
Abstract
The development of efficient and sustainable methods for carbon-phosphorus bond formation is of great importance due to the wide application of organophosphorus compounds in chemistry, material sciences and biology. Previous C–H phosphorylation reactions under nonelectrochemical or electrochemical conditions require directing groups, transition metal catalysts, or chemical oxidants and suffer from limited scope. Herein we disclose a catalyst- and external oxidant-free, electrochemical C–H phosphorylation reaction of arenes in continuous flow for the synthesis of aryl phosphorus compounds. The C–P bond is formed through the reaction of arenes with anodically generated P-radical cations, a class of reactive intermediates remained unexplored for synthesis despite intensive studies of P-radicals. The high reactivity of the P-radical cations coupled with the mild conditions of the electrosynthesis ensures not only efficient reactions of arenes of diverse electronic properties but also selective late-stage functionalization of complex natural products and bioactive compounds. The synthetic utility of the electrochemical method is further demonstrated by the continuous production of 55.0 grams of one of the phosphonate products.
Similar content being viewed by others
Introduction
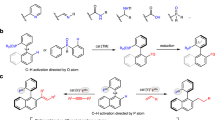
Aryl phosphorus compounds have wide applications in medicinal chemistry1, material science2, and catalysis as ligands and Lewis acid catalysts3,4. In addition, brigatinib, which contains a phenylphosphine oxide motif and serves as an anaplastic lymphoma kinase (ALK) inhibitor, has been achieved commercial success for treating metastatic non-small-cell lung cancer (NSCLC)5. Compared with the well-established Hirao reaction, which involves transition metal-catalyzed cross coupling of arylhalides with a P-donor6,7,8,9,10, C–H phosphorylation is more attractive and minimizes substrate prefunctionalization. In this context, several transition-metal catalyzed, directing group-assisted arene C–H phosphorylation reactions have been reported (Fig. 1a)11,12,13. The use of directing groups limits the scope of the arenes. Non-directed C(aryl)–H phosphorylation has also been reported and usually proceeds through a radical mechanism (Fig. 1b). In these reactions, either the arene or the phosphorus coupling partner is oxidized under photochemical14,15 or transition-metal catalyzed16,17 conditions to a radical intermediate, which then reacts with the other coupling partner to achieve C–P bond formation. These radical-based reactions are generally limited to electron-rich arenes or azoles18,19,20,21 due to the need to oxidize the arene to a radical cation and the moderate reactivity of the neutral P-radicals. In addition, excess arenes are frequently employed for the radical C–H phosphorylation reactions.

a Transition metal-catalyzed directed arene C–H phosphorylation. b Radical-mediated C–H phosphorylation of arenes. c Envisioned strategy for increasing the reactivity of P-radicals. d Electrochemical arene C–H phosphorylation in continuous flow (this work). DG directing group. BQ p-benzoquinone. EDG electron-donating group. EWG electron-withdrawing group.
Organic electrochemistry has been shown to be an enabling technology in addressing the challenges in sustainable synthesis22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37. In this context, electrochemical C–H phosphorylation of electron-rich heteroaromatics38,39 and arenes bearing directing groups40,41 have been reported. Reactions of electron-deficient arenes require metal catalysts and impractical three-electrode configuration in divided cells42,43,44,45,46,47.
Inspired by the observation that protonation of aminyl radicals to aminium radicals dramatically increases their reactivity toward π-systems48,49, we envision that the P-radical cation should be more reactive than the P-radicals and may react with electron-deficient arenes (Fig. 1c). The challenge is that the reactivity of P-radical cations toward electron-deficient arenes remains unknown and the trialkyl phosphite radical cations are known to react facilely with trialkyl phosphite precursor to form dimer radical cations50,51. Our interests in electrochemically driven radical reactions prompted us to investigate the reactivity of P-radical cations52. We surmise that the use of a continuous flow electrochemical reactor53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68 should be helpful in facilitating P-radical cation formation and its subsequent trapping reaction by the arenes because of the large ratio of electrode surface area to volume and efficient mass transfer in the flow reactor.
Herein we disclose a catalyst- and external oxidant-free, electrochemical C–H phosphorylation reaction of arenes in continuous flow for the synthesis of aryl phosphorus compounds (Fig. 1d). The electrochemical method is characterized by a broad scope, good functional group tolerance, easy scale-up, and compatibility with late-stage C–H functionalization of derivatives of complex natural products and drug molecules.
