
Abstract
Alcohol-associated liver disease (ALD) is a major global health challenge, with inflammation playing a central role in its progression. As inflammation emerges as a critical therapeutic target, ongoing research aims to unravel its underlying mechanisms. This review explores the immunological pathways of ALD, highlighting the roles of immune cells and their inflammatory mediators in disease onset and progression. We also examine the complex interactions between inflammatory cells and non-parenchymal liver cells, as well as their crosstalk with extra-hepatic organs, including the gut, adipose tissue, and nervous system. Furthermore, we summarize current clinical research on anti-inflammatory therapies and discuss promising therapeutic targets. Given the heterogeneity of ALD-associated inflammation, we emphasize the need for precision medicine to optimize treatment strategies and improve patient outcomes.
Similar content being viewed by others

Introduction
The widespread misuse of alcohol poses a major global health threat, leading to profound social, economic, and medical consequences. Rising alcohol consumption has been accompanied by a significant increase in disease burden and mortality rates worldwide [1, 2]. According to the World Health Organization, nearly 75 million people suffer from alcohol-related conditions, with ~3 million deaths annually attributed to alcohol consumption [3]. Furthermore, global alcohol intake continues to rise, with projections estimating an increase in average per capita consumption from 8 liters to 8.4 liters by 2025 [4, 5].
The liver is particularly vulnerable to alcohol-induced damage due to its central role in metabolizing alcohol and filtering toxins from the bloodstream [6, 7]. The byproducts of alcohol metabolism can cause significant liver injury, leading to alcohol-associated liver disease (ALD) [8, 9]. ALD encompasses a spectrum of conditions, ranging from simple steatosis to more severe forms such as steatohepatitis, cirrhosis, and fibrosis, ultimately progressing to hepatocellular carcinoma [10]. Among these, severe alcohol-associated hepatitis (sAH) is the most life-threatening, with 28-day and 90-day mortality rates of 20% and 30%, respectively [11, 12]. Despite advances in basic and translational research, the molecular mechanisms driving AH pathogenesis remain largely unclear.
Despite growing public awareness of alcohol-induced liver toxicity, alcohol consumption continues to rise [1, 13]. Currently, the U.S. Food and Drug Administration has not approved any pharmaceutical treatments for ALD, underscoring the urgent need for effective therapies [14]. While the precise pathophysiological mechanisms of ALD remain incompletely understood, strong evidence links inflammation to its progression. In ALD, liver inflammation results from both direct and indirect hepatic injury caused by excessive alcohol intake. Ethanol, acetaldehyde, and lipopolysaccharide (LPS) activate inflammatory cascades that drive alcohol-induced liver damage [15]. Although inflammation is a protective response to harmful stimuli, it also plays a central role in ALD pathogenesis [16, 17]. This review explores the role of inflammation in ALD development and progression and examines current and emerging therapeutic targets aimed at mitigating inflammation in this disease.
Causes and progression factors of liver inflammation
Direct damage caused by alcohol metabolites
Ethanol metabolism in the liver occurs through three primary pathways: the alcohol dehydrogenase (ADH) pathway in the hepatocyte cytosol, the cytochrome P450 2E1 (CYP2E1) pathway in the endoplasmic reticulum, and the catalase (CAT) pathway in peroxisomes [18]. These processes convert ethanol into the toxic intermediate acetaldehyde [2, 19]. Acetaldehyde is further metabolized into acetate by mitochondrial acetaldehyde dehydrogenase 2 (ALDH2) and cytosolic aldehyde dehydrogenase (ALDH1) [18]. The resulting acetate is released into the bloodstream and oxidized to carbon dioxide in extrahepatic tissues [7, 19]. Ethanol disrupts mitochondrial respiratory function, leading to structural and functional impairments [20]. Acetaldehyde is even more toxic than ethanol due to its high reactivity, forming covalent adducts with proteins [21, 22], phospholipids [23], and nucleic acids [24], thereby altering their biological functions. Notably, a recent study revealed that a significant portion of acetaldehyde produced in the liver is excreted into the intestine via bile, where it is further metabolized by intestinal epithelial ALDH2 [25]. This finding suggests that targeting ALDH2 in both the liver and intestine may offer a novel therapeutic approach for ALD.
Chronic alcohol consumption leads to increased hepatic levels of CYP2E1 [18, 26]. While enhanced alcohol metabolism may seem protective, elevated CYP2E1 accelerates ethanol oxidation, resulting in higher acetaldehyde production and excessive reactive oxygen species (ROS) generation, including hydroxyethyl radicals, superoxide anions, and hydroxyl radicals [27, 28]. The accumulation of ROS damages mitochondrial DNA and proteins, leading to glutathione depletion [29]. Notably, ROS can react with unsaturated lipids to form lipid peroxides such as malondialdehyde, 4-hydroxy-2-nonenal (4-HNE), and acrolein, further exacerbating oxidative stress [30, 31]. Hepatocyte injury caused by acetaldehyde and ROS generates damage-associated molecular patterns (DAMPs), which activate inflammatory signaling, drive apoptosis and necrosis, and contribute to ALD progression [32, 33].
Gut microbial disorders
Ethanol and acetaldehyde can impair the intestinal mucosa and alter gut microbiota composition, facilitating the translocation of pathogen-associated molecular patterns (PAMPs), such as LPS, peptidoglycan, and lipoproteins, from the gut to the liver [34]. Gut-derived antigens induce distinct immune activation in hepatic parenchymal and non-parenchymal cells through engagement of pattern recognition receptors (PRRs), including Toll-like receptors (TLRs) [35, 36], NOD-like receptors (NLRs) [37], and C-type lectin receptors (CLRs) [38]. A prototypical example involves LPS activation of Kupffer cells (KCs) through TLR4 signaling, which promotes the secretion of pro-inflammatory mediators such as CC-chemokine ligand 2 (CCL2), interleukin (IL)-8, tumor necrosis factor-α (TNF-α), IL-1β, and IL-6 [35, 39]. These cytokines and chemokines recruit circulating immune cells into the liver, disrupting immune homeostasis and promoting liver injury [35, 39].
Activation of immune responses
The migration of inflammatory cells and activation of the inflammatory cascade can indirectly contribute to liver injury. The pathogenic cascade is initiated by innate immune activation, characterized by KCs mobilization and neutrophil infiltration [8]. DAMPs released by dying hepatocytes, along with PAMPs, stimulate Kupffer cells to upregulate proinflammatory cytokines, leading to the recruitment of neutrophils and macrophages to the liver [40, 41]. Neutrophil migration into the parenchyma exacerbates hepatocellular damage through ROS generation and proteolytic enzyme release, ultimately contributing to alcohol-induced hepatocyte apoptosis [42]. Protein adducts and neoantigens generated through oxidative stress and lipid peroxidation further provoke an adaptive immune response, characterized by T-cell and B-cell infiltration [8]. Nevertheless, the precise mechanisms underlying adaptive immunity-mediated hepatocellular injury and inflammatory amplification in AH patients remain elusive. Chronic ethanol exposure not only induces hepatocyte death but also suppresses regenerative proliferation, thereby creating a pathological microenvironment conducive to ALD progression [43]. Additionally, liver injury activates hepatic stellate cells (HSCs), which secrete transforming growth factor-β (TGF-β) and collagen, ultimately driving liver fibrosis [44]. (Fig. 1).

Triggers of Immune Response in ALD. Acetaldehyde and reactive oxygen species (ROS) generated during alcohol metabolism activate inflammatory signaling pathways, leading to hepatocyte injury, apoptosis, and necrosis. These events trigger the release of inflammatory mediators and damage-associated molecular patterns (DAMPs). Chronic alcohol consumption disrupts the intestinal barrier, increasing permeability and facilitating gut-derived microbial products’ translocation to the liver via the portal vein, thereby initiating an immune response. The nervous system modulates hepatic immune cell phenotypes through sympathetic signaling and neurotransmitters. Additionally, adipose-liver crosstalk, mediated by cytokines, adipokines, and metabolic signals, further amplifies immune activation and promotes the release of pro-inflammatory cytokines and chemokines. Collectively, these inter-organ interactions drive liver inflammation, exacerbate hepatocellular damage, and contribute to the progression of ALD
Roles of immune cells
Macrophages
Macrophages in the liver can be broadly classified into two categories. The first group consists of KCs, the largest resident macrophage population in the liver [45]. They serve as the primary line of defense, clearing pathogens, endotoxins, and damaged cell debris from the bloodstream while also regulating liver immunity [45]. The second category comprises monocyte-derived macrophages (MoMFs), which migrate from the peripheral blood and differentiate into macrophages in response to liver injury or inflammation, playing key roles in immune regulation and tissue repair [46, 47].
Chronic alcohol intake disrupts gut microbiota and compromises intestinal barrier function, allowing bacterial endotoxins such as LPS to enter the bloodstream [48]. LPS binds to Toll-like receptor 4 (TLR4) on the surface of Kupffer cells, activating both the MyD88-dependent pathway, which mediates NF-κB signaling, and the MyD88-independent pathway involving interferon regulatory factor 3 (IRF3), ultimately upregulating TNF-α transcription [49, 50]. Additionally, TLR4 signaling triggers the extracellular signal-regulated kinase and p38 MAPK pathways, further increasing the expression of downstream molecules and promoting the production of TNF-α and other inflammatory cytokines and chemokines [51, 52].
During alcohol exposure, Kupffer cells are a major source of ROS, which contribute to the generation of TNF-α and IL-6 [53]. These inflammatory cytokines production amplifies immune cell activation and recruitment, perpetuating hepatic inflammation. Notably, Kupffer cells also play a critical role in liver regeneration. Through NF-κB and signal transducer and activator of transcription 3 activation, they release TNF-α and IL-6 that reduce hepatocyte apoptosis and promote hepatocyte proliferation, facilitating liver recovery after alcohol-induced injury [54,55,56]. Given their dual role in inflammation and regeneration, indiscriminate inhibition of Kupffer cells may not be an ideal therapeutic approach for ALD. The endoplasmic reticulum (ER) protein Nogo-B (also known as Reticulon 4B) promotes a pro-inflammatory state, exacerbating liver injury and steatosis [57]. The absence of Nogo-B increases ER stress, which shifts Kupffer cells/macrophages toward an anti-inflammatory phenotype, thereby reducing liver inflammation [57]. Targeting Nogo-B may present a potential therapeutic approach for ALD. In response to hepatocyte injury, Kupffer cells release CCL2, facilitating monocyte recruitment and migration to the liver, where they differentiate into pro-inflammatory macrophages [58]. The chemokine receptors CCR2 and CCR5 mediate interactions among intrahepatic immune cells, promoting the activation and migration of peripheral monocytes into the liver [59, 60]. Consequently, the upregulation of these key receptors is a hallmark of ALD [61]. Given their role in disease progression, modulating macrophage phenotypic transformation and simultaneously inhibiting CCR2 and CCR5 represent promising therapeutic strategies for ALD. However, it is important to note that the recent Phase III Randomized Study on Cenicriviroc (a dual CCR2/CCR5 antagonist) has not demonstrated the efficacy of Cenicriviroc in treating liver fibrosis in adult patients with NASH [62]. Moreover, considering issues such as the safety of the drug, the development of targeted therapies targeting CCR2/CCR5 for the treatment of ALD requires the cautious approach of relevant researchers.
The precise role of MoMFs in ALD remains incompletely understood. MoMFs (CD11bhiF4/80lntLy6C+) and KCs (CD11blowF4/80hiLy6C−) can be distinguished by their differential expression of cell surface markers [47]. Chronic alcohol consumption increases the number of recruited macrophages in the liver, promoting the differentiation of Ly6Chi monocytes into pro-inflammatory macrophages that contribute to tissue damage [47]. Additionally, the phagocytosis of apoptotic hepatocytes enables Ly6Chi monocytes/macrophages to transition into Ly6Clow monocytes/macrophages, which subsequently differentiate into tissue-protective macrophages [47]. The balance between these two subpopulations is thought to dictate the functional role of recruited macrophages in ALD pathogenesis [47, 63].
Neutrophils
Neutrophils play a critical role in the immune system, primarily mediating inflammatory responses. They contribute significantly to tissue repair and immune regulation [64, 65] by coordinating immune and inflammatory processes through phagocytosis, ROS generation, degranulation, cytokine and chemokine production, and the release of neutrophil extracellular traps (NETs) [66,67,68,69]. These mechanisms collectively influence alcohol-induced liver injury [66,67,68,69]. Both circulating and hepatic neutrophil counts are elevated in patients with AH [70], and the neutrophil-to-lymphocyte ratio correlates with AH-related mortality [71, 72]. A variety of CXC and CC chemokines are significantly upregulated in AH patients compared to healthy individuals, with their expression levels positively associated with neutrophil infiltration and the severity of liver injury [73, 74]. IL-8, a key neutrophil chemoattractant, binds to CXCR1 and CXCR2 receptors on neutrophil surfaces, promoting their chemotaxis and infiltration into the liver [75]. Moreover, liver sinusoidal endothelial cells (LSECs) contribute to neutrophil recruitment by upregulating the expression of adhesion molecules such as E-selectin [76].
