
Abstract
The electrocatalytic valorization of polyethylene terephthalate-derived ethylene glycol to valuable glycolic acid offers considerable economic and environmental benefits. However, conventional methods face scalability issues due to rapid activity decay of noble metal electrocatalysts. We demonstrate that a dynamic potential cycling approach, which alternates the electrode potential between oxidizing and reducing values, significantly mitigates surface deactivation of noble metals during electrochemical oxidation of ethylene glycol. This method enhances catalyst activity by 20 times compared to a constant-potential approach, maintaining this performance for up to 60 h with minimal deactivation. In situ Raman and X-ray absorption spectroscopy show that this effectiveness results from efficient removal of surface oxide during the reaction. The strategy is applicable to polyethylene terephthalate hydrolysates and various noble metals, such as palladium, gold, and platinum, with palladium showing a high conversion rate in recent studies. Our approach offers an efficient and durable method for electrochemical upcycling of biomass-derived compounds.
Similar content being viewed by others

Introduction
In recent years, the recycling of polyethylene terephthalate (PET), through both mechanical1,2,3,4 and chemical pathways5,6,7,8,9, has garnered increasing attention due to environmental and the economic advantages. This heightened interest stems from the acknowledgment that derivatives of PET, such as ethylene glycol (EG), can serve as green platform chemicals. They facilitate the preparation of a diverse array of value-added products through various catalytic pathways, making a substantial contribution to sustainability. In contrast to the widely-adopted thermocatalytic pathways10,11, electrochemical oxidation based on renewable energy has emerged as an appealing and sustainable approach to upgrade EG to value-added chemicals such as glycolic acid (GA), formic acid, and oxalic acid12,13,14,15,16,17. Among these, GA holds particular economic significance, being widely used in cosmetics, textile industry, and plastic manufacturing, as it serves as the monomer of polyglycolic acid, a biodegradable polymer18. In a typical electrocatalytic process, EG is electrochemically oxidized in an alkaline medium, employing either a constant-potential (CP) or a constant-current protocol, with noble metal catalysts like Pd, Pt, and Au widely used for their high selectivity in converting EG to GA due to their moderate C-C bond-breaking capabilities19,20,21,22.
However, under these conditions (e.g., CP electrochemical conversion), the activities of noble metal catalysts usually experience rapid decays, characterized by a swift decrease in current density. For instance, the current density can drop to half of the initial value within one hour22,23,24,25,26. This presents a significant challenge for the application of these catalysts toward EGOR, especially in long-term operation. Various spectroscopic evidence suggests that the quick deactivation of noble metal catalysts during alkaline EGOR should be attributed to the catalyst surface poisoning or the formation of surface oxide27,28,29,30,31,32,33. In order to alleviate the current decay of noble metal catalysts during long-term EGOR, researchers have invested significant efforts in engineering catalyst morphology34,35,36,37, composition38,39,40,41 and synergy effect42,43,44, but this also inevitably imposes stringent technical requirements on concurrent catalyst design and preparations. Recent studies on the electrocatalytic conversion of benzyl alcohol indicate that intermittent potential strategies can markedly extend the lifespan of the electrocatalytic process, while inevitable activity decay still persists in constant potential tests33. This suggests that a similar strategy can be applied in EGOR, also urging a re-examination of the underlying chemistry behind the deactivation of noble metal catalysts.
In this study, through in situ Raman spectroscopy and in situ X-ray absorption spectroscopy (XAS), we successfully identify the oxide formation on the noble metal catalysts, aligning with catalyst deactivation during CP-EGOR, highlighting the crucial role of surface oxidation in catalyst deactivation. Inspired by recent reports utilizing dynamic potential control for electrochemical reactions45,46,47,48,49, we further introduce an innovative electrochemical potential cycling (EPC) strategy to address the rapid activity decay of noble metal catalysts during conventional EGOR. In this approach, EGOR occurs under a triangle potential waveform: at more oxidizing potentials, EGOR turns on as expected and catalysts get oxidized; at more reducing potentials, the oxide on catalysts’ surface gets reduced, supported by in situ spectroscopic measurements. Our EPC-EGOR strategy exhibits a significantly enhanced catalytic rate for EG-to-GA conversion and allows noble metal catalysts to maintain a high catalytic activity during long-term EGOR operation (for up to 60 h with no apparent deactivation thereafter). In comparison, the same electrode during CP-EGOR suffers deactivation within minutes. Specifically, using nickel foam-supported Pd catalyst (Pd/NF) prepared by a simple replacement-deposition method, we can achieve a 20-fold higher conversion rate with EPC-EGOR than with CP-EGOR in a ten-hour measurement, with a high Faradaic efficiency (FE) of above 95% for GA. We observe that, to achieve a same conversion rate by Pd/NF, EPC-EGOR allows the use of about 90% less noble metal catalysts compared to CP-EGOR. Notably, with optimized EPC parameters in a flow-cell system, our Pd/NF catalysts exhibit a high conversion rate (1.70 mmol h−1 mg−1) of EG to GA among recent literature, where meticulously crafted electrocatalysts have been developed. Furthermore, we show that this EPC strategy is also applicable to PET hydrolysates and other noble metal catalysts (such as Au and Pt) for enhanced EGOR performance. In addition to advancing our understanding of mechanisms related to EGOR catalyst deactivation, the introduction of the EPC strategy opens avenues for a more sustainable and efficient electrochemical upcycling of polymer-derived compounds, laying the foundation for a greener and more economically viable future in the field of polymer valorization.
