandra, A. et al. The GDI-like solubilizing factor PDEdelta sustains the spatial organization and signalling of Ras family proteins. Nat. Cell Biol. 14, 148–158 (2011).
Grant, B. M. M. et al. Calmodulin disrupts plasma membrane localization of farnesylated KRAS4b by sequestering its lipid moiety. Sci. Signal. 13, https://doi.org/10.1126/scisignal.aaz0344 (2020).
Sperlich, B., Kapoor, S., Waldmann, H., Winter, R. & Weise, K. Regulation of K-Ras4B membrane binding by calmodulin. Biophys. J. 111, 113–122 (2016).
Zhou, M. et al. VPS35 binds farnesylated N-Ras in the cytosol to regulate N-Ras trafficking. J. Cell Biol. 214, 445–458 (2016).
Dharmaiah, S. et al. Structural basis of recognition of farnesylated and methylated KRAS4b by PDEd delta. Proc. Natl. Acad. Sci. USA 113, E6766–E6775 (2016).
Fansa, E. K., Kosling, S. K., Zent, E., Wittinghofer, A. & Ismail, S. PDE6delta-mediated sorting of INPP5E into the cilium is determined by cargo-carrier affinity. Nat. Commun. 7, 11366 (2016).
Grant, B. M. M., Enomoto, M., Ikura, M. & Marshall, C. B. A non-canonical calmodulin target motif comprising a polybasic region and lipidated terminal residue regulates localization. Int. J. Mol. Sci. 21, https://doi.org/10.3390/ijms21082751 (2020).
Alvarez-Moya, B., Lopez-Alcala, C., Drosten, M., Bachs, O. & Agell, N. K-Ras4B phosphorylation at Ser181 is inhibited by calmodulin and modulates K-Ras activity and function. Oncogene 29, 5911–5922 (2010).
Cho, K. J. et al. AMPK and endothelial nitric oxide synthase signaling regulates K-Ras plasma membrane interactions via Cyclic GMP-dependent protein kinase 2. Mol. Cell Biol. 36, 3086–3099 (2016).
Kaya, P. et al. An improved PDE6D inhibitor combines with sildenafil to inhibit KRAS mutant cancer cell growth. J. Med. Chem. 67, 8569–8584 (2024).
Hait, W. N. et al. The effect of calmodulin inhibitors with bleomycin on the treatment of patients with high-grade gliomas. Cancer Res. 50, 6636–6640 (1990).
Manoharan, G. B., Kopra, K., Eskonen, V., Harma, H. & Abankwa, D. High-throughput amenable fluorescence-assays to screen for calmodulin-inhibitors. Anal. Biochem. 572, 25–32 (2019).
Okutachi, S. et al. A covalent calmodulin inhibitor as a tool to study cellular mechanisms of K-Ras-driven stemness. Front. Cell Dev. Biol. 9, 665673 (2021).
Manoharan, G. B., Laurini, C., Bottone, S., Ben Fredj, N. & Abankwa, D. K. K-Ras binds calmodulin-related centrin1 with potential implications for K-Ras driven cancer cell stemness. Cancers 15, https://doi.org/10.3390/cancers15123087 (2023).
Abraham, S. J., Nolet, R. P., Calvert, R. J., Anderson, L. M. & Gaponenko, V. The hypervariable region of K-Ras4B is responsible for its specific interactions with calmodulin. Biochemistry 48, 7575–7583 (2009).
Villalonga, P. et al. Calmodulin binds to K-Ras, but not to H- or N-Ras, and modulates its downstream signaling. Mol. Cell Biol. 21, 7345–7354 (2001).
Li, C. J. et al. Dynamic redistribution of calmodulin in HeLa cells during cell division as revealed by a GFP-calmodulin fusion protein technique. J. Cell Sci. 112, 1567–1577 (1999).
Yu, Y. Y. et al. The association of calmodulin with central spindle regulates the initiation of cytokinesis in HeLa cells. Int. J. Biochem. Cell Biol. 36, 1562–1572 (2004).
Martin-Gago, P., Fansa, E. K., Wittinghofer, A. & Waldmann, H. Structure-based development of PDEdelta inhibitors. Biol. Chem. 398, 535–545 (2017).
Siddiqui, F. A. et al. PDE6D inhibitors with a new design principle selectively block K-Ras activity. ACS Omega 5, 832–842 (2020).
Fansa, E. K. & Wittinghofer, A. Sorting of lipidated cargo by the Arl2/Arl3 system. Small GTPases 7, 222–230 (2016).
Lauth, M. et al. DYRK1B-dependent autocrine-to-paracrine shift of Hedgehog signaling by mutant RAS. Nat. Struct. Mol. Biol. 17, 718–725 (2010).
Yelland, T. et al. Stabilization of the RAS:PDE6D complex is a novel strategy to inhibit RAS signaling. J. Med. Chem. 65, 1898–1914 (2022).
Chen, C. & Yamashita, Y. M. Centrosome-centric view of asymmetric stem cell division. Open Biol. 11, 200314 (2021).
Thomas, S. et al. A homozygous PDE6D mutation in Joubert syndrome impairs targeting of farnesylated INPP5E protein to the primary cilium. Hum. Mutat. 35, 137–146 (2014).
Arora, C. et al. The landscape of cancer-rewired GPCR signaling axes. Cell Genom. 4, 100557 (2024).
Wang, F. et al. Gliotransmission and adenosine signaling promote axon regeneration. Dev. Cell 58, 660–676 e667 (2023).
Patterson, V. L. & Burdine, R. D. Swimming toward solutions: using fish and frogs as models for understanding RASopathies. Birth Defects Res. 112, 749–765 (2020).
Palla, A. R. et al. Primary cilia on muscle stem cells are critical to maintain regenerative capacity and are lost during aging. Nat. Commun. 13, 1439 (2022).
Yohe, M. E. et al. MEK inhibition induces MYOG and remodels super-enhancers in RAS-driven rhabdomyosarcoma. Sci. Transl. Med. 10, https://doi.org/10.1126/scitranslmed.aan4470 (2018).
Dajee, M., Tarutani, M., Deng, H., Cai, T. & Khavari, P. A. Epidermal Ras blockade demonstrates spatially localized Ras promotion of proliferation and inhibition of differentiation. Oncogene 21, 1527–1538 (2002).
Acknowledgements
Apologies to all authors who could not be cited due to space constraints. This work was supported by the Luxembourg National Research Fund (FNR) grants AFR/23/17112420/Bil_ABANKWA_SPREDCanUL2 and INTER/FWO/23/18086068 molGluRAS2 to DKA.
Author information
Authors and Affiliations
Contributions
AGM—Prepared text sections, reviewed the literature, compiled compound Table S1 and prepared the figure. ESR—Prepared text sections, reviewed the literature, compiled compound Table S2 and prepared the figure. DKA—Wrote and edited the text. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schaffner-Reckinger, E., Gómez-Mulas, A. & Abankwa, D.K. Drugging Ras trafficking—are there new roads to travel?. npj Drug Discov. 2, 9 (2025). https://doi.org/10.1038/s44386-025-00012-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s44386-025-00012-7