The activation of neutrophils in ALD is primarily driven by toxic metabolic byproducts of alcohol metabolism and oxidative stress responses [45, 77, 78]. Upon activation, neutrophils release various inflammatory mediators, including cytokines, chemokines, and ROS, which exacerbate liver inflammation [45, 77, 78]. Alcohol exposure induces neutrophils to release inflammatory cytokines such as TNF-α and IL-1β, while also enhancing the production of additional chemokines, such as IL-8/CXCL8 [75, 79]. Alcohol-induced phosphorylation of the Bruton’s tyrosine kinase plays a key role in neutrophil activation, increased granulopoiesis, and neutrophil-mediated liver damage [80]. NETs, which serve to capture and eliminate pathogens, can contribute to tissue damage and perpetuate inflammatory responses when produced in excessive amounts [78]. Intestinal-derived endotoxins can activate neutrophils via the TLR4 pathway, promoting their migration and NET release [68]. Platelet activation also stimulates NET formation, which plays a key role in liver injury and inflammation [68]. Additionally, histones within NETs can bind to TLR9 on HSCs, activating them and stimulating their proliferation and extracellular matrix synthesis, thus accelerating liver fibrosis [81]. While neutrophils contribute to the progression of ALD, neutrophil-specific miR-223 appears to exert a protective effect against liver injury. Studies have shown that serum miR-223 levels are elevated in patients with AH, and miR-223 deficiency exacerbates ethanol-induced inflammation, oxidative stress, and liver damage [82]. The contrasting effects of neutrophils in ALD may be partly due to the heterogeneity within the neutrophil population [83, 84].
Recent research suggests that neutrophils can be categorized into two subtypes: high-density neutrophils (HDNs) and low-density neutrophils (LDNs) [84]. HDNs exhibit a hyperactivated phenotype, capable of producing more ROS and NETs, which aggravate liver injury [84]. In contrast, LDNs display a functionally exhausted phenotype, with reduced chemotactic and immune functions, impairing their ability to eliminate pathogens and necrotic cells [84]. HDN-derived NETs can promote the retention of LDNs in the liver, preventing their clearance by macrophages. The accumulation of LDNs further suppresses immune responses and increases the risk of infection [84]. Further exploration of neutrophil heterogeneity is essential for gaining a deeper understanding of their role in the pathogenesis of AH.
Recently, a study utilized single-cell RNA sequencing to identify a distinct population of IL-8+ neutrophils in human sAH samples [75]. Upon infiltrating the liver, peripheral neutrophils from sAH patients are activated by the inflammatory milieu, including mediators such as TNF-α and IL-1β, leading to elevated expression of IL-8 [75]. The resultant production of IL-8+ neutrophils exacerbates systemic inflammation and further recruits additional neutrophils to the liver. This self-perpetuating recruitment of IL-8+ neutrophils in sAH patients may culminate in uncontrolled hepatic inflammation and subsequent liver failure [75]. This significant discovery suggests that targeting IL-8+ neutrophils could be a promising therapeutic strategy, potentially mitigating the inflammatory response specific to sAH without impacting other hepatic conditions [75, 85].
B cells
Acetaldehyde and other intermediate products produced during alcohol metabolism are immunogenic and can bind to proteins in hepatocytes, forming new antigenic epitopes [86]. B cells, as antigen-presenting cells, can recognize and internalize these antigens, process them, and present them to T cells, thereby activating T-cell-mediated immune responses [87, 88]. Antibodies produced by B cells bind to acetaldehyde-protein adducts and other antigens, forming immune complexes [89]. These complexes can deposit on hepatocyte membranes or LSECs, activating the complement system and triggering local inflammatory responses, leading to hepatocyte damage [89]. Ahmadi et al. identified unique antibodies in the liver of sAH, which could not only recognize bacterial (Escherichia coli) antigens but also cross-react with numerous human antigens. The deposition of a considerable amount of antibodies, along with complement activation and immune cell activation, might result in acute liver failure due to antibody-mediated inflammation [90].
Due to alcohol-induced inhibition of B lymphocyte differentiation, patients with ALD experience a reduction in B lymphocytes, which are essential for humoral immune responses [91]. Elevated levels of immunoglobulin A in circulation are a characteristic feature of alcohol-related cirrhosis, arising from TLR9 activation in B cells [92]. B cells play a complex, dual role in the onset and progression of ALD. On one hand, B cells contribute to immune surveillance and inflammatory responses in the liver by recognizing antigens and secreting cytokines and antibodies, which facilitate hepatocyte damage and liver fibrosis [92]. On the other hand, IgA synthesized by B cells in the liver plays a crucial role in clearing antigens from the gut and is essential for safeguarding the body against pathogens [93]. Targeting B cells and their associated mechanisms may offer novel therapeutic strategies for treating ALD.
T cells
Individuals with ALD show significant infiltration and activation of CD3+ T cells in hepatic tissue [94]. Circulating T cells exhibit elevated expression of activation markers, such as CD69 and CD38, but there is a notable reduction in interferon-γ production upon stimulation [95, 96]. An extensive immediate Th1 response is observed in ALD patients, which may exacerbate inflammatory responses and contribute to tissue damage [97]. Th17 cells produce interleukin-17 (IL-17), which recruits and activates neutrophils, exacerbating liver inflammation and fibrosis [98]. Additionally, Th17 cells may aid in liver repair by producing more IL-22 [99]. The number of gut microbiota-specific Th17 cells, such as those specific to Candida albicans, increases during ALD, and these cells migrate to the liver, further amplifying the inflammatory response [100]. The cytokines secreted by Th1 and Th17 cells (e.g., IFN-γ and IL-17) can directly act on hepatic stellate cells, stimulating their activation and proliferation, which leads to the synthesis of extracellular matrix components like collagen and contributes to liver fibrosis [101, 102]. Recent studies have highlighted that CD8+ T cells exhibit selective depletion in the duodenum of patients with ALD, resulting in compromised intestinal barrier function and exacerbation of ALD pathology. Conversely, a striking accumulation of CD8+ T cells is observed in hepatic tissues. These reciprocal alterations in CD8+ T cell distribution suggest that therapeutic interventions targeting cellular survival mechanisms and functional restoration of both intestinal and intrahepatic CD8+ T cell populations may represent a novel therapeutic strategy for ALD management [103].
Natural killer T (NKT) cells can recognize lipid antigens, including specific lipid metabolic products generated by the intestinal microbiota [30, 104]. In ALD, dysbiosis of the intestinal microbiota allows these lipid antigens to enter the liver, where they activate NKT cells [105,106,107]. Type I NKT cells, including invariant NKT cells, are recruited and activated by IL-1β produced by Kupffer cells, and they typically adopt a pro-inflammatory phenotype in ALD [108, 109]. In contrast, type II NKT cells are activated by sulfonamide compounds and can inhibit the activity of type I NKT cells [110]. The cytokine production profile of NKT cells in patients with severe AH is altered, with notable reductions in IL-22 levels [104]. However, in contrast to the rich population of NKT cells observed in murine livers, the relative scarcity of these immune cells in human hepatic tissue underscores the need for deeper investigation into their potential involvement in the pathogenic mechanisms of ALD.
In contrast, the human liver is rich in mucosal-associated invariant T (MAIT) cells, which comprise 20%-50% of intrahepatic T cells [111]. MAIT cells are a unique subset of T cells predominantly located on mucosal surfaces, capable of recognizing specific metabolic products produced by the gut microbiota (e.g., riboflavin derivatives) [112]. These cells present antigens through their invariant T cell receptor (TCR) to the major histocompatibility complex-related protein 1 (MR1) [113]. MAIT cells help restrict bacterial translocation by producing antimicrobial peptides and cytokines (such as IFN-γ) [112]. However, in patients with severe AH, circulating MAIT cells are rapidly depleted, excessively activated, and exhibit impaired antimicrobial and cytotoxic responses, increasing the risk of bacterial infections [114]. Abnormal activation of MAIT cells may lead to excessive production of inflammatory mediators, further exacerbating liver inflammation [115]. Additionally, MAIT cells may influence the activation and fibrotic processes of HSCs by secreting cytokines such as IFN-γ and IL-17A [116]. Nevertheless, the precise role of MAIT cells in ALD remains to be fully understood.
NK and Type 1 Innate Lymphoid Cells (ILC1s)
Natural killer (NK) cells can inhibit liver fibrosis progression by directly targeting HSCs or by secreting cytokines such as IFN-γ [115, 117]. However, in patients with ALD, the number of NK cells is reduced, and their function is impaired, with alcohol directly hindering the development of CD11b+CD27+ NK cells [118]. Chronic alcohol consumption leads to a decrease in the hepatic population of conventional NK cells and impairs their cytolytic function, as evidenced by the reduced expression of natural killer group 2, member D (NKG2D), tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), and IFN-γ [119,120,121]. Recent studies have shown that after prolonged alcohol exposure, NK cells (but not ILC1s) in the livers of mice undergo apoptosis, leading to a predominance of ILC1s in group 1 innate lymphoid cells (ILCs) and the upregulation of IL-17A, which exacerbates inflammation and steatosis in ALD [122]. Restoring the ILC1/NK cell balance through NK cell transfer has been shown to provide significant protection against alcohol-induced steatohepatitis [122]. Further research is needed to fully understand the distinct types of NK cells and their respective roles in the progression of ALD.
Roles of cytokines and chemokines
TNF-α
TNF-α is a pro-inflammatory cytokine primarily produced by macrophages and monocytes, playing a critical role in inflammatory and immune responses [123]. In animal models of ALD, serum and hepatic concentrations of TNF-α are significantly elevated in patients with sAH and closely correlate with the extent of liver injury [17]. However, no significant elevation in TNF-α levels was observed in patients with alcohol use disorder (AUD) who exhibited normal serum biochemical profiles [124, 125]. Acetaldehyde and ROS generated during alcohol metabolism can directly stimulate KCs and HSCs, promoting the transcription and secretion of TNF-α [124, 125]. Chronic alcohol consumption compromises intestinal barrier integrity, allowing bacteria and endotoxins to translocate to the liver [48, 126]. These endotoxins activate KCs through the TLR4 signaling pathway, further increasing TNF-α production [127, 128]. TNF-α can activate TAK1, which subsequently triggers MAPKs and IκB kinases (IKKs), leading to the activation of NF-κB and, ultimately, inflammation and hepatocyte apoptosis [127, 128]. Additionally, alcohol-induced oxidative stress exacerbates lipid peroxidation and cell membrane damage, further enhancing TNF-α gene expression by activating NF-κB [129]. The resulting imbalance in the cytokine network contributes to the progression of liver injury.
IL-1β
The ingestion of alcohol can activate KCs in the liver, which in turn stimulate the NLRP3 inflammasome and caspase-1, promoting the maturation and release of IL-1β [81, 130]. Upon ligand binding, the IL-1 receptor (IL-1R) subunits form oligomers and engage MyD88, activating NF-κB and MAPKs, thereby initiating pro-inflammatory responses [131]. In individuals with ALD, elevated levels of IL-1β, IL-18, and caspase-1 in the liver are positively correlated with the severity of the condition [132]. IL-1β induces the recruitment of invariant natural killer T cells, leading to liver injury through TNF-α production and the recruitment of neutrophils to the liver [108]. Persistent elevation of IL-1β levels following both alcohol exposure and withdrawal can result in sustained liver inflammation and impaired hepatocyte regeneration, suggesting that IL-1β may serve as a promising therapeutic target for ALD [133]. In preclinical animal models, administration of IL-1β inhibitors, including IL-1 receptor antagonists (IL-1RA) or anti-IL-1β antibodies, has been shown to mitigate liver inflammation, steatosis, and fibrosis [134]. Unfortunately, a clinical trial evaluating drugs targeting the IL-1β pathway, such as anakinra, for the treatment of AH, yielded negative results [135].
IL-8 and CXCL1
Neutrophil migration is a hallmark of ALD, with IL-8 and CXCL1 being the key factors in recruiting neutrophils to liver tissue [66, 75]. Elevated serum and hepatic levels of IL-8 and CXCL1 are directly correlated with the severity and mortality of AH [16]. Research has shown that circulating IL-8 levels are markedly increased in patients with severe AH, serving as superior indicators of short-term mortality compared to traditional prognostic markers [136]. Recent studies have further revealed that neutrophils are the primary source of IL-8 in AH [75]. In ALD, neutrophils are recruited to the liver and release IL-8 through autocrine and paracrine mechanisms, acting as a chemotactic factor that attracts additional neutrophils to the liver, thus perpetuating a vicious cycle [75]. Studies using animal models of alcohol consumption have demonstrated that blocking the IL-8 receptor (CXCR1/2) effectively reduces liver injury, inflammation, and mortality in mice [137, 138]. CXCL1 was significantly upregulated in mice subjected to a high-fat diet (HFD) and ethanol exposure, while both CXCL1 knockout and anti-CXCL1 treatment protected these mice from neutrophil-mediated liver injury [66]. Consequently, inhibiting IL-8/CXCL1 to reduce neutrophil migration to the liver may represent a promising therapeutic strategy for AH.
IL-17A
Studies have shown that individuals with AH and cirrhosis exhibit significantly higher levels of IL-17A [98, 139]. IL-17A has the notable ability to stimulate the expression of a broad range of pro-inflammatory cytokines and chemokines, including IL-6, TNF-α, and IL-8, which further amplify the inflammatory response within the liver [98]. The elevation of these factors leads to the recruitment and activation of neutrophils, thereby exacerbating hepatocyte damage. Additionally, IL-17A stimulates the proliferation and collagen synthesis of HSCs and upregulates the expression of matrix metalloproteinases (MMPs), influencing extracellular matrix remodeling and accelerating the progression of liver fibrosis [140].
IL-6
IL-6 is a multifunctional cytokine produced by immune cells and hepatocytes [141]. Studies have shown that serum IL-6 levels are significantly elevated in patients with ALD and are directly linked to liver dysfunction and the severity of ALD complications [142, 143]. Recent research highlights IL-6’s diverse biological effects, demonstrating its role in inducing the expression of various pro-inflammatory cytokines and chemokines, as well as regulating anti-apoptotic gene transcription and liver regeneration [144]. In mouse hepatocytes, IL-6-mediated activation of the JAK/PI3K/Akt signaling pathway has been shown to inhibit hepatocyte apoptosis [145]. Furthermore, IL-6 activates mtDNA repair enzymes and repair mitochondrial DNA damage in liver cells following chronic alcohol consumption, promoting hepatic cell regeneration and repair [146]. These findings suggest that further investigation into the mechanisms by which IL-6 influences ALD is necessary. Moreover, intervention strategies targeting IL-6 may produce varying outcomes depending on the disease’s inflammatory stage.