Results and discussion
As Pd is known as a highly active catalyst toward EGOR17,35,36,37,50, a nickel foam (NF)-supported Pd catalysts (Pd/NF) prepared through a simple spontaneous displacement reaction was used in this work. As this straightforward method is widely employed in electrocatalysis, it helps establish a robust basis for comparison with existing literature17,42,50. The prepared Pd/NF is named according to the concentration of H2PdCl4 used; for instance, Pd/NF-1 is derived from 1 mM H2PdCl4 solution (further details are available in the “Methods” Section). Scanning electron microscopy (SEM) and corresponding energy-dispersive spectroscopy (EDS) mapping reveal the formation of a Pd deposited layer on the NF surface, consistent with literature findings50. For Pd/NF-1, the stacking of nanosheets formed a Pd layer with a thickness of approximately 0.21 ± 0.05 μm (Fig. 1a and Fig. S1). With the catalyst loading increased, scanning transmission electron microscopy-secondary electron (STEM-SE) images depict a composite Pd network consisting of nanosheet arrays and aggregates (approximately urchin-like nanoparticles) on Pd/NF-10, and the Pd layer thickness increases to about 1.0 ± 0.1 μm (Fig. S2). X-ray diffraction (XRD) patterns (Fig. 1b) confirm the polycrystalline nature of metallic Pd/NF, as peaks at 40.1°, 46.7°, and 68.1° match the diffraction pattern of Pd (COD no. 900-8478). The surface of Pd is primarily metallic, with minor oxidation, as evidenced by high-resolution X-ray photoelectron spectroscopy (XPS) (Fig. S3). Consistent with the literature50, the Pd/NF-1 catalyst prepared in this work indeed exhibits a significant EGOR current on cyclic voltammograms (CV) in an electrolyte of 1 M KOH and 0.3 M EG (Fig. S4), with an onset potential of about +0.53 V and a peak current density of 208 mA cm−2 at around 1.08 V. To assess its long-term catalytic performance, an EGOR experiment was conducted at a CP mode, which was widely adopted in the field51,52,53,54. Under this condition, the current density drops consistently over time, signaling the decay of the EGOR activity, consistent with the observations in other studies22,23,24,25,26. As shown in Fig. 1c, at a CP of 0 V, the current density of Pd/NF-1 for EGOR drops to nearly zero abruptly after only about 15 sec. The current density decays slower at a less oxidizing potential50,55 but at the cost of the electrocatalytic activity (i.e., the initial EGOR current becomes smaller as shown in Fig. 1c), and we find the highest conversion rate in the CP mode is achieved at +0.93 V (Fig. 1d). In a 10-hour electrolysis, our Pd/NF-1 catalyst exhibits a conversion rate of 0.06 mmol h−1 at +0.93 V in the first five hours, but the overall conversion rate in ten hours is almost half of this value, suggesting a very low EGOR activity in the second five hours (Fig. 1d), consistent with the EG consumption data (Fig. 1e). The reaction products are found to be mostly glycolic acid (FE: 96.45%), with a small amount of oxalic acid and formic acid (Fig. 1f). The consumption of EG at +0.93 V is less than 4% in a 15-hour long test (Fig. 1e). Similarly, other noble metals, such as Pt and Au, also exhibit low activities during CP-EGOR (Fig. S7), despite their active EGOR in CV measurements. This highlights the dilemma of applying noble metal catalysts to EGOR.