Results
Reaction development
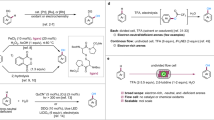
The C–H phosphorylation of benzoate 1 with triethyl phosphite [P(OEt)3] was chosen as a model reaction for the optimization of electrolysis conditions (Table 1 and Supplementary Table 1). The electrolysis was conducted in an undivided continuous flow electrochemical cell equipped with a graphite anode, a Pt cathode, and a flow channel cut in a fluorinated ethylene propylene spacer with 0.25 mm thickness (see the Supplementary Methods for details)62. An optimal 70% yield of phosphonate 2 was obtained when a solution of 1 (1 equiv), P(OEt)3 (5 equiv), HBF4 (2 equiv) and H2O (2 equiv) in MeCN was passed through the flow cell at 0.2 mL min−1. The C–H functionalization occurred exclusively at the position ortho to the ester group because the steric hindrance of tBu group prevented reaction at its ortho position. HBF4 (entry 2) and H2O (entry 3) were critical for success as 2 was not formed without either one. Increasing the amount of P(OEt)3 to 7 equiv (entry 4) or decreasing to 3 equiv (entry 5) or replacing P(OEt)3 with diethyl phosphite [HPO(OEt)2] (entry 6) were all detrimental to the formation of 2. A high concentration of P(OEt)3 probably led to competitive trapping of the P-radical cation with P(OEt)3. The use of other acids such as TFA (entry 7), AcOH (entry 8), or TfOH (entry 9), or Lewis acid Sc(OTf)3 (entry 10) instead of HBF4 resulted in either no product formation or low yield and recovery of most 1. Reaction in a batch reactor with the same amount of charge afforded the desired 2 in 36% yield (entry 11).
Evaluation of substrate scope
Under the optimal reaction conditions, the electrochemical C–H phosphorylation with P(OEt)3 tolerated arenes of various electronic properties (Fig. 2), including the highly electron-deficient dimethyl terephthalate (4) and dimethyl phthalate (5). The site selectivity of the C–P bond formation was like Friedel-Crafts reactions and favored reaction at ortho- or para- to the electron-releasing groups if there were no severe steric hindrance. Besides benzene and its derivatives, 2-substituted thiophenes reacted successfully at 4-position to give the corresponding products (29 and 30). Trialkyl phosphites bearing primary alkyl chains of different lengths reacted smoothly to give the corresponding phosphonate products (31–36). The electrochemical method was also suitable for late-stage functionalization of relatively complex natural products and bioactive compounds (37–46). Compounds that contained multiple aryl rings reacted at the most electron-rich one (38, 40, 41, and 46).

Reaction on 0.2 mmol of arene. All yields are isolated yields. aThe ratio of regioisomers was determined by NMR analysis of crude reaction mixture. bIsomers were separable by chromatography. rt room temperature. Ac acetyl.
Scale-up of continuous flow electrosynthesis is usually achieved through scale-out (passing more material through the reactor) or numbering up using parallel reactors without the need to change reaction conditions69. Continuous production can be achieved if the electrochemical system is stable without electrode passivation or tubing blockage. To test the stability of the electrochemical system for the continuous production of aryl phosphonates, a solution of mesitylene and P(OEt)3 in MeCN was mixed in-line with a solution of HBF4 and H2O in MeCN and passed through a reactor system consisting of two parallel electrochemical flow cells for 231 h (Fig. 3). The reaction produced 55.0 g (214 mmol) of the desired mesitylphosphonate 27 in 83% yield, which was even higher than the small-scale reaction (70%) and suggested that there was no reduction in reaction efficiency over time. The increased yield was probably due to the in situ mixing of HBF4 and P(OEt)3, the latter of which is known to undergo acid-promoted decomposition70. In fact, premixing of the reactants with HBF4 caused yield reduction after a few hours. It is fortunate that the residence time in the reactor is short (tr = 75 s) and in situ mixing is possible with continuous flow technology, showcasing the advantage of continuous flow electrosynthesis.