IL-22
IL-22 is a cytokine with hepatoprotective properties, primarily produced by immune cells, including helper T, NK, and NKT cell subsets [147, 148]. Upon binding to its receptor complex, IL-22 activates the JAK/STAT signaling pathway, promoting hepatocyte regeneration and repair [149, 150]. In various acute and chronic liver injury models in mice, IL-22 has been shown to reduce hepatocyte damage and enhance liver regeneration by upregulating the expression of antioxidant and anti-apoptotic genes, such as heme oxygenase-1 (HO-1) and B-cell lymphoma-extra large (Bcl-xL) [151]. In mouse models, ethanol-induced intestinal dysbiosis and reduced levels of associated metabolites lead to decreased intestinal IL-22 expression [152]. This reduction results in diminished intestinal REG3G (Regenerating Family Member 3 Gamma) expression and increased bacterial translocation to the liver, exacerbating liver inflammation [152]. Given that IL-22 receptor expression is limited to epithelial cells and not detectable in immune cells [151], this specificity is expected to reduce potential side effects of IL-22 in clinical applications, making it a promising therapeutic target for ALD [153].
Other cytokines and chemokines
CCL20 is one of the most significantly upregulated chemokines in patients with AH [73] and is associated with endotoxemia, liver fibrosis, and short-term mortality [154]. Recent RNA sequencing and ELISA findings have shown that several neutrophil chemokines, such as CXCL1, CXCL5, and CXCL6, are also upregulated in severe AH livers. Notably, single-cell RNA sequencing (scRNA-seq) analysis of severe AH livers revealed that hepatic stellate cells produce CXCL5, while hepatocytes express CXCL6 [75]. Serum CCL2 levels have a significant correlation with liver cirrhosis scores in both animal models and clinical patient populations [155]. CCL2 knockout mice show protection against alcohol-induced liver injury by inhibiting inflammatory cytokine production and enhancing lipid oxidation [156].
IL-10 is a potent anti-inflammatory cytokine produced by immune cells, such as macrophages and NK cells, following alcohol consumption or LPS stimulation [157, 158]. IL-10 suppresses the secretion of inflammatory cytokines by macrophages and monocytes [159] and reduces the antigen-presenting capacity of B cells and dendritic cells [158]. Additionally, IL-10 promotes liver regeneration and mitigates liver injury through activation of the JAK-STAT signaling pathway and its downstream effectors [160]. Preclinical research has demonstrated that IL-10 deficiency exacerbates hepatic inflammation and hepatocellular injury in mice, while stimulating IL-10 production in HSCs and KCs can prevent alcohol-induced liver damage [161, 162]. Therefore, IL-10 represents a promising therapeutic target for ALD.
Interactions of hepatocytes or nonparenchymal cells with immune cells
Hepatocytes and hepatokines
Hepatocytes play a significant role in shaping the immune environment in ALD. Damaged hepatocytes release a variety of chemokines [163, 164] and DAMPs [165, 166], which promote immune cell infiltration in ALD. Research has shown that alcohol stimulates hepatocytes to release extracellular vesicles containing CD40L in a caspase-3-dependent manner [167], thereby activating macrophages and increasing the production of inflammatory cytokines. Macrophage migration inhibitory factor (MIF), a pleiotropic cytokine/chemokine that signals through interactions with the CD74 receptor and its auxiliary receptors CXCR2, CXCR4, and CXCR7 [168, 169], is elevated in ALD patients [170] and in mice chronically exposed to ethanol [171]. MIF released from injured hepatocytes may act as a DAMP during ALD progression, leading to the recruitment of innate immune cells to the liver and the activation of inflammatory pathways. Targeting MIF release or signaling could offer a viable therapeutic strategy for ALD. Additionally, damaged hepatocytes can produce neutrophil chemotactic factors [172], which are significantly correlated with mortality in AH patients. Notably, miR-223, which is specifically released by neutrophils, has a protective effect on hepatocytes. Neutrophils recruited to the liver release proteases, ROS, and NETs, which are the primary mechanisms underlying liver injury [84, 173].
Hepatocytes are capable of synthesizing and secreting various biological signaling molecules, collectively known as hepatokines. Hepatocyte-derived growth differentiation factor 15 (GDF15) exerts multifaceted effects on ALD [174]. GDF15 regulates the metabolic pathways of macrophages, acquiring an anti-inflammatory function dependent on oxidative phosphorylation, reducing the expression of pro-inflammatory factors, and decreasing the infiltration of monocytes and neutrophils into the liver, thereby alleviating liver inflammation [174, 175]. Leukocyte Cell Derived Chemotaxin-2 (LECT2), which is elevated in patients with metabolic dysfunction-associated steatohepatitis (MASH), selectively promotes JNK phosphorylation in KCs, leading to liver inflammation [176]. Our recent work indicates that LECT2 levels are significantly increased in the serum of AH patients and that LECT2, produced by stressed hepatocytes, plays a crucial role in regulating the inflammatory phenotype of ALD. LECT2 inhibition ameliorates neutrophil NET formation and reduces liver injury in animal models [177]. Thus, targeting hepatokines to modulate inflammation represents a potential therapeutic approach.
Liver sinusoidal endothelial cells (LSECs)
LSECs play a crucial role in maintaining liver homeostasis and regulating hepatic microcirculation [178]. In response to inflammatory signals, these endothelial cells activate the NF-κB signaling pathway, increasing the expression of inflammatory mediators and adhesion molecules [179]. The chemokines they release, such as CXCL1 and CXCL2, attract monocytes and neutrophils to the liver, where they contribute to the localization and amplification of inflammatory responses [180]. During prolonged alcohol consumption, the expression of intercellular adhesion molecule-1 (ICAM-1) on LSECs is upregulated [173, 181]. This upregulation facilitates the binding of ICAM-1 to the integrin CD11/CD18 complex on neutrophils, thereby enhancing leukocyte adhesion [173, 181]. E-selectin levels are elevated in the livers of both mice and humans with ALD [76, 182]. In models of chronic and binge ethanol exposure, the ablation of E-selectin reduces hepatic neutrophil infiltration and liver injury [76]. LSECs also influence macrophage phenotypes through paracrine signaling of IL-10 and TGF-β [183]. They are a critical component of the liver’s immunosuppressive microenvironment, which dampens KC responsiveness to gut-derived toxins. Hepatic endothelial cells thus play a pivotal role in modulating inflammatory responses in ALD by regulating the recruitment, activation, and adhesion of inflammatory cells.
Hepatic stellate cells (HSCs)
HSCs exert significant influence over ALD by secreting cytokines such as TGF-β, PDGF, and TIMP-1 [45]. These cytokines orchestrate the recruitment, activation, and fibrotic processes of inflammatory cells, engaging signaling pathways like NF-κB and Smad, along with oxidative stress mechanisms and cellular interactions [184, 185]. Activated HSCs promote macrophage activation and migration by releasing IL-6 and CCL2 [186, 187], while IL-1 and TNF-α secreted by macrophages enhance HSC viability through the activation of NF-κB signaling [188]. Neutrophils also contribute to HSC activation by producing ROS in vitro, and chemokines released by activated HSCs help attract neutrophils to the liver [137, 189]. Interactions between HSCs and various cell types create a positive feedback loop that is closely associated with alcohol-induced liver fibrosis.
Crosstalk between extrahepatic organs and inflammation within the liver
Gut and gut microbiota
Research on the role of the microbiota in ALD dates back to 1995. Studies have demonstrated that intestinal decontamination with polymyxin B and neomycin significantly reduces levels of LPS, aspartate aminotransferase (AST), and liver histopathological scores in male Wistar rats that were given alcohol via gavage for 3 weeks [190]. Alcohol administration in mice in the NIAAA model resulted in a significant reduction in the genus Akkermansia [191] and oral administration of Akkermansia muciphila in the Lieber-DeCarli model ameliorated features of ALD [192]. In mice fed the Lieber–DeCarli alcohol liquid diet for 6 weeks, chronic alcohol consumption reduced the abundance of Bacteroidetes and Firmicutes [193]. These changes in the gut microbiota were associated with increased plasma endotoxins and liver inflammation [194]. Similarly, over half of patients with alcohol misuse exhibit intestinal barrier dysfunction and dysbiosis [195], with patients with AUD also showing gut microbiota dysbiosis and elevated serum endotoxins of intestinal origin [196, 197]. The abundance of Enterococcus faecalis and Candida in the gut of patients with AH is strongly correlated with liver disease severity and mortality [198]. In general, gut microbiota dysbiosis leads to increased systemic inflammatory mediators, ammonia, endotoxemia, enhanced intestinal permeability, and alcohol craving [199]. Thus, understanding which aspects of gut microbiota are dysregulated after alcohol consumption and whether these changes influence the progression of ALD could be critical for developing prevention or treatment strategies for ALD [193].
Alcohol-induced dysbiosis primarily affects intestinal permeability and immune cell function through microbial components (such as LPS, peptidoglycan, flagellin, cytolysin, and β-glucan) and metabolic products (including bile acids, short-chain fatty acids (SCFAs), indole derivatives, and vitamin B), thereby exacerbating liver inflammation [200,201,202,203,204]. Peptidoglycan not only stimulates TLR2 receptors on lymphocytes and monocytes but also interacts with NOD2 and NLRP3 [205]. β-glucan, a cell wall polysaccharide found in most fungi, including Candida species, circulates to the liver at elevated concentrations and induces the production of mature IL-1β via C-type lectin ___domain family 7 member A (CLEC7A) on KCs [206]. The reduced abundance of beneficial gut microbiota results in decreased SCFA production, which, in conjunction with alcohol, increases intestinal permeability and allows more PAMPs to enter the liver [207]. Some bile acids bind to the G protein-coupled receptor TGR5 and inhibit TNF production by Kupffer cells via the TGR5-cAMP-dependent pathway. This interaction between bile acids and TGR5 reduces NLRP3 inflammasome activation, inhibits NF-κB signaling, and induces IL-10 production via CREB [208]. Indole-3-acetic acid (IAA) promotes the expression of IL-22 and Reg3γ through the aryl hydrocarbon receptor (AHR), enhancing intestinal mucosal integrity and mitigating alcohol-induced liver inflammation [209].
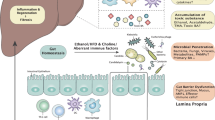
In addition to fecal microbiota transplantation, other interventions aim to modify the gut microbiome composition through diet or bacteriophage therapy. Modulation of the gut microbiota in ALD mouse models using the prebiotic pectin has been shown to alter the gut microbiome and metabolome of alcohol-fed mice [210]. This intervention results in enhanced tryptophan metabolite production and a reduction in liver injury and inflammation following microbial transplantation [210]. Recent studies have demonstrated that soluble dietary fiber can alleviate alcohol-induced liver injury in mice by regulating conjugated bile acid levels in the gut through Bacteroides acidifaciens, thereby modulating the FXR-FGF15 signaling pathway [211]. Additionally, the application of bacteriophages targeting Enterococcus faecalis has been shown to reduce alcohol-induced liver injury in humanized mice, providing a novel approach for the targeted treatment of ALD [198]. In summary, strategies targeting the gut microbiota, such as probiotics, prebiotics, or specific phages, offer promising therapeutic avenues for ALD (Fig. 2).

Interaction Between Gut Microbiome and both Hepatocytes and Hepatic Immune Cells. Chronic alcohol consumption and gut dysbiosis synergistically impair intestinal barrier integrity through suppressing the expression of critical defense factors, including antimicrobial peptides, regenerating islet-derived protein 3α (REG3α), and mucins, thereby exacerbating pathological permeability in the gut-liver axis. Gut microbial components, including pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharide, flagellin, peptidoglycan, cytolysin, and β-glucan, can translocate across a compromised intestinal barrier and activate inflammatory signaling pathways in hepatic immune cells, particularly Kupffer cells. This activation triggers an inflammatory cascade that contributes to liver injury and disease progression. Conversely, prebiotics and probiotics may exert hepatoprotective effects by strengthening gut barrier integrity and modulating immune responses through their metabolites, potentially mitigating inflammation and liver damage
Adipose tissue
Adipose tissue is primarily composed of adipocytes [45], witch store excess energy by taking up circulating non-esterified fatty acids (NEFA) and subsequently esterifying them into triglycerides [212]. Long time alcohol consumption significantly triggers adipocyte apoptosis within adipose tissue in mice, enhances lipolysis, and promotes the liberation of NEFAs [213], along with the generation of proinflammatory cytokines [214]. Increased delivery of NEFA to the liver leads to hepatic steatosis, and facilitate alcohol-induced liver injury by activating KCs [215]. Additionally, saturated NEFAs have a proinflammatory effect by activating myeloid cells, promoting cytokine release and endothelial adhesion [216]. TNFα secreted from adipose tissue can induce hepatocyte apoptosis and exacerbate inflammation through activation of the NF-κB and JNK pathways, among other pathways [217, 218].