a SEM image (top) and high magnification STEM-secondary electron (STEM-SE) image (bottom) of Pd/NF-1. b XRD pattern of a series of Pd/NF catalysts. The a.u. of the ordinate represents arbitrary units. c Current density (j) of Pd/NF-1 at various CPs, with corresponding long-time running shown in the inset. d The conversion rates of EGOR under different CPs. e Faradaic efficiency (FE) and the EG consumption measured after holding the Pd/NF-1 at +0.93 for different time periods. The error bars for catalyst evaluation results are from three independent experiments and include the error caused by HPLC detection, as in other figures. f FE and selectivity of different products obtained from CP fixed at +0.93 and +0.83 V in a 5 h test, respectively. For simplicity and comparison, the product salts obtained under alkaline conditions are labeled as acids, as are other figures. Reaction conditions: 0.3 M EG in 1 M KOH. Source data are provided as a Source Data file.
Deactivation mechanisms of catalysts under CP-EGOR
To gain insightful understanding of the catalyst deactivation mentioned above, we initially rule out the possibility of catalyst detachment or dissolution, as the results from inductively coupled plasma atomic emission spectroscopy (ICP-AES) indicate the loss of metal into the electrolyte is less than 0.1% by mass (Table S2). To gain insights of the catalyst deactivation, we choose Au nanoparticles deposited by ion sputtering on Ti substrates for 5 min (about 58 nm thick, Fig. S8) as the EGOR catalyst for in situ Raman measurements, given its high activity for surface-enhanced Raman scattering and similar EGOR behaviors to other noble metals (Fig. S7). We detected noble metal oxide formation, but not COad (CO-like intermediate species) poisoning, during CP-EGOR. In the Raman spectra of the electrode surface collected at different time during the oxidation of EG at CP (Fig. 2a, b), a strong band of around ~730 cm−1 is observed at the open-circuit potential (Fig. 2b), which is present regardless of the presence of EG (Fig. S9d). We assign this peak to the Au-OH stretching based on literature results56. When a constant potential of +1.43 V is applied, the EGOR current density attenuates over time, and the Au-OH stretching band gradually diminishes, accompanied by the emergence of a new band at ~570 cm−1 when the current density drops to minimal (maker F in Fig. 2b). The same band also appears when the electrode is biased at +1.43 V without EG in the electrolyte, where surface oxidation of Au is taking place according to CV (Fig. S9a), and thus we assign this band to the surface oxide AuOx (Fig. S9c)56,57. Previous spectroscopic studies report a COad band at around 2000 cm−1 on noble metal surfaces, concluding that the activity decay during alcohol oxidation is due to COad poisoning58,59. However, this band was not observed in our Raman spectra during CP-EGOR (Fig. S10). While our Raman data indicate the presence of the oxide layer when EGOR is deactivated, we further find with XAS that the initial presence of the oxide layer is before the deactivation. To identify detailed information related to the chemical state and the coordination environment of noble metal catalysts under the working conditions, we carried out operando measurements with X-ray absorption near edge structure spectroscopy (XANES) and extended X-ray absorption fine structure spectroscopy (EXAFS) (Fig. 2c, d) while EGOR is on-going (Fig. S11). The XANES white-line intensity of the Au L3-edge is generally used as a marker for its oxidation state: a higher intensity indicates that Au is more oxidized60. We observe that in the presence of EG, the normalized white-line intensity of Au at +1.33 V is higher than that at +0.43 V, suggesting that the Au surface is indeed oxidized during EGOR. The same conclusion can also be drawn from EXAFS results, which show that the signal intensity for the Au-O peak grows when EGOR takes place (Fig. 2c, d). Similarly, the increase in the intensity of the Pd-O coordination peak and the Pd white line peak indicates that the surface of Pd undergoes oxidation during EGOR (Fig. S12). Unfortunately, the exact forms of PdOx and AuOx are difficult to determine, as the thickness of the oxide layer calculated from the CV curve is only about 0.025 nm (Fig. S13 and Table S4), that is, the thickness of only one single atomic layer. Moreover, at the oxidation potential, the oxide layer does not grow rapidly over time, remaining around 0.04 nm (Table S4). This poses a significant challenge for characterization.