Reaction conditions: amesitylene (260 mmol), P(OEt)3 (5 equiv), HBF4 (2 equiv), H2O (2 equiv), MeCN, rt, 231 h. bTMSBr, MeCN. cSOCl2, DMF, reflux. dPhMgBr, THF. eHOCH2CH2CH2NH2, Et3N, THF. f(HOCH2)2CMe2, Et3N, THF. g(MeNHCH2)2, Et3N, THF. TMSBr trimethylsilyl bromide. THF tetrahydrofuran.
Diethyl mesitylphosphonate 27 could be hydrolyzed completely to phosphonic acid 47 (Fig. 3). One of the OEt groups of 27 could be replaced with different nucleophiles through a two-step process involving the intermediacy of ethyl mesitylphosphonochloridate to produce phosphinate 48, phosphonamidate 49, or mixed phosphonate 50. Similar two-step procedures were also employed to convert 47 to triarylphosphine oxide 51 and P-heterocycles 52–54.
Mechanistic studies
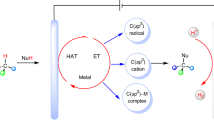
We next investigated the reaction mechanism. The cyclic voltammograms of P(OEt)3, compound 1, and HPO(OEt)2 indicated that P(OEt)3 (Ep/2 = 1.50 V vs SCE) was the easiest to oxidize among the three (Fig. 4a). The crude reaction mixture for the reaction of 1 with P(OEt)3 with (standard conditions) or without H2O (conditions I) was analyzed by 31P-NMR spectra (Fig. 4b). These experiments showed that P(OEt)3 decomposed into several phosphorus species in the absence of H2O. In contrast, under the standard conditions, the 31P-NMR spectrum was much cleaner with three major phosphorus species that were assigned to compound 2, HPO(OEt)2, and triethyl phosphate [OP(OEt)3]. Since trialkyl phosphites underwent rapid acid-promoted hydrolysis to H-phosphonates70, we thus conducted the phosphorylation reaction of 1 in the presence of both P(OEt)3 and HPO(OEt)2 without adding H2O (conditions II and III). These reactions produced 2 in 58% yield in the presence of 2 equiv of HPO(OEt)2 and a better yield of 78% with 0.2 equiv of HPO(OEt)2 (Fig. 4b). Under these conditions, the 31P-NMR spectra of the crude reaction mixtures were similar to those under the standard conditions.

a Cyclic voltammograms. b Exploring the potential role of H2O with 31P-NMR. The reaction mixture was treated with saturated aqueous NaHCO3 before recording NMR spectra. c Electrolysis in the presence of P(OEt)3 and HPO(OnBu)2. d 18O-labeling experiment. e Kinetic isotope effect experiment. f Proposed mechanism. SCE saturated calomel electrode.
The electrolysis reaction of 1 in the presence of P(OEt)3 and HPO(OnBu)2 produced 2 in 51% yield with an only trace amount of 55 (Fig. 4c), suggesting that the PO(OR)2 moiety of phosphonate product was derived from P(OR)3 instead of HPO(OR)2. Consistent with these results, 18O was not incorporated into 2 when the reaction was carried out with H218O (Fig. 4d). These results pointed out that HPO(OR)2 was critical for the C–H phosphorylation reaction. However, its exact role remained unclear. Finally, a kinetic isotope effect (KIE) of 1.0 was obtained from an intermolecular competition experiment with benzene and benzene-d6 (Fig. 4e), suggesting that C(aryl)–H bond cleavage was not involved in the rate-limiting step of the electrochemical reaction.
Based on the above studies, a possible mechanism for the electrochemical C–H phosphorylation reaction is proposed (Fig. 4f). The reaction starts with the oxidation of trialkyl phosphite 56 on the anode to generate P-radical cation 57, which reacts with arene 58 to give distal radical cation 59. The latter is oxidized further on the anode and then deprotonated to produce phosphonium 60, which loses an alkyl group to a nucleophilic species in the reaction mixture such as H2O or alcohol produced from the hydrolysis of P(OR)3 or during the workup to afford the final phosphonate product 61. Under these acidic conditions, protons, which are the most easily reduced species in the reaction mixture, accept electrons at the Pt cathode to generate H2. In addition to promote hydrolysis of P(OR)3, the acidic additive HBF4 serves as the supporting electrolyte and a proton source for H2 evolution, avoiding unwanted cathodic reduction of electron-deficient species such as 60 and 61. The HPO(OR)2 formed in situ through the hydrolysis of P(OR)3 likely forms reversibly with radical cation 57 an adduct 62, which reduces the decomposition of 57 and buys more time for its reaction with the arene.