Adipose tissue modulates homeostatic functions in both adipose and multiple non-adipose organs through the secretion of diverse bioactive molecules, collectively termed adipokines [219]. Adiponectin, the most abundant adipokine in circulation [220], exerts its biological effects via two primary receptors (AdipoR1 and AdipoR2) [221], these receptors exhibit differential expression patterns across hepatic cell populations, including hepatocytes, HSCs, and KCs, mediating beneficial effects such as anti-inflammatory responses and anti-fibrotic actions [222, 223]. Chronic inflammation of adipose tissue caused by alcohol inhibits the release of adiponectin, which in turn impairs lipid metabolism in the liver and exacerbates the progression of injury [224]. In contrast, elevated serum leptin levels in alcoholic cirrhosis demonstrate a positive correlation with the severity of hepatic pathology [225]. Alcohol abuse-induced hyperleptinemia exacerbates hepatic fibrosis by promoting HSC activation [226]. Furthermore, leptin potentiates hepatic inflammation through TNF-α release from Kupffer cells and stimulates CCL2 production in HSCs [227].
Gut-brain-liver axis
The central nervous system (CNS) represents another critical target of alcohol-induced toxicity and neurodegeneration. Ethanol exerts dual pathological effects through direct neurotoxic actions and activation of neuroimmune signaling pathways, driving alcohol abuse-associated neuroinflammation, neuronal apoptosis, cerebral functional impairment, and addictive behaviors [3]. While abstinence remains the cornerstone intervention for alcohol-associated liver disease (ALD), the refractory nature and high relapse propensity of AUD necessitate deeper investigations into fundamental regulatory mechanisms [14].
Emerging evidence highlights bidirectional communication between the gut microbiota and the CNS via neuroendocrine and neuroimmune pathways, collectively termed the gut-brain axis [228]. Dysregulation of the gut microbiota has been linked to alcohol addiction, withdrawal symptoms, and inflammatory responses [229,230,231]. Day et al. demonstrated a significant correlation between intestinal Candida albicans colonization density and alcohol consumption patterns in murine models, providing experimental validation for microbial involvement in AUD pathogenesis [232]. Notably, sodium valerate (a microbiota-derived metabolite structurally analogous to γ-aminobutyric acid (GABA)), has been identified as a novel therapeutic candidate. This compound modulates gut microbial composition, reduces microbial-derived neuroactive metabolites through GABA degradation pathways, and attenuates excessive alcohol intake and anxiety-like/approach-avoidance behaviors in male mice [233]. These findings position probiotic-mediated microbial homeostasis restoration as a promising strategy for AUD management.
Beyond AUD specific interventions, alcohol exposure disrupts circadian rhythm regulation by altering mRNA expression of clock genes in the suprachiasmatic nucleus. Intriguingly, non-invasive 40 Hz light flicker exposure activates hypothalamic SIRT1 expression, mitigating ethanol-induced hepatotoxicity and hepatic steatosis through central clock modulation [234]. Blockade of IL-17 signaling pathway effectively reverses alcohol-associated hepatic injury and compulsive drinking behaviors in dependent mice [140, 235], suggesting therapeutic potential of inflammatory cytokine targeting along the brain-liver axis. The hepatic vagus nerve, a critical component of the nervous system, intricately innervates the liver and plays an essential role in liver-brain interactions [236]. The nervous system precisely discharges acetylcholine from the terminals of the vagus nerve [236]. This neurotransmitter acts by inhibiting TNFα synthesis via the α7 nicotinic acetylcholine receptors (α7nAChRs) present on the surface of hepatic KCs, demonstrating neural regulation of hepatic inflammation [237]. These mechanistic insights underscore the importance of understanding alcohol’s multiorgan pathological network for developing comprehensive therapeutic strategies against ALD [15, 219].
Advances in targeting inflammation for the treatment of alcohol-associated liver disease
Existing effective interventions for ALD include nutritional support, alcohol abstinence therapy, hormonal treatment, and liver transplantation [238,239,240]. Additionally, medications such as naltrexone, and acamprosate have proven efficacy in AUD [241, 242]. These drugs modulate opioid pathways within the CNS [243]. Corticosteroids can improve short-term survival rates, typically around 28 days, but do not enhance long-term survival rates, which are usually measured at 6 months [244]. Additionally, corticosteroids may lead to concurrent infections [244]. Pentoxifylline, a selective phosphodiesterase inhibitor, can reduce pro-inflammatory cytokine levels and is considered a potential therapeutic option for AH [245]. However, clinical trials have shown that pentoxifylline is not effective in improving the survival rate of AH patients [246]. The following section summarizes various therapeutic targets that have been clinically tested, with a focus on targeting inflammation, and discusses potential anti-inflammatory strategies for treating ALD (Fig. 3).

Emerging Therapeutic Targets and Clinical Trials for ALD. Three main therapeutic strategies are currently being explored: (1) Protecting hepatocytes by reducing apoptosis, alleviating oxidative stress, and promoting liver regeneration; (2) Targeting inflammatory pathways to mitigate liver and systemic inflammation; and (3) Modulating the gut-liver axis through strategies such as lowering LPS levels, probiotic supplementation, and fecal microbiota transplantation. These approaches aim to address key mechanisms driving ALD progression and improve clinical outcomes
Targeting inflammasomes
Therapies targeting inflammasomes are a subject of extensive research. Since the inhibition of ASK1 (apoptosis signal-regulation kinase 1) in NLRP3 (NOD-like receptor thermal protein ___domain associated protein 3) mutant mice can reduce liver fibrosis, hepatocyte death, and liver TNF-α expression, it suggests the application prospects of ASK1 inhibitor selonsertib in liver diseases. A phase 2 clinical trial, conducted in a double-blind, placebo-controlled manner, aimed to assess the safety and efficacy of selonsertib in combination with prednisolone against a placebo paired with prednisolone (NCT02854631). The study revealed no significant differences in the Lille response on day 7, alterations in the MELD score on day 28, or mortality rates on day 28 between the treatment and control groups [247]. The overall infection rate did not significantly differ between the groups. Caspase 1 is known to trigger inflammatory responses, whereas caspase 8 is implicated in regulating apoptosis and subsequent necrosis across various hepatic conditions. The pan-caspase inhibitor emricasan has been under investigation for patients with severe AH (NCT01912404), but the trial was halted prematurely due to concerns regarding suboptimal drug bioavailability, which led to a reduction in the safe dosage.
Antioxidant therapies
N-acetylcysteine (NAC), an antioxidant agent, has the capacity to replenish glutathione levels in hepatocytes and alleviate oxidative stress-induced liver damage [248,249,250]. However, NAC treatment did not provide survival benefits at the 6-month mark for patients with severe AH compared with a placebo (NCT00962442) [250]. Furthermore, when NAC was used as an adjunct to prednisolone, it did not increase the 6-month survival rate compared with prednisolone monotherapy (NCT00863785) [249]. S-adenosylmethionine (SAMe), which serves as a direct precursor to glutathione, is currently being evaluated for its safety, efficacy, and effectiveness in an ongoing phase 4 randomized clinical trial (NCT02024295) for patients with AH and patients with alcohol-associated cirrhosis (NCT04250259).
Targeting LPS
LPS is a key inducer of inflammatory responses [251]. Studies have shown that IMM-124E is safe for use in patients with severe AH, but it does not reduce circulating LPS levels or mortality (NCT01968382). TAK-242, a TLR4 inhibitor, is currently being tested in an RCT for alcohol-associated cirrhosis and ACLF (NCT04620148) [252]. Combining LPS neutralization with inhibitors of downstream signaling pathways may represent a promising approach for treating ALD.
Targeting IL-1, IL-22, and chemokine pathways
Despite evidence that IL-1 significantly contributes to the progression of ALD in experimental models, clinical trials using IL-1 receptor antagonists [253] (anakinra, NCT01809132) and monoclonal antibodies [254] (canakinumab, NCT03775109) for severe AH have not demonstrated positive outcomes. In preclinical ALD models, the dual chemokine receptor CCR2/CCR5 antagonist cenicriviroc has shown promise in preventing macrophage infiltration and protecting the liver from inflammatory damage [255].
Preclinical studies indicate that IL-22 effectively mitigates liver injury associated with ALD [256, 257]. F-652, a recombinant fusion protein composed of human IL-22 and IgG2-Fc, targets IL-22R1 on epithelial cells, exerting minimal effects on immune cells and protecting tissues from damage and inflammation while promoting tissue repair [258]. An open-label phase II study of F-652 showed significant improvement in MELD scores for 18 patients by day 4 [259]. Further clinical trials are needed to confirm the benefit of F-652 in severe AH. The autocrine IL-8 loop enhances neutrophil recruitment and activation in severe AH, sustaining liver inflammation through p38 MAPK activation [75]. Thus, targeting IL-8 or CXCR1/2 holds promise as a treatment strategy for sAH [85]. Overall, therapies targeting cytokines or chemokines show potential for ALD treatment but remain under investigation. Additional research is required to establish their efficacy, safety, and to develop more effective treatment strategies.
Gut microbiota modulation
Probiotics and prebiotics are used to correct the imbalance of the gut microbiota in alcohol-induced liver damage [207]. Recent animal studies have shown that strains such as Lactobacillus plantarum and Lactobacillus rhamnosus GG can alleviate alcohol-associated hepatic steatosis, liver injury, and gut microbiota dysbiosis [194, 260, 261]. A diet high in soluble fiber has also been found to reduce alcohol-induced liver injury by increasing the abundance of Bacteroides acidifaciens [211]. A randomized, placebo-controlled study demonstrated that 1 month of oral Lactobacillus rhamnosus GG significantly reduced liver injury in patients with ALD (NCT01922895) [239]. Another double-controlled randomized prospective clinical trial is ongoing to investigate the effects of oral supplementation with Lactobacillus rhamnosus R0011/Lactobacillus acidophilus R0052 in patients with AH (NCT02335632) [262].
Currently, several comprehensive clinical trials are investigating the efficacy of fecal microbiota transplantation (FMT) in patients with AH (e.g., NCT04758806, NCT03091010) and liver cirrhosis (e.g., NCT04932577). These trials aim to assess various aspects of FMT treatment, including efficacy, potential side effects, long-term outcomes, and the mechanisms by which FMT might exert therapeutic effects in these specific conditions [239, 262]. FMT is currently considered a safe and effective treatment method, but potential risks remain, such as infections, allergic reactions, and gastrointestinal dysfunction. Prior to FMT treatment, it is crucial to assess the patient’s condition and risk factors carefully and to select an appropriate donor and treatment plan.
Conclusion and perspective
Inflammation plays a pivotal role in the progression of ALD, driving the pathological continuum from steatosis to fibrosis and beyond. While neutrophil infiltration is a hallmark of severe AH, the inflammatory landscape in ALD is more complex, involving macrophages, T cells, and NKT cells. These immune cells interact with hepatocytes and nonparenchymal liver cells, orchestrating an intricate inflammatory response through the secretion of various mediators [45].
However, inflammation is not inherently detrimental—it also plays a critical role in liver repair and antimicrobial defense, particularly against bacterial infections. Many inflammatory mediators, such as TNF-α, exhibit dual roles in ALD pathogenesis, contributing to liver injury and fibrosis while simultaneously supporting liver regeneration and immune defense. This complexity may, in part, explain why anti-inflammatory therapies targeting these mediators have yielded disappointing results in clinical trials. The widespread prevalence of alcohol consumption makes mitigating ALD progression particularly challenging. Despite decades of research on inflammation in AH, clinical trials of anti-inflammatory therapies have largely failed to achieve meaningful improvements in patient outcomes. The heterogeneity of inflammatory responses among AH patients likely contributes to these setbacks, underscoring the need for a more individualized approach. Fortunately, advancements in precision medicine and high-resolution inflammatory profiling are shedding new light on disease mechanisms. By leveraging these insights, the future of AH treatment may shift toward targeted, patient-specific interventions that overcome the translational research deadlock and offer new hope for effective therapies.
References
Zakhari S, Li TK. Determinants of alcohol use and abuse: impact of quantity and frequency patterns on liver disease. Hepatology. 2007;46:2032–9.
Hyun J, Han J, Lee C, Yoon M., Jung Y. Pathophysiological aspects of alcohol metabolism in the liver. Int J Mol Sci. 2021;22:5717.
World Health Organization. Global status report on alcohol and health 2018. Geneva: World Health Organization; 2018.
Hendriks HFJ. Alcohol and human health: what is the evidence?. Annu Rev Food Sci Technol. 2020;11:1–21.
Rehm J, Baliunas D, Borges GL, Graham K, Irving H, Kehoe T, et al. The relation between different dimensions of alcohol consumption and burden of disease: an overview. Addiction. 2010;105:817–43.
Lieber CS. Alcoholic liver disease: new insights in pathogenesis lead to new treatments. J Hepatol. 2000;32:113–28.
Cederbaum AI. Alcohol metabolism. Clin Liver Dis. 2012;16:667–85.
Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141:1572–85.
Holford NH. Clinical pharmacokinetics of ethanol. Clin Pharmacokinet. 1987;13:273–92.
Liangpunsakul S, Haber P, McCaughan GW. Alcoholic liver disease in Asia, Europe, and North America. Gastroenterology. 2016;150:1786–97.
Morales-Arráez D, Ventura-Cots M, Altamirano J, Abraldes JG, Cruz-Lemini M, Thursz MR, et al. The MELd score is superior to the maddrey discriminant function score to predict short-term mortality in alcohol-associated hepatitis: a global study. Am J Gastroenterol. 2022;117:301–10.
Parker R, Aithal GP, Becker U, Gleeson D, Masson S, Wyatt JI, et al. Natural history of histologically proven alcohol-related liver disease: a systematic review. J Hepatol. 2019;71:586–93.
Hadland SE, Xuan Z, Blanchette JG, Heeren TC, Swahn MH, Naimi TS. Alcohol policies and alcoholic cirrhosis mortality in the United States. Prev Chronic Dis. 2015;12:E177.
Rodriguez WE, Wahlang B, Wang Y, Zhang J, Vadhanam MV, Joshi-Barve S, et al. Phosphodiesterase 4 inhibition as a therapeutic target for alcoholic liver disease: from bedside to bench. Hepatology. 2019;70:1958–71.