a EGOR current density (j) over time (green solid line) measured at +1.43 V during in situ Raman spectrum collection using an Au/Ti electrode. b In situ Raman spectra collected at different time during the I-t measurement in (a). The a.u. of the ordinate represents arbitrary units. The acquisition time for each spectrum was 20 s. The capital letters in (a) and (b) represent the collection points of Raman spectra. c The Au L3-edge XANES spectra and (d) EXAFS spectra of an Au/Ti electrode collected at +0.43 V and +1.33 V, compared with reference samples (gold oxide and Au metal). Electrolyte: 1 M KOH with 0.3 M EG. e EGOR current density over time measured with EG injection at different time: 0 s (black dashed line), 150 s (red solid line), and 1800 s (purple solid line). A sample of Pd/NF-10 was initially biased at +1.23 V in a pure 1 M KOH solution, and EG was then quickly injected under vigorous stirring at different time to achieve a concentration of 0.3 M. Insets show the details of the current change upon EG injection. f Schematic illustration of the EGOR deactivation over noble metal (M=Pd, Pt, Au) catalyst during CP electrolysis. Source data are provided as a Source Data file.
Through electrochemical impedance spectroscopy (EIS), we observe that the oxidation of catalyst surfaces during EGOR leads to increased charge-transfer resistance (Rct) at the electrode-electrolyte interface (Fig. S14 and Table S5). When Pd catalysts are measured at different potentials, Rct is minimal at +0.83 V, approximately 20 Ω, which align with the optimal conversion rate of Pd/NF-1 at a CP of around +0.83 V (Fig. 1d). However, further increasing the bias leads to a significantly larger Rct. For example, Rct at +1.23 V is 1585 Ω, creating a substantial barrier for electron transfer in EGOR. This phenomenon is consistent with Au as an EGOR catalyst, which shows the smallest Rct at +1.13 V (approximately 14 Ω), and the Rct is 4679 Ω when the potential is further increased to +1.33 V (Fig. S15 and Table S6). Based on the aforementioned spectroscopic data, we attribute the large Rct at more positive potentials to the presence of oxide layers, which severely deteriorate the catalyst activity for EGOR. As shown in Fig. 2e, when we add EG into the electrolyte at different time (0, 150, and 1800 s), the current spike due to EGOR decreases significantly if EG is added at a later time when the oxide layers grow more in an EG-free solution. Additionally, commercial PdO demonstrated no capability to oxidize ethylene glycol (Fig. S16b). It only exhibited weak EGOR activity after a small amount of PdO was reduced at the lower potentials during CV (Fig. S16c).
Figure 2f schematically summarizes our findings of how the metal oxide layer develops on noble metal catalysts during CP-EGOR: the initial metallic catalyst surface favors the adsorption of OH and EG (Fig. S17). As EGOR takes place at the CP mode (i.e., EG gets oxidized at the catalyst surface), part of the OHads is also oxidized, forming a partially oxidized metal surface that is inactive toward EGOR, resulting in the observed current decay. Once the metal surface becomes completely oxidized, the catalyst loses its ability to perform EGOR. The reasons for the detrimental effects of the oxide layer on EGOR can be summarized as follows: 1) Once the metal surface is fully oxidized, the catalyst no longer adsorbs EG32, leading to EGOR deactivation. This is evidenced by the EGOR inactivity of PdO (Fig. S16). 2) After the metal surface oxidation, we observed an increase in interfacial charge transfer resistance (Fig. S14), which is unfavorable for EGOR. 3) The formation of the oxide layer weakens the catalyst’s ability to oxidize water or OH−61, thereby not generating the key adsorbate OHads necessary for removing reaction intermediates during EGOR, which also inhibits EGOR.
Interestingly, we notice that the catalytic activity of deactivated catalysts can be fully restored when the electrode is kept at the open-circuit condition for a certain amount of time (Fig. S18), accompanied by a reduced Rct (Fig. S19–S20 and Table S7, S8) and the disappearance of the metal oxide Raman band (Fig. S21). Two possible reasons can be responsible for this observation. (1) According to the Pourbaix diagram of Pd62, at pH 14, when the potential is at an open-circuit potential of about 0.2 V (Fig. S18a), Pd oxides and hydroxides are not the thermodynamically stable phase, and thus they are expected to turn into the metallic phase. (2) Using OCP as an indicator of surface oxidation states, we observed that the presence of EG accelerates the decomposition of the metal oxide (Fig. S22), indicating a reductive effect of EG on the oxide, consistent with previous literature33. In short, our experiments indicate that metal oxides formed on noble metal catalysts are not stable, and the catalyst surfaces will convert back to metallic states when the applied bias is lifted.