In summary, we have developed an electrochemical method for the direct C–H phosphorylation of arenes with trialkyl phosphites without the need for any molecular catalysts or conventional chemical oxidants. Key to the success is to include H2O as an additive and conduct the electrolysis with a continuous flow cell. The continuous flow electrosynthesis is compatible with electron-rich and electron-deficient arenes, providing rapid and scalable access to various aryl phosphonates from easily available materials.
Methods
Representative procedure for the electrochemical C–H phosphorylation
The electrolysis was conducted using a flow electrolytic cell equipped with a graphite anode and a Pt cathode with an exposed surface area of 10 cm2 and interelectrode distance of 0.25 mm. The solution containing arene substrate (0.05 M, 1 equiv), P(OEt)3 (5 equiv), HBF4•Et2O (2 equiv) and H2O (2 equiv) in dry MeCN was pushed using a syringe pump to pass through the flow electrolytic cell operated with a flow rate of 0.2 mL min-1 and a constant current (45–55 mA). 4 mL of the outlet solution was collected once the reactor stabilized. The reaction was quenched with saturated NaHCO3 and extracted with ethyl acetate. The organic extracts were combined and concentrated under reduced pressure. The residue was chromatographed through silica gel eluting with ethyl acetate/hexanes or methanol/dichloromethane to give the target product. All new compounds were fully characterized (see the Supplementary Methods).
Data availability
The X-ray crystallographic coordinates for compound 44 have been deposited in the Cambridge Crystallographic Data Centre (CCDC), under deposition number 2079646 [www.ccdc.cam.ac.uk/data_request/cif]. The data supporting the findings of this study are available within the article and in the Supplementary Information file.
References
Finkbeiner, P., Hehn, J. P. & Gnamm, C. Phosphine oxides from a medicinal chemist’s perspective: physicochemical and in vitro parameters relevant for drug discovery. J. Med. Chem. 63, 7081–7107 (2020).
Gagnon, K. J., Perry, H. P. & Clearfield, A. Conventional and unconventional metal–organic frameworks based on phosphonate ligands: MOFs and UMOFs. Chem. Rev. 112, 1034–1054 (2012).
Surry, D. S. & Buchwald, S. L. Biaryl phosphane ligands in palladium-catalyzed amination. Angew. Chem. Int. Ed. 47, 6338–6361 (2008).
Bayne, J. M. & Stephan, D. W. Phosphorus Lewis acids: emerging reactivity and applications in catalysis. Chem. Soc. Rev. 45, 765–774 (2016).
Markham, A. Brigatinib: first global approval. Drugs 77, 1131–1135 (2017).
Schwan, A. L. Palladium catalyzed cross-coupling reactions for phosphorus–carbon bond formation. Chem. Soc. Rev. 33, 218–224 (2004).
Demmer, C. S., Krogsgaard-Larsen, N. & Bunch, L. Review on modern advances of chemical methods for the introduction of a phosphonic acid group. Chem. Rev. 111, 7981–8006 (2011).
Wang, S., Yang, C., Sun, S. & Wang, J. Catalyst-free phosphorylation of aryl halides with trialkyl phosphites through electrochemical reduction. Chem. Commun. 55, 14035–14038 (2019).
Bai, Y. et al. Nickel-catalyzed electrochemical phosphorylation of aryl bromides. Org. Lett. 21, 6835–6838 (2019).
Zhu, C., Yue, H., Nikolaienko, P. & Rueping, M. Merging electrolysis and nickel catalysis in redox neutral cross-coupling reactions: experiment and computation for electrochemically induced C–P and C–Se bonds formation. CCS Chem. 2, 179–190 (2020).
Feng, C. G., Ye, M., Xiao, K. J., Li, S. & Yu, J. Q. Pd(II)-catalyzed phosphorylation of aryl C-H bonds. J. Am. Chem. Soc. 135, 9322–9325 (2013).
Li, C., Yano, T., Ishida, N. & Murakami, M. Pyridine-directed palladium-catalyzed phosphonation of C(sp2)-H bonds. Angew. Chem. Int. Ed. 52, 9801–9804 (2013).
Min, M., Kang, D., Jung, S. & Hong, S. Rhodium-catalyzed direct C-H phosphorylation of (Hetero)arenes suitable for late-stage functionalization. Adv. Synth. Catal. 358, 1296–1301 (2016).