Gao B, Ahmad MF, Nagy LE, Tsukamoto H. Inflammatory pathways in alcoholic steatohepatitis. J Hepatol. 2019;70:249–59.
Gao B, Tsukamoto H. Inflammation in alcoholic and nonalcoholic fatty liver disease: friend or foe?. Gastroenterology. 2016;150:1704–9.
Wang HJ, Gao B, Zakhari S, Nagy LE. Inflammation in alcoholic liver disease. Annu Rev Nutr. 2012;32:343–68.
Jiang Y, Zhang T, Kusumanchi P, Han S, Yang Z, Liangpunsakul S. Alcohol metabolizing enzymes, microsomal ethanol oxidizing system, cytochrome P450 2E1, catalase, and aldehyde dehydrogenase in alcohol-associated liver disease. Biomedicines. 2020;8:8.
Zakhari S. Overview: how is alcohol metabolized by the body?. Alcohol Res Health. 2006;29:245–54.
Thoudam T, Chanda D, Lee JY, Jung MK, Sinam IS, Kim BG, et al. Enhanced Ca(2+)-channeling complex formation at the ER-mitochondria interface underlies the pathogenesis of alcohol-associated liver disease. Nat Commun. 2023;14:1703.
Donohue TM Jr., Tuma DJ, Sorrell MF. Acetaldehyde adducts with proteins: binding of [14 C]acetaldehyde to serum albumin. Arch Biochem Biophys. 1983;220:239–46.
Stevens VJ, Fantl WJ, Newman CB, Sims RV, Cerami A, Peterson CM. Acetaldehyde adducts with hemoglobin. J Clin Invest. 1981;67:361–9.
Kenney WC. Acetaldehyde adducts of phospholipids. Alcohol Clin Exp Res. 1982;6:412–6.
Brooks PJ, Zakhari S. Acetaldehyde and the genome: beyond nuclear DNA adducts and carcinogenesis. Environ Mol Mutagen. 2014;55:77–91.
Fu Y, Mackowiak B, Lin YH, Maccioni L, Lehner T, Pan H, et al. Coordinated action of a gut-liver pathway drives alcohol detoxification and consumption. Nat Metab. 2024;6:1380–96.
Liangpunsakul S, Kolwankar D, Pinto A, Gorski JC, Hall SD, Chalasani N. Activity of CYP2E1 and CYP3A enzymes in adults with moderate alcohol consumption: a comparison with nonalcoholics. Hepatology. 2005;41:1144–50.
Wu D, Cederbaum AI. Alcohol, oxidative stress, and free radical damage. Alcohol Res Health. 2003;27:277–84.
Tuma DJ, Thiele GM, Xu D, Klassen LW, Sorrell MF. Acetaldehyde and malondialdehyde react together to generate distinct protein adducts in the liver during long-term ethanol administration. Hepatology. 1996;23:872–80.
Scarlata G, Colaci C, Scarcella M, Dallio M, Federico A, Boccuto L, et al. The role of cytokines in the pathogenesis and treatment of alcoholic liver disease. Diseases. 2024;12:12.
Osna NA, Rasineni K, Ganesan M, Donohue TM Jr, Kharbanda KK. Pathogenesis of alcohol-associated liver disease. J Clin Exp Hepatol. 2022;12:1492–513.
Chen WY, Zhang J, Ghare S, Barve S, McClain C, Joshi-Barve S. Acrolein is a pathogenic mediator of alcoholic liver disease and the scavenger hydralazine is protective in mice. Cell Mol Gastroenterol Hepatol. 2016;2:685–700.
Xie G, Zhong W, Zheng X, Li Q, Qiu Y, Li H, et al. Chronic ethanol consumption alters mammalian gastrointestinal content metabolites. J Proteome Res. 2013;12:3297–306.
Louvet A, Mathurin P. Alcoholic liver disease: mechanisms of injury and targeted treatment. Nat Rev Gastroenterol Hepatol. 2015;12:231–42.
Szabo G. Gut-liver axis in alcoholic liver disease. Gastroenterology. 2015;148:30–6.
Heymann F, Tacke F. Immunology in the liver-from homeostasis to disease. Nat Rev Gastroenterol Hepatol. 2016;13:88–110.
Nakamoto N, Kanai T. Role of toll-like receptors in immune activation and tolerance in the liver. Front Immunol. 2014;5:221.
Xu T, Du Y, Fang XB, Chen H, Zhou DD, Wang Y, et al. New insights into Nod-like receptors (NLRs) in liver diseases. Int J Physiol Pathophysiol Pharm. 2018;10:1–16.
Wu J, Wu D, Ma K, Wang T, Shi G, Shao J, et al. Paeonol ameliorates murine alcohol liver disease via mycobiota-mediated Dectin-1/IL-1β signaling pathway. J Leukoc Biol. 2020;108:199–214.
Stadlbauer V, Mookerjee RP, Wright GA, Davies NA, Jürgens G, Hallström S, et al. Role of toll-like receptors 2, 4, and 9 in mediating neutrophil dysfunction in alcoholic hepatitis. Am J Physiol Gastrointest Liver Physiol. 2009;296:G15–22.
Michelena J, Altamirano J, Abraldes JG, Affò S, Morales-Ibanez O, Sancho-Bru P, et al. Systemic inflammatory response and serum lipopolysaccharide levels predict multiple organ failure and death in alcoholic hepatitis. Hepatology. 2015;62:762–72.
Abenavoli L, Scarlata GGM, Paravati MR, Boccuto L., Luzza F., Scarpellini E. Gut microbiota and liver transplantation: immune mechanisms behind the rejection. Biomedicines. 2023;11:1792.
Ramaiah SK, Jaeschke H. Hepatic neutrophil infiltration in the pathogenesis of alcohol-induced liver injury. Toxicol Mech Methods. 2007;17:431–40.
Saso K, Moehren G, Higashi K, Hoek JB. Differential inhibition of epidermal growth factor signaling pathways in rat hepatocytes by long-term ethanol treatment. Gastroenterology. 1997;112:2073–88.
Cassard AM, Ciocan D. Microbiota, a key player in alcoholic liver disease. Clin Mol Hepatol. 2018;24:100–7.
Gao H, Jiang Y, Zeng G, Huda N., Thoudam T., Yang Z., et al. Cell-to-cell and organ-to-organ crosstalk in the pathogenesis of alcohol-associated liver disease. eGastroenterology 2024;2:e100104.
Li Z, Weinman SA. Regulation of hepatic inflammation via macrophage cell death. Semin Liver Dis. 2018;38:340–50.
Wang M, You Q, Lor K, Chen F, Gao B, Ju C. Chronic alcohol ingestion modulates hepatic macrophage populations and functions in mice. J Leukoc Biol. 2014;96:657–65.
Liangpunsakul S, Toh E, Ross RA, Heathers LE, Chandler K, Oshodi A, et al. Quantity of alcohol drinking positively correlates with serum levels of endotoxin and markers of monocyte activation. Sci Rep. 2017;7:4462.
Bala S, Csak T, Kodys K, Catalano D, Ambade A, Furi I, et al. Alcohol-induced miR-155 and HDAC11 inhibit negative regulators of the TLR4 pathway and lead to increased LPS responsiveness of Kupffer cells in alcoholic liver disease. J Leukoc Biol. 2017;102:487–98.
Schilling JD, Machkovech HM, He L, Sidhu R, Fujiwara H, Weber K, et al. Palmitate and lipopolysaccharide trigger synergistic ceramide production in primary macrophages. J Biol Chem. 2013;288:2923–32.
Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. 2017;17:306–21.
Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. 2017;66:1300–12.
Kono H, Rusyn I, Yin M, Gäbele E, Yamashina S, Dikalova A, et al. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J Clin Investig. 2000;106:867–72.
Abshagen K, Eipel C, Kalff JC, Menger MD, Vollmar B. Loss of NF-kappaB activation in Kupffer cell-depleted mice impairs liver regeneration after partial hepatectomy. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1570–7.
Yang K, Du C, Cheng Y, Li Y, Gong J, Liu Z. Augmenter of liver regeneration promotes hepatic regeneration depending on the integrity of Kupffer cell in rat small-for-size liver transplantation. J Surg Res. 2013;183:922–8.
Yoshiya S, Shirabe K, Imai D, Toshima T, Yamashita Y, Ikegami T, et al. Blockade of the apelin-APJ system promotes mouse liver regeneration by activating Kupffer cells after partial hepatectomy. J Gastroenterol. 2015;50:573–82.
Park JK, Shao M, Kim MY, Baik SK, Cho MY, Utsumi T, et al. An endoplasmic reticulum protein, Nogo-B, facilitates alcoholic liver disease through regulation of kupffer cell polarization. Hepatology. 2017;65:1720–34.
Ju C, Tacke F. Hepatic macrophages in homeostasis and liver diseases: from pathogenesis to novel therapeutic strategies. Cell Mol Immunol. 2016;13:316–27.
Mitchell C, Couton D, Couty JP, Anson M, Crain AM, Bizet V, et al. Dual role of CCR2 in the constitution and the resolution of liver fibrosis in mice. Am J Pathol. 2009;174:1766–75.
Seki E, de Minicis S, Inokuchi S, Taura K, Miyai K, van Rooijen N, et al. CCR2 promotes hepatic fibrosis in mice. Hepatology. 2009;50:185–97.
Ambade A, Mandrekar P. Oxidative stress and inflammation: essential partners in alcoholic liver disease. Int J Hepatol. 2012;2012:853175.
Anstee QM, Neuschwander-Tetri BA, Wai-Sun Wong V, Abdelmalek MF, Rodriguez-Araujo G, Landgren H, et al. Cenicriviroc Lacked Efficacy to Treat Liver Fibrosis in Nonalcoholic Steatohepatitis: AURORA Phase III Randomized Study. Clin Gastroenterol Hepatol. 2024;22:124–134.e1.
Dou L, Shi X, He X, Gao Y. Macrophage phenotype and function in liver disorder. Front Immunol. 2019;10:3112.
Németh T, Sperandio M, Mócsai A. Neutrophils as emerging therapeutic targets. Nat Rev Drug Discov. 2020;19:253–75.
Liu K, Wang FS, Xu R. Neutrophils in liver diseases: pathogenesis and therapeutic targets. Cell Mol Immunol. 2021;18:38–44.
Chang B, Xu MJ, Zhou Z, Cai Y, Li M, Wang W, et al. Short- or long-term high-fat diet feeding plus acute ethanol binge synergistically induce acute liver injury in mice: an important role for CXCL1. Hepatology. 2015;62:1070–85.
Das S, Maras JS, Hussain MS, Sharma S, David P, Sukriti S, et al. Hyperoxidized albumin modulates neutrophils to induce oxidative stress and inflammation in severe alcoholic hepatitis. Hepatology. 2017;65:631–46.
Bukong TN, Cho Y, Iracheta-Vellve A, Saha B, Lowe P, Adejumo A, et al. Abnormal neutrophil traps and impaired efferocytosis contribute to liver injury and sepsis severity after binge alcohol use. J Hepatol. 2018;69:1145–54.
Cho Y, Szabo G. Two faces of neutrophils in liver disease development and progression. Hepatology. 2021;74:503–12.
Bautista AP. Neutrophilic infiltration in alcoholic hepatitis. Alcohol. 2002;27:17–21.
Vaz K, Little R, Majeed A, Kemp W, Roberts SK. Determinants of short- and long-term outcomes of an australian cohort of patients admitted with alcoholic hepatitis. Dig Dis Sci. 2022;67:3356–65.
Feng D, Hwang S, Guillot A, Wang Y, Guan Y, Chen C, et al. Inflammation in alcohol-associated hepatitis: pathogenesis and therapeutic targets. Cell Mol Gastroenterol Hepatol. 2024;18:101352.
Affò S, Dominguez M, Lozano JJ, Sancho-Bru P, Rodrigo-Torres D, Morales-Ibanez O, et al. Transcriptome analysis identifies TNF superfamily receptors as potential therapeutic targets in alcoholic hepatitis. Gut. 2013;62:452–60.
Dominguez M, Miquel R, Colmenero J, Moreno M, García-Pagán JC, Bosch J, et al. Hepatic expression of CXC chemokines predicts portal hypertension and survival in patients with alcoholic hepatitis. Gastroenterology. 2009;136:1639–50.
Guan Y, Peiffer B, Feng D, Parra M.A., Wang Y., Fu Y., et al. IL-8+ neutrophils drive inexorable inflammation in severe alcohol-associated hepatitis. J Clin Investig. 2024;134:e178616.
Bertola A, Park O, Gao B. Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: a critical role for E-selectin. Hepatology. 2013;58:1814–23.
Wang J, Wang X, Peng H, Dong Z, Liangpunsakul S, Zuo L, et al. Platelets in alcohol-associated liver disease: interaction with neutrophils. Cell Mol Gastroenterol Hepatol. 2024;18:41–52.
Rycyk-Bojarzynska A, Kasztelan-Szczerbinska B, Cichoz-Lach H, Surdacka A., Rolinski J. Neutrophil PAD4 expression and its pivotal role in assessment of alcohol-related liver disease. Int J Mol Sci. 2024;25:7597.
Khan RS, Lalor PF, Thursz M, Newsome PN. The role of neutrophils in alcohol-related hepatitis. J Hepatol. 2023;79:1037–48.
Nagesh PT, Cho Y, Zhuang Y, Babuta M, Ortega-Ribera M, Joshi R, et al. In vivo Bruton’s tyrosine kinase inhibition attenuates alcohol-associated liver disease by regulating CD84-mediated granulopoiesis. Sci Transl Med. 2024;16:eadg1915.