EGOR Performance with the EPC strategy
Clearly, to unlock the full potential of noble metal catalysts toward EGOR, it is crucial to inhibit the growth of surface oxides or efficiently remove them during the reaction. Instead of waiting for the time-consuming disappearance of the surface oxide at the open-circuit condition (Fig. S18), we demonstrate here an electrochemical potential cycling (EPC) strategy in which the working electrode potential is continuously cycling between two values: one positive enough to oxidize EG, and the other negative enough to reduce surface oxides on catalysts. We expect that the active potential control of the working electrode helps maintain the metallic state of the catalyst surfaces, allowing EGOR to proceed more effectively than it does at the CP mode. Indeed, as shown in Fig. 3a, when we cycle the working electrode potential between +0.33 and +1.23 V ( +0.33 → +1.23 → +0.33 V is referred to as one cycle) at a scan rate of 0.1 V/s, the EGOR peak current density is maintained above 100 mA·cm−2 for 10 h (2000 cycles, Fig. 3b), in sharp contrast to the fast current decay in the CP mode (Fig. 1c). As a result, the EG conversion rate (Fig. 3c) and the amount of EG consumed (Fig. S23) at the EPC mode are 10 times higher than that at the CP mode. We analyze the reaction products by HPLC and find a very similar product distribution between the two modes: GA is the dominant product, with FEs of 97.84% and 96.45% for EPC and CP modes, respectively (Fig. 3d). The significantly enhanced conversion rate at the EPC mode is independent of the catalyst substrate, NF, as we observed an EG consumption of less than 0.1% using a bare NF (Fig. S24) for a same test. Therefore, we demonstrate that the EPC strategy excels in EGOR by effectively modulating the catalyst surface state through dynamic potential control.

a Representative potential and current density (j) data over time in an EPC-EGOR experiment. Each cycle consists of a positive sweep from +0.33 V to +1.23 V and a negative sweep from +1.23 V to +0.33 V at a scan rate of 0.1 V/s. b CVs of different cycles of EPC-EGOR. c The conversion rates of EGOR under EPC and CP (an optimal potential of +0.93 V), respectively. d The corresponding Faradaic efficiency (FE) of diverse products obtained from EPC and CP. Reaction conditions: 0.3 M EG in 1 M KOH. Source data are provided as a Source Data file.
A gradual decrease of the EGOR peak current density in EPC mode is observed (Fig. 3b), but this does not indicate the activity loss of the catalyst, because the original catalytic activity of the catalyst can be fully recovered when the electrolyte is replaced with fresh solutions (1 M KOH with 0.3 M EG, Fig. S25a), suggesting that the decreasing EG concentration is responsible for the EGOR peak current density decay over time (Fig. S25b).
More promisingly, our EPC strategy is also applicable to other noble metal catalyst for improved EGOR performance. As shown in Fig. S26, compared with the highest conversion rate under the CP mode, EPC mode (at a scan rate of 0.1 V/s) improves the EG conversion rate by a factor of 4.2 for Pt and 5.7 for Au, respectively. Results from in situ Raman spectroscopy corroborate that EPC enhances the noble metal EGOR activity by suppressing metal oxide formation. Scanning the potential of an Au/Ti electrode from +0.53 V to +2.08 V at a scan rate of 2 mV/s (Fig. S27) results in a decrease in the Au-OH band intensity due to the continuous consumption of OHads (Fig. 4a and Fig. S17), consistent with the observation in the CP mode. As the potential rises to +1.53 V, the AuOx band at 570 cm−1 emerges (Fig. 4a), accompanied by a significant decrease in EGOR current density (Fig. S27). However, in contrast to the CP mode, this oxide layer can be reduced when the potential scan is reversed in the EPC mode. As shown in Fig. 4b, the AuOx band disappears when the potential is reversely scanned to +1.18 V, accompanied by the reappearing of Au-OH band. As summarized in Fig. 4c, these findings indicate that during EPC-EGOR, the surface oxide on noble metal catalysts formed in the positive scan can be reduced in the negative scan, thus allowing EGOR to actively proceed with these catalysts. The fact that the hydrogen adsorption/desorption region of Pd/NF-1 shows no significant change even after 500 cycles of potential cycling further demonstrates that the metal active sites are continuously regenerated during the EPC process (Fig. S28).

a In situ Raman spectra collected during (a) a positive scan and (b) a negative scan on Au/Ti under EGOR conditions. EPC runs at +0.53 ~ +2.08 V with a scan rate of 2 mV/s. The acquisition time for each spectrum was 45 s. The a.u. of the ordinate represents arbitrary units. Condition: 1 M KOH with 0.3 M EG. c Schematic illustration of the EGOR deactivation over noble metal (M = Pd, Pt, Au) catalyst during EPC electrolysis. Source data are provided as a Source Data file.