Niu, L. B. et al. Visible-light-induced external oxidant-free oxidative phosphonylation of C(sp(2))-H bonds. ACS Catal. 7, 7412–7416 (2017).
Shaikh, R. S., Ghosh, I. & Konig, B. Direct C-H phosphonylation of electron-rich arenes and heteroarenes by visible-light photoredox catalysis. Chem. Eur. J. 23, 12120–12124 (2017).
Berger, O. & Montchamp, J. L. Manganese-catalyzed and mediated synthesis of arylphosphinates and related compounds. J. Org. Chem. 84, 9239–9256 (2019).
Berger, O. & Montchamp, J. L. Manganese-mediated homolytic aromatic substitution with phosphinylidenes. Chem. Rec. 17, 1203–1212 (2017).
Luo, K., Chen, Y. Z., Yang, W. C., Zhu, J. & Wu, L. Cross-coupling hydrogen evolution by visible light photocatalysis toward C(sp(2))-P formation: metal-free C-H functionalization of thiazole derivatives with diarylphosphine oxides. Org. Lett. 18, 452–455 (2016).
Mu, X.-J., Zou, J.-P., Qian, Q.-F. & Zhang, W. Manganese(III) acetate promoted regioselective phosphonation of heteroaryl compounds. Org. Lett. 8, 5291–5293 (2006).
Peng, P. et al. Visible light mediated aerobic radical C–H phosphorization toward arylphosphonates. Org. Chem. Front. 3, 749–752 (2016).
Zhang, H. J., Lin, W., Wu, Z., Ruan, W. & Wen, T. B. Silver-mediated direct phosphorylation of benzothiazoles and thiazoles with diarylphosphine oxides. Chem. Commun. 51, 3450–3453 (2015).
Francke, R. & Little, R. D. Redox catalysis in organic electrosynthesis: basic principles and recent developments. Chem. Soc. Rev. 43, 2492–2521 (2014).
Siu, J. C., Fu, N. & Lin, S. Catalyzing electrosynthesis: a homogeneous electrocatalytic approach to reaction discovery. Acc. Chem. Res. 53, 547–560 (2020).
Nutting, J. E., Rafiee, M. & Stahl, S. S. Tetramethylpiperidine N-oxyl (TEMPO), phthalimide N-oxyl (PINO), and related N-oxyl species: electrochemical properties and their use in electrocatalytic reactions. Chem. Rev. 118, 4834–4885 (2018).
Yan, M., Kawamata, Y. & Baran, P. S. Synthetic organic electrochemical methods since 2000: on the verge of a renaissance. Chem. Rev. 117, 13230–13319 (2017).
Wiebe, A. et al. Electrifying organic synthesis. Angew. Chem. Int. Ed. 57, 5594–5619 (2018).
Ackermann, L. & Metalla-electrocatalyzed, C.-H. Activation by earth-abundant 3d metals and beyond. Acc. Chem. Res. 53, 84–104 (2020).
Karkas, M. D. Electrochemical strategies for C-H functionalization and C-N bond formation. Chem. Soc. Rev. 47, 5786–5865 (2018).
Yuan, Y. & Lei, A. Electrochemical oxidative cross-coupling with hydrogen evolution reactions. Acc. Chem. Res. 52, 3309–3324 (2019).
Jiao, K. J., Xing, Y. K., Yang, Q. L., Qiu, H. & Mei, T. S. Site-selective C-H functionalization via synergistic use of electrochemistry and transition metal catalysis. Acc. Chem. Res. 53, 300–310 (2020).
Ye, Z. & Zhang, F. Recent advances in constructing nitrogen-containing heterocycles via electrochemical dehydrogenation. Chin. J. Chem. 37, 513–528 (2019).
Moeller, K. D. Using physical organic chemistry to shape the course of electrochemical reactions. Chem. Rev. 118, 4817–4833 (2018).
Jing, Q. & Moeller, K. D. From molecules to molecular surfaces. exploiting the interplay between organic synthesis and electrochemistry. Acc. Chem. Res. 53, 135–143 (2020).
Wang, F. & Stahl, S. S. Electrochemical oxidation of organic molecules at lower overpotential: accessing broader functional group compatibility with electron−proton transfer mediators. Acc. Chem. Res. 53, 561–574 (2020).