Babuta M, Morel C, de Carvalho Ribeiro M, Calenda C, Ortega-Ribera M, Thevkar Nagesh P, et al. Neutrophil extracellular traps activate hepatic stellate cells and monocytes via NLRP3 sensing in alcohol-induced acceleration of MASH fibrosis. Gut. 2024;73:1854–69.
Li M, He Y, Zhou Z, Ramirez T, Gao Y, Gao Y, et al. MicroRNA-223 ameliorates alcoholic liver injury by inhibiting the IL-6-p47(phox)-oxidative stress pathway in neutrophils. Gut. 2017;66:705–15.
Hassani M, Hellebrekers P, Chen N, van Aalst C, Bongers S, Hietbrink F, et al. On the origin of low-density neutrophils. J Leukoc Biol. 2020;107:809–18.
Cho Y, Bukong TN, Tornai D, Babuta M, Vlachos IS, Kanata E, et al. Neutrophil extracellular traps contribute to liver damage and increase defective low-density neutrophils in alcohol-associated hepatitis. J Hepatol. 2023;78:28–44.
Guan Y, Feng D, Maccioni L, Wang Y., Gao B. New therapeutic target for alcohol-associated hepatitis (AH): AH-associated IL-8(+) neutrophils. eGastroenterology. 2024;2:e100166.
Niemela O. Distribution of ethanol-induced protein adducts in vivo: relationship to tissue injury. Free Radic Biol Med. 2001;31:1533–8.
Andrade MC, Albernaz MJ, Araújo MS, Santos BP, Teixeira-Carvalho A, Faria AM, et al. Short-term administration of ethanol in mice deviates antigen presentation activity towards B cells. Scand J Immunol. 2009;70:226–37.
Parra D, Takizawa F, Sunyer JO. Evolution of B cell immunity. Annu Rev Anim Biosci. 2013;1:65–97.
Rodriguez MA, Montano JD, Williams RC. Immunoglobulin production by peripheral blood mononuclear cells in patients with alcoholic liver disease. Clin Exp Immunol. 1984;55:369–76.
Ahmadi AR, Song G, Gao T, Ma J, Han X, Hu MW, et al. Discovery and characterization of cross-reactive intrahepatic antibodies in severe alcoholic hepatitis. Elife 2023;12:RP86678.
Wang H, Zhou H, Mahler S, Chervenak R, Wolcott M. Alcohol affects the late differentiation of progenitor B cells. Alcohol Alcohol. 2011;46:26–32.
Massonnet B, Delwail A, Ayrault JM, Chagneau-Derrode C, Lecron JC, Silvain C. Increased immunoglobulin A in alcoholic liver cirrhosis: exploring the response of B cells to Toll-like receptor 9 activation. Clin Exp Immunol. 2009;158:115–24.
Moro-Sibilot L, Blanc P, Taillardet M, Bardel E, Couillault C, Boschetti G, et al. Mouse and human liver contain immunoglobulin A-secreting cells originating from peyer’s patches and directed against intestinal antigens. Gastroenterology. 2016;151:311–23.
Matos LC, Batista P, Monteiro N, Ribeiro J, Cipriano MA, Henriques P, et al. Lymphocyte subsets in alcoholic liver disease. World J Hepatol. 2013;5:46–55.
Chedid A, Mendenhall CL, Moritz TE, French SW, Chen TS, Morgan TR, et al. Cell-mediated hepatic injury in alcoholic liver disease. Veterans Affairs Cooperative Study Group 275. Gastroenterology. 1993;105:254–66.
Li W, Amet T, Xing Y, Yang D, Liangpunsakul S, Puri P, et al. Alcohol abstinence ameliorates the dysregulated immune profiles in patients with alcoholic hepatitis: a prospective observational study. Hepatology. 2017;66:575–90.
Song K, Coleman RA, Alber C, Ballas ZK, Waldschmidt TJ, Mortari F, et al. TH1 cytokine response of CD57 + T-cell subsets in healthy controls and patients with alcoholic liver disease. Alcohol. 2001;24:155–67.
Lemmers A, Moreno C, Gustot T, Maréchal R, Degré D, Demetter P, et al. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology. 2009;49:646–57.
Gao B, Xiang X. Interleukin-22 from bench to bedside: a promising drug for epithelial repair. Cell Mol Immunol. 2019;16:666–7.
Zeng S, Rosati E, Saggau C, Messner B, Chu H, Duan Y, et al. Candida albicans-specific Th17 cell-mediated response contributes to alcohol-associated liver disease. Cell Host Microbe. 2023;31:389–404.e7.
Lin F, Taylor NJ, Su H, Huang X, Hussain MJ, Abeles RD, et al. Alcohol dehydrogenase-specific T-cell responses are associated with alcohol consumption in patients with alcohol-related cirrhosis. Hepatology. 2013;58:314–24.
Meng F, Wang K, Aoyama T, Grivennikov SI, Paik Y, Scholten D, et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology. 2012;143:765–776.e3.
Maccioni L, Guan Y, Kim M, Parra M.A., Peiffer B., Fu Y., et al. Opposite regulation of intestinal and intrahepatic CD8(+) T cells controls alcohol-associated liver disease progression. Gut. 2025.
Marrero I, Maricic I, Morgan TR, Stolz AA, Schnabl B, Liu ZX, et al. Differential activation of unconventional T cells, including iNKT cells, in alcohol-related liver disease. Alcohol Clin Exp Res. 2020;44:1061–74.
Gao B, Radaeva S, Park O. Liver natural killer and natural killer T cells: immunobiology and emerging roles in liver diseases. J Leukoc Biol. 2009;86:513–28.
Wang H, Feng D, Park O, Yin S, Gao B. Invariant NKT cell activation induces neutrophil accumulation and hepatitis: opposite regulation by IL-4 and IFN-γ. Hepatology. 2013;58:1474–85.
Yin S, Wang H, Bertola A, Feng D, Xu MJ, Wang Y, et al. Activation of invariant natural killer T cells impedes liver regeneration by way of both IFN-γ- and IL-4-dependent mechanisms. Hepatology. 2014;60:1356–66.
Cui K, Yan G, Xu C, Chen Y, Wang J, Zhou R, et al. Invariant NKT cells promote alcohol-induced steatohepatitis through interleukin-1β in mice. J Hepatol. 2015;62:1311–8.
Mathews S, Feng D, Maricic I, Ju C, Kumar V, Gao B. Invariant natural killer T cells contribute to chronic-plus-binge ethanol-mediated liver injury by promoting hepatic neutrophil infiltration. Cell Mol Immunol. 2016;13:206–16.
Maricic I, Sheng H, Marrero I, Seki E, Kisseleva T, Chaturvedi S, et al. Inhibition of type I natural killer T cells by retinoids or following sulfatide-mediated activation of type II natural killer T cells attenuates alcoholic liver disease in mice. Hepatology. 2015;61:1357–69.
Kurioka A, Walker LJ, Klenerman P, Willberg CB. MAIT cells: new guardians of the liver. Clin Transl. Immunology. 2016;5:e98.
Li W, Lin EL, Liangpunsakul S, Lan J, Chalasani S, Rane S, et al. Alcohol abstinence does not fully reverse abnormalities of mucosal-associated invariant T cells in the blood of patients with alcoholic hepatitis. Clin Transl Gastroenterol. 2019;10:e00052.
López-Sagaseta J, Dulberger CL, Crooks JE, Parks CD, Luoma AM, McFedries A, et al. The molecular basis for Mucosal-Associated Invariant T cell recognition of MR1 proteins. Proc Natl Acad Sci USA. 2013;110:E1771–8.
Riva A, Patel V, Kurioka A, Jeffery HC, Wright G, Tarff S, et al. Mucosa-associated invariant T cells link intestinal immunity with antibacterial immune defects in alcoholic liver disease. Gut. 2018;67:918–30.
Gao B, Ma J, Xiang X. MAIT cells: a novel therapeutic target for alcoholic liver disease?. Gut. 2018;67:784–6.
Hegde P, Weiss E, Paradis V, Wan J, Mabire M, Sukriti S, et al. Mucosal-associated invariant T cells are a profibrogenic immune cell population in the liver. Nat Commun. 2018;9:2146.
Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z, Gao B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology. 2006;130:435–52.
Zhang F, Little A, Zhang H. Chronic alcohol consumption inhibits peripheral NK cell development and maturation by decreasing the availability of IL-15. J Leukoc Biol. 2017;101:1015–27.
Ben-Eliyahu S, Page GG, Yirmiya R, Taylor AN. Acute alcohol intoxication suppresses natural killer cell activity and promotes tumor metastasis. Nat Med. 1996;2:457–60.
Jeong WI, Park O, Gao B. Abrogation of the antifibrotic effects of natural killer cells/interferon-gamma contributes to alcohol acceleration of liver fibrosis. Gastroenterology. 2008;134:248–58.
Laso FJ, Madruga JI, Girón JA, López A, Ciudad J, San Miguel JF, et al. Decreased natural killer cytotoxic activity in chronic alcoholism is associated with alcohol liver disease but not active ethanol consumption. Hepatology. 1997;25:1096–100.
Cheng C, Zhang Q, Li Y, Jiang J, Xie L, Shen H, et al. Interplay between liver type 1 innate lymphoid cells and NK cells drives the development of alcoholic steatohepatitis. Cell Mol Gastroenterol Hepatol. 2023;15:261–74.
Tan Q, Liu Y, Deng X, Chen J, Tsai PJ, Chen PH, et al. Autophagy: a promising process for the treatment of acetaminophen-induced liver injury. Archives Toxicol. 2020;94:2925–38.
Bird GL, Sheron N, Goka AK, Alexander GJ, Williams RS. Increased plasma tumor necrosis factor in severe alcoholic hepatitis. Ann Intern Med. 1990;112:917–20.
Khoruts A, Stahnke L, McClain CJ, Logan G, Allen JI. Circulating tumor necrosis factor, interleukin-1 and interleukin-6 concentrations in chronic alcoholic patients. Hepatology. 1991;13:267–76.
Walline CC, Blum JS, Linton T, Mangiacarne D, Liangpunsakul S. Early activation of peripheral monocytes with hallmarks of M1 and M2 monocytic cells in excessive alcohol drinkers: a pilot study. J Investig Med. 2018;66:1–4.
Molteni M, Gemma S, Rossetti C. The role of toll-like receptor 4 in infectious and noninfectious inflammation. Mediators Inflamm. 2016;2016:6978936.
Shafaghati L, Razaghi-Moghadam Z, Mohammadnejad J. A systems biology approach to understanding alcoholic liver disease molecular mechanism: the development of static and dynamic models. Bull Math Biol. 2017;79:2450–73.
Shen Z, Ajmo JM, Rogers CQ, Liang X, Le L, Murr MM, et al. Role of SIRT1 in regulation of LPS- or two ethanol metabolites-induced TNF-alpha production in cultured macrophage cell lines. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1047–53.
Ibáñez F, Montesinos J, Ureña-Peralta JR, Guerri C, Pascual M. TLR4 participates in the transmission of ethanol-induced neuroinflammation via astrocyte-derived extracellular vesicles. J Neuroinflammation. 2019;16:136.
Tsutsui H, Cai X, Hayashi S. Interleukin-1 family cytokines in liver diseases. Mediators Inflamm. 2015;2015:630265.
Voican CS, Njiké-Nakseu M, Boujedidi H, Barri-Ova N, Bouchet-Delbos L, Agostini H, et al. Alcohol withdrawal alleviates adipose tissue inflammation in patients with alcoholic liver disease. Liver Int. 2015;35:967–78.
Iracheta-Vellve A, Petrasek J, Gyogyosi B, Bala S, Csak T, Kodys K, et al. Interleukin-1 inhibition facilitates recovery from liver injury and promotes regeneration of hepatocytes in alcoholic hepatitis in mice. Liver Int. 2017;37:968–73.
Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Investig. 2012;122:3476–89.
Gawrieh S, Dasarathy S, Tu W, Kamath PS, Chalasani NP, McClain CJ, et al. Randomized trial of anakinra plus zinc vs. prednisone for severe alcohol-associated hepatitis. J Hepatol. 2024;80:684–93.
Patel OP, Noor MT, Kumar R, Thakur BS. Serum interleukin 8 and 12 levels predict severity and mortality in patients with alcoholic hepatitis. Indian J Gastroenterol. 2015;34:209–15.
Roh YS, Zhang B, Loomba R, Seki E. TLR2 and TLR9 contribute to alcohol-mediated liver injury through induction of CXCL1 and neutrophil infiltration. Am J Physiol Gastrointest Liver Physiol. 2015;309:G30–41.
Wieser V, Adolph TE, Enrich B, Kuliopulos A, Kaser A, Tilg H, et al. Reversal of murine alcoholic steatohepatitis by pepducin-based functional blockade of interleukin-8 receptors. Gut. 2017;66:930–8.
Støy S, Sandahl TD, Dige AK, Agnholt J, Rasmussen TK, Grønbæk H, et al. Highest frequencies of interleukin-22-producing T helper cells in alcoholic hepatitis patients with a favourable short-term course. PLoS One. 2013;8:e55101.
Xu J, Ma HY, Liu X, Rosenthal S., Baglieri J., McCubbin R., et al. Blockade of IL-17 signaling reverses alcohol-induced liver injury and excessive alcohol drinking in mice. JCI Insight 2020;5:e131277.
Norris CA, He M, Kang LI, Ding MQ, Radder JE, Haynes MM, et al. Synthesis of IL-6 by hepatocytes is a normal response to common hepatic stimuli. PLoS One. 2014;9:e96053.
Gudowska-Sawczuk M, Wrona A, Gruszewska E, Cylwik B, Panasiuk A, Flisiak R, et al. Serum level of interleukin-6 (IL-6) and N-terminal propeptide of procollagen type I (PINP) in patients with liver diseases. Scand J Clin Lab Investig. 2018;78:125–30.