Optimization of EPC parameters for efficient EGOR
To optimize the performance of EGOR in the EPC mode, we studied the effect of various parameters, including catalysts loading, potential range, and scan rate. First, we compare the EGOR performance between EPC and CP modes with different Pd loading masses43,63. As previously described, Pd/NF catalysts of different Pd loading masses were obtained by varying the concentration of H2PdCl4 during Pd deposition. When comparing the EG conversion rates of these electrodes in CP-EGOR and in EPC-EGOR, we identified the optimal rates in both conditions. Specifically, since the conversion rates in CP-EGOR are potential dependent, the optimal value was identified by conducting tests at different potentials (Fig. S29). As for EPC-EGOR, we found that the EPC potential range strongly affects the performance of high catalyst-loading samples (more discussion will be provided later), and therefore we adjusted the scan range for these electrodes to identify their optimal EG conversion rates (Fig. S30). Figure 5a shows that, under optimized conditions, the EG conversion rates from EPC are higher than those from CP for all the catalyst loadings investigated, although in both cases the EG conversion rates increases with the amount of the catalyst. On the other hand, with a same electrolysis duration and a same potential range, the EPC mode consistently exhibits higher conversion rates of EG than the CP mode up to a Pd loading of 8.5 mg (Fig. S29). Further increasing the loading to 10 mg (Pd/NF-10), CP then exceeds EPC in the EG conversion rate (Fig. S29n), which can be overturned by expanding the EPC potential range as shown in Fig. 5a. Notably, the optimal EG conversion rate of Pd/NF-1 with EPC is comparable to that of Pd/NF-8.5 with CP (Fig. 5a), suggesting that, to achieve a same conversion rate, EPC can reduce the Pd loading by up to 90% (Table S1).

a Conversion rates of ethylene glycol oxidized by optimal EPC and optimal CP potentials with varying Pd loading masses. All the optimal EPC conversion rates of each Pd/NF were obtained at a scan rate of 0.1 V/s. The optimal EPC conversion rates of Pd/NF-1 to Pd/NF-4, Pd/NF-7 to Pd/NF-8.5, and Pd/NF-10 were obtained in the potential ranges of +0.33 V to +1.23 V, +0.33 V to +1.43 V, and +0.33 V to +1.83 V, respectively (Fig. S21). The optimal conversion rates of CP-EGOR of each Pd/NF are obtained from Fig. S20. b Conversion rate of ethylene glycol by Pd/NF-10 at different ranges of EPC potentials. c Conversion rate of Pd/NF-2 for EGOR at various scan rates within the same EPC potential range. d The conversion rates of Pd/NF-1 for EGOR under EPC (potential range: +0.63 ~ +1.28 V, scan rate: 2.0 V/s) and CP (an optimal potential of +0.93 V), respectively. Conversion rates (e) and corresponding Faradaic efficiency (FE) (f) from six EPC-EGOR tests (potential range: +0.33 ~ +1.23 V, scan rate: 0.1 V/s) using Pd/NF-2. Condition: 1 M KOH with 0.3 M EG. Source data are provided as a Source Data file.
In Fig. 3, we demonstrate the high EGOR activity by EPC using a potential range of +0.33 to +1.23 V for EPC. However, we find that this range may not be the most effective one for unlocking the full potential of EPC for EGOR. For example, the EG conversion rate of Pd/NF-10 can be doubled when the upper limit of the potential range is expanded from +1.23 to +1.83 V (Fig. 5b). The enhancement is elucidated through the corresponding CV curves (Fig. S30g), where raising the upper potential limit of EPC allows Pd/NF-10 to generate more EGOR current (until a peak current density is observed), leading to a higher conversion rate of EG. This effect, however, is less pronounced at lower loadings, where current decay (or catalyst deactivation) turns on at a less positive potential (Fig. S31), and therefore, expanding the upper potential limit does not contribute to a higher conversion rate for low catalyst-loading electrodes. Concerns may arise regarding the oxidation of NF and its impact on EGOR when the upper limit potential of EPC is increased. As depicted in Fig. S32a, NF undergoes significant oxidation above +1.43 V in an EG-free KOH solution (1 M). Under the same EGOR testing conditions for Pd/NF-10, NF achieves an EPC conversion rate of 0.212 mmol h−1 (Fig. S32b) within the potential range of +0.33 to +1.83 V (scan rate: 0.1 V/s), which is approximately five times lower than that of Pd/NF-10. Besides exhibiting lower EPC activity, NF also displays different selectivity compared to Pd/NF-10, with the main product being formic acid (selectivity: 76.08%, Fig. S32c). Since the selectivity of Pd/NF-10 toward formic acid stays consistently at about 2% across different EPC potential ranges (Fig. S31i), we conclude that, even at more oxidizing potentials, NF contributes minimally during the EPC-EGOR test of Pd/NF-10.