Röckl, J. L., Pollok, D., Franke, R. & Waldvogel, S. R. A decade of electrochemical dehydrogenative C,C-coupling of aryls. Acc. Chem. Res. 53, 45–61 (2020).
Gandeepan, P., Finger, L. H., Meyer, T. H. & Ackermann, L. 3d metallaelectrocatalysis for resource economical syntheses. Chem. Soc. Rev. 49, 4254–4272 (2020).
Jiang, Y., Xu, K. & Zeng, C. Use of electrochemistry in the synthesis of heterocyclic structures. Chem. Rev. 118, 4485–4540 (2018).
Yuan, Y. et al. Exogenous-oxidant-free electrochemical oxidative C-H phosphonylation with hydrogen evolution. Chem. Commun. 55, 4230–4233 (2019).
Wang, S., Xue, Q., Guan, Z., Ye, Y. & Lei, A. Mn-catalyzed electrooxidative undirected C–H/P–H cross-coupling between aromatics and diphenyl phosphine oxides. ACS Catal. 11, 4295–4300 (2021).
Wu, Z.-J. et al. Scalable rhodium(III)-catalyzed aryl C−H phosphorylation enabled by anodic oxidation induced reductive elimination. Angew. Chem. Int. Ed. 58, 16770–16774 (2019).
Zhang, S.-K. et al. Electrocatalytic C-H phosphorylation through nickel(III/IV/II) catalysis. Chem 7, 1379–1392 (2021).
Strekalova, S. O. et al. Electrochemical phosphorylation of arenes catalyzed by cobalt under oxidative and reductive conditions. Phosphorus Sulfur Silicon Relat. Elem. 194, 506–509 (2019).
Khrizanforov, M. N. et al. Novel approach to metal-induced oxidative phosphorylation of aromatic compounds. Catal. Today 279, 133–141 (2017).
Nikitin, E. V. et al. Electrochemical synthesis of aryl phosphonates. Russ. Chem. Bull. 32, 566–569 (1983).
Yurko, E. O., Gryaznova, T. V. & Budnikova, Y. H. Electrochemical C-H phosphonation of caffeine. Phosphorus Sulfur Silicon Relat. Elem. 194, 343–344 (2019).
Khrizanforov, M. N. et al. Various ways of C-P bonds formation via selective electrochemical phosphorylation of aromatic C-H bonds. Phosphorus Sulfur Silicon Relat. Elem. 191, 1491–1493 (2016).
Budnikova, Y. H. & Dudkina, Y. B. Progress of electrochemical С(sp2)-H phosphonation. Phosphorus Sulfur Silicon Relat. Elem. 194, 415–419 (2019).
Ruffoni, A. et al. Practical and regioselective amination of arenes using alkyl amines. Nat. Chem. 11, 426–433 (2019).
Ganley, J. M., Murray, P. R. D. & Knowles, R. R. Photocatalytic generation of aminium radical cations for C horizontal line N bond formation. ACS Catal. 10, 11712–11738 (2020).
Gara, W. B. & Roberts, B. P. Electrochemical generation of the radical cations [X3PPX3]⨥.J. Chem. Soc. Chem. Commun. 1975, 949–950 (1975).
Kerr, C. M. L., Webster, K. & Williams, F. Electron spin resonance studies of.gamma.-irradiated phosphite and phosphate esters. Identification of phosphinyl, phosphonyl, phosphoranyl, and phosphine dimer cation radicals. J. Phys. Chem. 79, 2650–2662 (1975).
Xiong, P. & Xu, H.-C. Chemistry with electrochemically generated N-centered radicals. Acc. Chem. Res. 52, 3339–3350 (2019).
Pletcher, D., Green, R. A. & Brown, R. C. D. Flow electrolysis cells for the synthetic organic chemistry laboratory. Chem. Rev. 118, 4573–4591 (2018).
Atobe, M., Tateno, H. & Matsumura, Y. Applications of flow microreactors in electrosynthetic processes. Chem. Rev. 118, 4541–4572 (2018).
Noel, T., Cao, Y. & Laudadio, G. The Fundamentals Behind the Use of Flow Reactors in Electrochemistry. Acc. Chem. Res. 52, 2858–2869 (2019).
Elsherbini, M. & Wirth, T. Electroorganic synthesis under flow conditions. Acc. Chem. Res. 52, 3287–3296 (2019).