Kasztelan-Szczerbińska B, Surdacka A, Celiński K, Roliński J, Zwolak A, Miącz S, et al. Prognostic significance of the systemic inflammatory and immune balance in alcoholic liver disease with a focus on gender-related differences. PLoS One. 2015;10:e0128347.
Naseem S, Hussain T, Manzoor S. Interleukin-6: a promising cytokine to support liver regeneration and adaptive immunity in liver pathologies. Cytokine Growth Factor Rev. 2018;39:36–45.
Chou CH, Lai SL, Chen CN, Lee PH, Peng FC, Kuo ML, et al. IL-6 regulates Mcl-1L expression through the JAK/PI3K/Akt/CREB signaling pathway in hepatocytes: implication of an anti-apoptotic role during liver regeneration. PLoS ONE. 2013;8:e66268.
Zhang X, Tachibana S, Wang H, Hisada M, Williams GM, Gao B, et al. Interleukin-6 is an important mediator for mitochondrial DNA repair after alcoholic liver injury in mice. Hepatology. 2010;52:2137–47.
Radaeva S, Sun R, Pan HN, Hong F, Gao B. Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology. 2004;39:1332–42.
Witte E, Witte K, Warszawska K, Sabat R, Wolk K. Interleukin-22: a cytokine produced by T, NK and NKT cell subsets, with importance in the innate immune defense and tissue protection. Cytokine Growth Factor Rev. 2010;21:365–79.
Carmo RF, Cavalcanti MSM, Moura P. Role of Interleukin-22 in chronic liver injury. Cytokine. 2017;98:107–14.
Dudakov JA, Hanash AM, van den Brink MR. Interleukin-22: immunobiology and pathology. Annu Rev Immunol. 2015;33:747–85.
Kong X, Feng D, Mathews S, Gao B. Hepatoprotective and anti-fibrotic functions of interleukin-22: therapeutic potential for the treatment of alcoholic liver disease. J Gastroenterol Hepatol. 2013;28:56–60.
Hendrikx T, Duan Y, Wang Y, Oh JH, Alexander LM, Huang W, et al. Bacteria engineered to produce IL-22 in intestine induce expression of REG3G to reduce ethanol-induced liver disease in mice. Gut. 2019;68:1504–15.
Gao B, Shah VH. Combination therapy: new hope for alcoholic hepatitis?. Clin Res Hepatol Gastroenterol. 2015;39:S7–s11.
Affò S, Morales-Ibanez O, Rodrigo-Torres D, Altamirano J, Blaya D, Dapito DH, et al. CCL20 mediates lipopolysaccharide induced liver injury and is a potential driver of inflammation and fibrosis in alcoholic hepatitis. Gut. 2014;63:1782–92.
Degré D, Lemmers A, Gustot T, Ouziel R, Trépo E, Demetter P, et al. Hepatic expression of CCL2 in alcoholic liver disease is associated with disease severity and neutrophil infiltrates. Clin Exp Immunol. 2012;169:302–10.
Mandrekar P, Ambade A, Lim A, Szabo G, Catalano D. An essential role for monocyte chemoattractant protein-1 in alcoholic liver injury: regulation of proinflammatory cytokines and hepatic steatosis in mice. Hepatology. 2011;54:2185–97.
Mollazadeh H, Cicero AFG, Blesso CN, Pirro M, Majeed M, Sahebkar A. Immune modulation by curcumin: The role of interleukin-10. Crit Rev Food Sci Nutr. 2019;59:89–101.
Rutz S, Ouyang W. Regulation of Interleukin-10 Expression. Adv Exp Med Biol. 2016;941:89–116.
Kawaratani H, Tsujimoto T, Douhara A, Takaya H, Moriya K, Namisaki T, et al. The effect of inflammatory cytokines in alcoholic liver disease. Mediators Inflamm. 2013;2013:495156.
Neumann C, Scheffold A, Rutz S. Functions and regulation of T cell-derived interleukin-10. Semin Immunol. 2019;44:101344.
Miller AM, Horiguchi N, Jeong WI, Radaeva S, Gao B. Molecular mechanisms of alcoholic liver disease: innate immunity and cytokines. Alcohol Clin Exp Res. 2011;35:787–93.
Byun JS, Suh YG, Yi HS, Lee YS, Jeong WI. Activation of toll-like receptor 3 attenuates alcoholic liver injury by stimulating Kupffer cells and stellate cells to produce interleukin-10 in mice. J Hepatol. 2013;58:342–9.
Lazaro R, Wu R, Lee S, Zhu NL, Chen CL, French SW, et al. Osteopontin deficiency does not prevent but promotes alcoholic neutrophilic hepatitis in mice. Hepatology. 2015;61:129–40.
Wang W, Xu MJ, Cai Y, Zhou Z, Cao H, Mukhopadhyay P, et al. Inflammation is independent of steatosis in a murine model of steatohepatitis. Hepatology. 2017;66:108–23.
Ge X, Antoine DJ, Lu Y, Arriazu E, Leung TM, Klepper AL, et al. High mobility group box-1 (HMGB1) participates in the pathogenesis of alcoholic liver disease (ALD). J Biol Chem. 2014;289:22672–91.
Cai Y, Xu MJ, Koritzinsky EH, Zhou Z., Wang W., Cao H., et al. Mitochondrial DNA-enriched microparticles promote acute-on-chronic alcoholic neutrophilia and hepatotoxicity. JCI Insight. 2017;2:e92634.
Verma VK, Li H, Wang R, Hirsova P, Mushref M, Liu Y, et al. Alcohol stimulates macrophage activation through caspase-dependent hepatocyte derived release of CD40L containing extracellular vesicles. J Hepatol. 2016;64:651–60.
Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, et al. MIF signal transduction initiated by binding to CD74. J Exp Med. 2003;197:1467–76.
van der Vorst EP, Döring Y, Weber C. Chemokines and their receptors in Atherosclerosis. J Mol Med (Berl). 2015;93:963–71.
Kumagi T, Akbar F, Horiike N, Onji M. Increased serum levels of macrophage migration inhibitory factor in alcoholic liver diseases and their expression in liver tissues. Clin Biochem. 2001;34:189–93.
Barnes MA, McMullen MR, Roychowdhury S, Pisano SG, Liu X, Stavitsky AB, et al. Macrophage migration inhibitory factor contributes to ethanol-induced liver injury by mediating cell injury, steatohepatitis, and steatosis. Hepatology. 2013;57:1980–91.
Joshi-Barve S, Barve SS, Butt W, Klein J, McClain CJ. Inhibition of proteasome function leads to NF-kappaB-independent IL-8 expression in human hepatocytes. Hepatology. 2003;38:1178–87.
Jaeschke H. Neutrophil-mediated tissue injury in alcoholic hepatitis. Alcohol. 2002;27:23–7.
Kim HH, Shim YR, Choi SE, Kim MH, Lee G, You HJ, et al. Catecholamine induces Kupffer cell apoptosis via growth differentiation factor 15 in alcohol-associated liver disease. Exp Mol Med. 2023;55:158–70.
Li X, Huai Q, Zhu C, Zhang X, Xu W, Dai H, et al. GDF15 Ameliorates liver fibrosis by metabolic reprogramming of macrophages to acquire anti-inflammatory properties. Cell Mol Gastroenterol Hepatol. 2023;16:711–34.
Takata N, Ishii KA, Takayama H, Nagashimada M, Kamoshita K, Tanaka T, et al. LECT2 as a hepatokine links liver steatosis to inflammation via activating tissue macrophages in NASH. Sci Rep. 2021;11:555.
Xu H, Wu Z, Qin J, Li X., Xu F., Wang W., et al. Stressed hepatocyte sustains alcohol-associated hepatitis progression by producing leukocyte cell-derived chemotaxin 2. Gut. 2025, gutjnl-2024-334318.
Gracia-Sancho J, Marrone G, Fernández-Iglesias A. Hepatic microcirculation and mechanisms of portal hypertension. Nat Rev Gastroenterol Hepatol. 2019;16:221–34.
Yeligar SM, Machida K, Tsukamoto H, Kalra VK. Ethanol augments RANTES/CCL5 expression in rat liver sinusoidal endothelial cells and human endothelial cells via activation of NF-kappa B, HIF-1 alpha, and AP-1. J Immunol. 2009;183:5964–76.
Liu M, Cao S, He L, Gao J, Arab JP, Cui H, et al. Super enhancer regulation of cytokine-induced chemokine production in alcoholic hepatitis. Nat Commun. 2021;12:4560.
Bautista AP. Chronic alcohol intoxication induces hepatic injury through enhanced macrophage inflammatory protein-2 production and intercellular adhesion molecule-1 expression in the liver. Hepatology. 1997;25:335–42.
Blaya D, Rubio-Tomás T, Rodrigo-Torres D, Lozano J, Coll M, Argemi J, et al. Endothelial dysfunction markers predict short-term mortality in patients with severe alcoholic hepatitis. Hepatol Int. 2021;15:1006–17.
Casey LM, Hughes KR, Saunders MN, Miller SD, Pearson RM, Shea LD. Mechanistic contributions of Kupffer cells and liver sinusoidal endothelial cells in nanoparticle-induced antigen-specific immune tolerance. Biomaterials. 2022;283:121457.
Svegliati-Baroni G, Saccomanno S, van Goor H, Jansen P, Benedetti A, Moshage H. Involvement of reactive oxygen species and nitric oxide radicals in activation and proliferation of rat hepatic stellate cells. Liver. 2001;21:1–12.
Casini A, Ceni E, Salzano R, Biondi P, Parola M, Galli A, et al. Neutrophil-derived superoxide anion induces lipid peroxidation and stimulates collagen synthesis in human hepatic stellate cells: role of nitric oxide. Hepatology. 1997;25:361–7.
Mauer J, Chaurasia B, Goldau J, Vogt MC, Ruud J, Nguyen KD, et al. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat Immunol. 2014;15:423–30.
Ehling J, Bartneck M, Wei X, Gremse F, Fech V, Möckel D, et al. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut. 2014;63:1960–71.
Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58:1461–73.
Sprenger H, Kaufmann A, Garn H, Lahme B, Gemsa D, Gressner AM. Induction of neutrophil-attracting chemokines in transforming rat hepatic stellate cells. Gastroenterology. 1997;113:277–85.
Adachi Y, Moore LE, Bradford BU, Gao W, Thurman RG. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology. 1995;108:218–24.
Lowe PP, Gyongyosi B, Satishchandran A, Iracheta-Vellve A, Ambade A, Kodys K, et al. Alcohol-related changes in the intestinal microbiome influence neutrophil infiltration, inflammation and steatosis in early alcoholic hepatitis in mice. PLoS ONE. 2017;12:e0174544.
Grander C, Adolph TE, Wieser V, Lowe P, Wrzosek L, Gyongyosi B, et al. Recovery of ethanol-induced Akkermansia muciniphila depletion ameliorates alcoholic liver disease. Gut. 2018;67:891–901.
Bull-Otterson L, Feng W, Kirpich I, Wang Y, Qin X, Liu Y, et al. Metagenomic analyses of alcohol induced pathogenic alterations in the intestinal microbiome and the effect of Lactobacillus rhamnosus GG treatment. PLoS One. 2013;8:e53028.
Forsyth CB, Farhadi A, Jakate SM, Tang Y, Shaikh M, Keshavarzian A. Lactobacillus GG treatment ameliorates alcohol-induced intestinal oxidative stress, gut leakiness, and liver injury in a rat model of alcoholic steatohepatitis. Alcohol. 2009;43:163–72.
Leclercq S, Matamoros S, Cani PD, Neyrinck AM, Jamar F, Stärkel P, et al. Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc Natl Acad Sci USA. 2014;111:E4485–93.
Bode C, Kolepke R, Schäfer K, Bode JC. Breath hydrogen excretion in patients with alcoholic liver disease-evidence of small intestinal bacterial overgrowth. Z Gastroenterol. 1993;31:3–7.
Lang S, Duan Y, Liu J, Torralba MG, Kuelbs C, Ventura-Cots M, et al. Intestinal fungal dysbiosis and systemic immune response to fungi in patients with alcoholic hepatitis. Hepatology. 2020;71:522–38.
Duan Y, Llorente C, Lang S, Brandl K, Chu H, Jiang L, et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature. 2019;575:505–11.
Temko JE, Bouhlal S, Farokhnia M, Lee MR, Cryan JF, Leggio L. The microbiota, the gut and the brain in eating and alcohol use disorders: a ‘Ménage à Trois’?. Alcohol Alcohol. 2017;52:403–13.
Molinero N, Ruiz L, Sánchez B, Margolles A, Delgado S. Intestinal bacteria interplay with bile and cholesterol metabolism: implications on host physiology. Front Physiol. 2019;10:185.
Park JW, Kim SE, Lee NY, Kim JH, Jung JH, Jang MK, et al. Role of microbiota-derived metabolites in alcoholic and non-alcoholic fatty liver diseases. Int J Mol Sci. 2021;23:426.
Miyamoto J, Igarashi M, Watanabe K, Karaki SI, Mukouyama H, Kishino S, et al. Gut microbiota confers host resistance to obesity by metabolizing dietary polyunsaturated fatty acids. Nat Commun. 2019;10:4007.
Zhu L, Wang Y, Pan CQ, Xing H. Gut microbiota in alcohol-related liver disease: pathophysiology and gut-brain cross talk. Front Pharm. 2023;14:1258062.
Shen H, Zhou L, Yang Y, Shu H, Wu D, Yang S, et al. The gut microbiota-produced vitamin B6 mitigates alcohol-associated liver disease by attenuating hepatic oxidative stress damage. Hepatol Commun. 2025;9:e0599.