We also evaluate the impact of scan rate on the EG conversion rate. As shown in Fig. 5c, EPC with a scan rate of above 0.1 V/s exhibits a higher conversion rate, with the maximum EG conversion rate achieved at a scanning rate of 1.5 V/s. When the EGOR duration is expanded to five hours, the enhancement factor of the EG conversion rate for EPC over CP increases from 3.4-fold at 0.1 V/s to 5.1-fold at 1.5 V/s (Fig. S33c). The impact of the scan rate on the EG conversion rate can be understood as follows: (1) At a low scan rate, the duration in each EGOR cycle (td) is sufficiently long, allowing oxide layer growth on catalysts, resulting in deactivation; (2) As the scan rate is increasing, td shortens, during which the oxide layer formation is suppressed and the catalyst can maintain a high activity toward EGOR, as characterized by the increased current density during the negative scan (Fig. S33a). (3) At excessively high scan rates, two factors may contribute to lowering the EG conversion rate. First, small td at high scan rates may render the reaction nonviable when td is smaller than the EGOR time constant. Second, the charging/discharging processes of the electrochemical double layer can be significant at high scan rates, which yield non-Faradaic current. This is indeed what we have observed in Fig. S33b, where the total FE decreases consistently at a higher scan rate, independent of the leaching of the catalyst (Table S9). Therefore, there exists an optimal scan rate for the best EPC-EGOR performance.
Through the above systematic investigations, we learn that: (1) raising the upper potential limit of EPC is necessary for high catalyst-loading electrodes for better EGOR performance; (2) by optimizing the EPC potential range, the EPC mode can outperform the CP mode in the EG conversion rate at all catalyst loadings investigated, and the advantage is more significant with a lower catalyst loading; (3) a higher EPC-EG conversion rate can be achieved with a higher scan rate of up to 1.5 V/s, although the FE decreases consistently when the scan rate is increased due to capacitive charging. In a ten-hour test with all the above parameters optimized (Fig. S34), the enhancement factor of the EG conversion rate for EPC over CP increases from 10-fold to 20-fold (Fig. 5d). We also prepared a PET hydrolysate solution by hydrolyzing commercial PET powers at 60 °C (Fig. S35a) and directly used the solution as the electrolyte for EGOR, where we find that the EG conversion rate for EPC was 23 times higher than that for CP over a 10-hour electrolysis period (Fig. S35d). Further increasing the OH− concentration in the electrolyte to 3 M, we are able to achieve an EG conversion rate of as high as 2.702 mmol h−1 with the EPC strategy using the Pd/NF-1 catalyst (Fig. S36), surpassing the previously reported value (2.24 mmol h−1) obtained from CP-EGOR using a carefully-engineered electrocatalyst under the same electrolyte condition (Table S10).
We further demonstrate the application of the EPC strategy in a flow cell under industrial relevant currents (Fig. 6a). At a flow rate of 20 mL min−1, Pd/NF-1 (geometric area: 4 cm2) exhibited rapid deactivation in CP modes, similar to that observed in an H-cell. As shown in Fig. S37b, Pd/NF-1 becomes completely deactivated within 120 seconds at a constant potential of +1.53 V. To reach a same charge consumption of 3200 C (equivalent to 12% EG consumption), EPC mode only takes 2 h while CP mode needs up to 30 h (Fig. 6b). More importantly, the EPC-EGOR conversion rate of Pd/NF-1 in the flow cell system reaches 1.7 mmol h−1 mg−1, a notable improvement compared to recent studies (Table S11). Consequently, the conversion rate of EG by Pd/NF-1 in EPC mode was about 14 times higher than in CP mode (Fig. 6c), suggesting that our EPC strategy can be extended to the flow cell system for enhanced EGOR performance.