Winterson, B. & Wirth, T. Electrochemistry in Flow for Drug Discovery 1–52 (Springer, Berlin).
Tanbouza, N., Ollevier, T. & Lam, K. Bridging lab and industry with flow electrochemistry. iScience 23, 101720 (2020).
Laudadio, G. et al. Sulfonyl fluoride synthesis through electrochemical oxidative coupling of thiols and potassium fluoride. J. Am. Chem. Soc. 141, 11832–11836 (2019).
Laudadio, G. et al. Sulfonamide synthesis through electrochemical oxidative coupling of amines and thiols. J. Am. Chem. Soc. 141, 5664–5668 (2019).
Folgueiras-Amador, A. A., Philipps, K., Guilbaud, S., Poelakker, J. & Wirth, T. An easy-to-machine electrochemical flow microreactor: efficient synthesis of isoindolinone and flow functionalization. Angew. Chem. Int. Ed. 56, 15446–15450 (2017).
Huang, C., Qian, X. Y. & Xu, H. C. Continuous-flow electrosynthesis of benzofused S-heterocycles by dehydrogenative C-S cross-coupling. Angew. Chem. Int. Ed. 58, 6650–6653 (2019).
Folgueiras-Amador, A. A., Qian, X. Y., Xu, H. C. & Wirth, T. Catalyst- and supporting-electrolyte-free electrosynthesis of benzothiazoles and thiazolopyridines in continuous flow. Chem. Eur. J. 24, 487–491 (2018).
Gütz, C., Stenglein, A. & Waldvogel, S. R. Highly modular flow cell for electroorganic synthesis. Org. Process Res. Dev. 21, 771–778 (2017).
Zhong, X. et al. Scalable flow electrochemical alcohol oxidation: maintaining high stereochemical fidelity in the synthesis of levetiracetam. Org. Process Res. Dev. https://doi.org/10.1021/acs.oprd.1c00036 (2021).
Tateno, H., Matsumura, Y., Nakabayashi, K., Senboku, H. & Atobe, M. Development of a novel electrochemical carboxylation system using a microreactor. RSC Adv. 5, 98721–98723 (2015).
Ošeka, M. et al. Electrochemical aziridination of internal alkenes with primary amines. Chem 7, 255–266 (2021).
Maljuric, S., Jud, W., Kappe, C. O. & Cantillo, D. Translating batch electrochemistry to single-pass continuous flow conditions: an organic chemist’s guide. J. Flow. Chem. 10, 181–190 (2020).
Dong, Z., Wen, Z., Zhao, F., Kuhn, S. & Noël, T. Scale-up of micro- and milli-reactors: An overview of strategies, design principles and applications. Chem. Eng. Sci. X 10, 100097 (2021).
Li, C. et al. Water determines the products: an unexpected Brønsted acid-catalyzed PO–R cleavage of P(iii) esters selectively producing P(O)–H and P(O)–R compounds. Green. Chem. 21, 2916–2922 (2019).
Acknowledgements
We acknowledge NSFC (No. 21971213, H.-C.X.) and Fundamental Research Funds for the Central Universities for support.
Author information
Authors and Affiliations
Contributions
H.L., C.H., Y.-T.Z., Z.-Y.L., L.H.J., J.S. and S.Z. performed the experiments and analyzed the data. H.-C.X. designed and directed the project and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Long, H., Huang, C., Zheng, YT. et al. Electrochemical C–H phosphorylation of arenes in continuous flow suitable for late-stage functionalization. Nat Commun 12, 6629 (2021). https://doi.org/10.1038/s41467-021-26960-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-26960-y
This article is cited by
-
Heterogeneous electrosynthesis of C–N, C–S and C–P products using CO2 as a building block
Nature Synthesis (2024)
-
Electrochemical synthesis of peptide aldehydes via C‒N bond cleavage of cyclic amines
Nature Communications (2024)
-
Electrochemistry in organics: a powerful tool for “green” synthesis
Journal of Solid State Electrochemistry (2024)
-
Lab-scale flow chemistry? Just do it yourself!
Journal of Flow Chemistry (2024)
-
Recent Advances in the Application of P(III)-Nucleophiles to Create New P−C Bonds through Michaelis–Arbuzov-Type Rearrangement
Topics in Current Chemistry (2024)