Leclercq S, De Saeger C, Delzenne N, de Timary P, Stärkel P. Role of inflammatory pathways, blood mononuclear cells, and gut-derived bacterial products in alcohol dependence. Biol Psychiatry. 2014;76:725–33.
Yang AM, Inamine T, Hochrath K, Chen P, Wang L, Llorente C, et al. Intestinal fungi contribute to development of alcoholic liver disease. J Clin Invest. 2017;127:2829–41.
Dukić M, Radonjić T, Jovanović I, Zdravković M, Todorović Z, Kraišnik N, et al. Alcohol, inflammation, and microbiota in alcoholic liver disease. Int J Mol Sci. 2023;24:3735.
Chu H, Duan Y, Yang L, Schnabl B. Small metabolites, possible big changes: a microbiota-centered view of non-alcoholic fatty liver disease. Gut. 2019;68:359–70.
Roager HM, Licht TR. Microbial tryptophan catabolites in health and disease. Nat Commun. 2018;9:3294.
Wrzosek L, Ciocan D, Hugot C, Spatz M, Dupeux M, Houron C, et al. Microbiota tryptophan metabolism induces aryl hydrocarbon receptor activation and improves alcohol-induced liver injury. Gut. 2021;70:1299–308.
Shen H, Zhou L, Zhang H, Yang Y, Jiang L, Wu D, et al. Dietary fiber alleviates alcoholic liver injury via Bacteroides acidifaciens and subsequent ammonia detoxification. Cell Host Microbe. 2024;32:1331–1346.e6.
Rutkowski JM, Stern JH, Scherer PE. The cell biology of fat expansion. J Cell Biol. 2015;208:501–12.
Boden G. Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes. 1997;46:3–10.
Eguchi A, Feldstein AE. Adipocyte cell death, fatty liver disease and associated metabolic disorders. Dig Dis. 2014;32:579–85.
Tang T, Sui Y, Lian M, Li Z, Hua J. Pro-inflammatory activated Kupffer cells by lipids induce hepatic NKT cells deficiency through activation-induced cell death. PLoS ONE. 2013;8:e81949.
Yeop Han C, Kargi AY, Omer M, Chan CK, Wabitsch M, O'Brien KD, et al. Differential effect of saturated and unsaturated free fatty acids on the generation of monocyte adhesion and chemotactic factors by adipocytes: dissociation of adipocyte hypertrophy from inflammation. Diabetes. 2010;59:386–96.
Jones BE, Lo CR, Liu H, Srinivasan A, Streetz K, Valentino KL, et al. Hepatocytes sensitized to tumor necrosis factor-alpha cytotoxicity undergo apoptosis through caspase-dependent and caspase-independent pathways. J Biol Chem. 2000;275:705–12.
Schwabe RF, Brenner DA. Mechanisms of Liver Injury. I. TNF-alpha-induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol. 2006;290:G583–9.
Parker R, Kim S-J, Gao B. Alcohol, adipose tissue and liver disease: mechanistic links and clinical considerations. Nature Rev Gastroenterol Hepatol. 2018;15:50–59.
Giannessi D, Maltinti M, Del Ry S. Adiponectin circulating levels: a new emerging biomarker of cardiovascular risk. Pharmacol Res. 2007;56:459–67.
Ramakrishnan N, Auger K, Rahimi N, Jialal I. Biochemistry, Adiponectin. StatPearls Publishing Copyright © 2025, StatPearls Publishing LLC.; 2025.
Xu H, Zhao Q, Song N, Yan Z, Lin R, Wu S, et al. AdipoR1/AdipoR2 dual agonist recovers nonalcoholic steatohepatitis and related fibrosis via endoplasmic reticulum-mitochondria axis. Nat Commun. 2020;11:5807.
Alzahrani B, Iseli T, Ramezani-Moghadam M, Ho V, Wankell M, Sun EJ, et al. The role of AdipoR1 and AdipoR2 in liver fibrosis. Biochim Biophys Acta Mol Basis Dis. 2018;1864:700–8.
Park PH, Sanz-Garcia C, Nagy LE. Adiponectin as an anti-fibrotic and anti-inflammatory adipokine in the liver. Curr Pathobiol Rep. 2015;3:243–52.
Leclercq IA, Farrell GC, Schriemer R, Robertson GR. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J Hepatol. 2002;37:206–13.
Shen J, Sakaida I, Uchida K, Terai S, Okita K. Leptin enhances TNF-alpha production via p38 and JNK MAPK in LPS-stimulated Kupffer cells. Life Sci. 2005;77:1502–15.
Aleffi S, Petrai I, Bertolani C, Parola M, Colombatto S, Novo E, et al. Upregulation of proinflammatory and proangiogenic cytokines by leptin in human hepatic stellate cells. Hepatology. 2005;42:1339–48.
Worth T. Could the gut give rise to alcohol addiction? Nature 2024.
Seo B, Jeon K, Moon S, Lee K, Kim WK, Jeong H, et al. Roseburia spp. abundance associates with alcohol consumption in humans and its administration ameliorates alcoholic fatty liver in mice. Cell Host Microbe. 2020;27:25–40.e6.
Xu Z, Wang C, Dong X, Hu T, Wang L, Zhao W, et al. Chronic alcohol exposure induced gut microbiota dysbiosis and its correlations with neuropsychic behaviors and brain BDNF/Gabra1 changes in mice. Biofactors. 2019;45:187–99.
Zhang J, Zhu S, Ma N, Johnston LJ, Wu C, Ma X. Metabolites of microbiota response to tryptophan and intestinal mucosal immunity: A therapeutic target to control intestinal inflammation. Med Res Rev. 2021;41:1061–88.
Day AW, Perez-Lozada J, DiLeo A, Blandino K, Maguire J, Kumamoto CA. Candida albicans Colonization Modulates Murine Ethanol Consumption and Behavioral Responses Through Elevation of Serum Prostaglandin E(2) and Impact on the Striatal Dopamine System. bioRxiv 2025.
Bokoliya SC, Russell J, Dorsett Y, Panier HA, Singh V, Daddi L, et al. Short-chain fatty acid valerate reduces voluntary alcohol intake in male mice. Microbiome. 2024;12:108.
Yao Y, Zhang W, Ming R, Deng Q, Zuo A, Zhang S, et al. Noninvasive 40-Hz light flicker rescues circadian behavior and abnormal lipid metabolism induced by acute ethanol exposure via improving SIRT1 and the circadian clock in the liver-brain axis. Front Pharm. 2020;11:355.
Ma HY, Xu J, Liu X, Zhu Y, Gao B, Karin M, et al. The role of IL-17 signaling in regulation of the liver-brain axis and intestinal permeability in Alcoholic Liver Disease. Curr Pathobiol Rep. 2016;4:27–35.
Hajiasgharzadeh K, Baradaran B. Cholinergic anti-inflammatory pathway and the liver. Adv Pharm Bull. 2017;7:507–13.
Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–8.
Hosseini N, Shor J, Szabo G. Alcoholic hepatitis: a review. Alcohol Alcohol. 2019;54:408–16.
Xu MJ, Zhou Z, Parker R, Gao B. Targeting inflammation for the treatment of alcoholic liver disease. Pharmacol Ther. 2017;180:77–89.
Louvet A, Labreuche J, Artru F, Bouthors A, Rolland B, Saffers P, et al. Main drivers of outcome differ between short term and long term in severe alcoholic hepatitis: a prospective study. Hepatology. 2017;66:1464–73.
Morley KC, Baillie A, Fraser I, Furneaux-Bate A, Dore G, Roberts M, et al. Baclofen in the treatment of alcohol dependence with or without liver disease: multisite, randomised, double-blind, placebo-controlled trial. Br J Psychiatry. 2018;212:362–9.
Miller PM, Book SW, Stewart SH. Medical treatment of alcohol dependence: a systematic review. Int J Psychiatry Med. 2011;42:227–66.
Burnette EM, Nieto SJ, Grodin EN, Meredith LR, Hurley B, Miotto K, et al. Novel agents for the pharmacological treatment of alcohol use disorder. Drugs. 2022;82:251–74.
Louvet A, Thursz MR, Kim DJ, Labreuche J, Atkinson SR, Sidhu SS, et al. Corticosteroids reduce risk of death within 28 days for patients with severe alcoholic hepatitis, compared with pentoxifylline or placebo-a meta-analysis of individual data from controlled trials. Gastroenterology. 2018;155:458–468.e8.
Lee YS, Kim HJ, Kim JH, Yoo YJ, Kim TS, Kang SH, et al. Treatment of severe alcoholic hepatitis with corticosteroid, pentoxifylline, or dual therapy: a systematic review and meta-analysis. J Clin Gastroenterol. 2017;51:364–77.
Thursz MR, Richardson P, Allison M, Austin A, Bowers M, Day CP, et al. Prednisolone or pentoxifylline for alcoholic hepatitis. N Engl J Med. 2015;372:1619–28.
Taru V, Szabo G, Mehal W, Reiberger T. Inflammasomes in chronic liver disease: hepatic injury, fibrosis progression and systemic inflammation. Journal Hepatol. 2024;81:895–910.
Abdelmegeed MA, Banerjee A, Jang S, Yoo SH, Yun JW, Gonzalez FJ, et al. CYP2E1 potentiates binge alcohol-induced gut leakiness, steatohepatitis, and apoptosis. Free Radic Biol Med. 2013;65:1238–45.
Nguyen-Khac E, Thevenot T, Piquet MA, Benferhat S, Goria O, Chatelain D, et al. Glucocorticoids plus N-acetylcysteine in severe alcoholic hepatitis. N Engl J Med. 2011;365:1781–9.
Moreno C, Langlet P, Hittelet A, Lasser L, Degré D, Evrard S, et al. Enteral nutrition with or without N-acetylcysteine in the treatment of severe acute alcoholic hepatitis: a randomized multicenter controlled trial. J Hepatol. 2010;53:1117–22.
Basauri A, González-Fernández C, Fallanza M, Bringas E, Fernandez-Lopez R, Giner L, et al. Biochemical interactions between LPS and LPS-binding molecules. Crit Rev Biotechnol. 2020;40:292–305.
Engelmann C, Habtesion A, Hassan M, Kerbert AJ, Hammerich L, Novelli S, et al. Combination of G-CSF and a TLR4 inhibitor reduce inflammation and promote regeneration in a mouse model of ACLF. J Hepatol. 2022;77:1325–38.
Szabo G, Mitchell M, McClain CJ, Dasarathy S, Barton B, McCullough AJ, et al. IL-1 receptor antagonist plus pentoxifylline and zinc for severe alcohol-associated hepatitis. Hepatology. 2022;76:1058–68.
Vergis N, Patel V, Bogdanowicz K, Czyzewska-Khan J, Fiorentino F, Day E, et al. IL-1 Signal Inhibition In Alcoholic Hepatitis (ISAIAH): a study protocol for a multicentre, randomised, placebo-controlled trial to explore the potential benefits of canakinumab in the treatment of alcoholic hepatitis. Trials. 2021;22:792.
Ambade A, Lowe P, Kodys K, Catalano D, Gyongyosi B, Cho Y, et al. Pharmacological inhibition of CCR2/5 signaling prevents and reverses alcohol-induced liver damage, steatosis, and inflammation in mice. Hepatology. 2019;69:1105–21.
Scheiermann P, Bachmann M, Goren I, Zwissler B, Pfeilschifter J, Mühl H. Application of interleukin-22 mediates protection in experimental acetaminophen-induced acute liver injury. Am J Pathol. 2013;182:1107–13.
Xiang X, Feng D, Hwang S, Ren T, Wang X, Trojnar E, et al. Interleukin-22 ameliorates acute-on-chronic liver failure by reprogramming impaired regeneration pathways in mice. J Hepatol. 2020;72:736–45.
Khawar MB, Azam F, Sheikh N, Abdul Mujeeb K. How does interleukin-22 mediate liver regeneration and prevent injury and fibrosis?. J Immunol Res. 2016;2016:2148129.
Arab JP, Sehrawat TS, Simonetto DA, Verma VK, Feng D, Tang T, et al. An open-label, dose-escalation study to assess the safety and efficacy of IL-22 agonist F-652 in patients with alcohol-associated hepatitis. Hepatology. 2020;72:441–53.
Chang B, Sang L, Wang Y, Tong J, Zhang D, Wang B. The protective effect of VSL#3 on intestinal permeability in a rat model of alcoholic intestinal injury. BMC Gastroenterol. 2013;13:151.
Tian F, Chi F, Wang G, Liu X, Zhang Q, Chen Y, et al. Lactobacillus rhamnosus CCFM1107 treatment ameliorates alcohol-induced liver injury in a mouse model of chronic alcohol feeding. J Microbiol. 2015;53:856–63.
Yoon EL, Kim W. Current and future treatment for alcoholic-related liver diseases. J Gastroenterol Hepatol. 2023;38:1218–26.
Acknowledgements
The authors used BioRender.com to create the illustrations. This work was supported by the Ministry of Science and Technology of the People’s Republic of China (grant no. 2023YFA1800800 to HW); National Natural Science Foundation of China (grant no. 82225008 to HW); China Postdoctoral Science Foundation - Anhui Joint Support Program (grant no. 2024T027AH to HS).
Author information
Authors and Affiliations
Contributions
HS wrote the section on causes of inflammation, cytokines and chemokines, extrahepatic organs, and clinical therapies; SL thoroughly reviewed and enhanced the full text; YI and GS wrote the section on immune cells; and HW initiated and supervised the paper writing process and edited the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shen, H., Liangpunsakul, S., Iwakiri, Y. et al. Immunological mechanisms and emerging therapeutic targets in alcohol-associated liver disease. Cell Mol Immunol (2025). https://doi.org/10.1038/s41423-025-01291-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41423-025-01291-w