a Digital photos of flow-cell systems. b The charge (Q)-time curves of Pd/NF-1 in the flow-cell are obtained under EPC-EGOR and CP-EGOR conditions, respectively. c The conversion rates of Pd/NF-1 for EGOR with the same charge consumption under EPC (potential range: +0.63 ~ +2.03 V, scan rate: 2.0 V/s,) and CP (an optimal potential of +0.93 V, 30 h), respectively. Condition: 1 M KOH and 0.3 M EG, the volume of anolyte is 200 mL, flow rate: 20 mL min−1. Source data are provided as a Source Data file.
We also demonstrate the catalyst stability with the EPC strategy by performing multiple tests with one single Pd/NF electrode for a total test time of 60 h with no apparent deactivation thereafter. As shown in Fig. 5e, the conversion rates obtained from six consecutive tests at 5 h and 10 h are nearly the same, with almost identical product selectivity and FEs (Fig. 5f) (see Fig. S38 for representative current-potential curves and EG consumption data). Compared with CP-EGOR in a 5-hours test, the leaching of Pd after EGOR in EPC mode is relatively high, but less than 1%, which may be due to the continuous redox process on the catalyst surface and the relatively high reaction currents (Table S2). Nevertheless, the high EGOR activity and the great catalyst stability enabled by EPC make this method attractive for electrochemical upcycling of biomass-derived compounds.
In summary, this study presents an innovative EPC approach to enhance the catalytic activity of noble metal catalysts (Pd, Au, Pt) toward the electrochemical conversion of EG to GA. In comparison to conventional constant-potential methods, our EPC strategy demonstrates a remarkable 20-fold increase in the EG conversion rate, maintaining a high GA selectivity (> 95%) without the need for intricate catalyst design. In situ Raman and XAS characterization attributes the enhancement under the EPC condition to the suppression of surface oxide on the noble metal catalysts: in the anodic scan, noble metal catalysts oxidize EG and develop a layer of metal oxide on the surface that eventually deactivates EGOR; in the cathodic scan, the metal oxide layer gets reduced, regenerating the metallic surface for EGOR in the next cycle. We show that EPC-EGOR can achieve a same conversion rate with about 90% less noble metal catalysts compared to CP-EGOR. The dynamic control of the catalyst surface state by EPC also enables the catalysts to maintain great stability for up to 60 h, with no signs of deactivation thereafter. In comparison, when tested with the conventional constant-potential method, the electrocatalytic activity of the same electrode under the same condition drops to zero within minutes. Furthermore, we confirmed that this dynamic potential control strategy is applicable to flow cell systems, exhibiting a high conversion rate (1.70 mmol h−1 mg−1) of EG to GA among recent literature. The notable catalytic activity and stability were realized with the straightforwardly prepared catalyst, implying the immense potential of this approach for further enhancement through deliberate catalyst design and modification. Our study demonstrates the significance of catalyst surface engineering through dynamic potential control for EGOR, offering valuable insights and a pioneering methodology with broad implications for the electrochemical conversion of diverse chemicals.
Methods
Materials
All chemical reagents were used as received unless otherwise noted. Glycolic acid (99%) and glyoxylic acid (ca. 50% in water) were bought from Adamas-beta. Palladium chloride (PdCl2, 98%) and ethylene glycol (EG) were purchased from Aladdin. Alcohol (AR), hydrogen peroxide (H2O2, 37% aqueous solution), hydrochloric acid (HCl, AR) and sulfuric acid (H2SO4, GR) were obtained from Sinopharm Chemical Reagent Co. Ltd. Formic acid (≥ 99%), oxalic acid (99.8%), and potassium hydroxide (KOH, AR) were supplied by Macklin. Chloroplatinic acid hexahydrate (H2PtCl6 · 6H2O, ACS reagent, ≥ 37.50% Pt basis), gold(III) chloride trihydrate (HAuCl4 · 3H2O), PdO powder (99.97%) and trisodium citrate dihydrate were all procured from Sigma-Aldrich. Ni foam (NF, thickness: 1.7 mm; porosity: ~98%) and titanium foil (thickness 0.1 mm; purity: ≥ 99.99%) were purchased from Kunshan Guangjiayuan New Material Co., Ltd. All solutions were prepared with ultrapure water (18.2 MΩ·cm).
Prepared of Pd/NF and Pt/NF
Before preparing Pd/NF, NF needs to be cleaned to remove impurities and oxide layers from the surface. NF was first sonicated with ethanol and water for 20 min respectively. After sonicating in 3 M HCl for 20 min, cleaned by ultrapure water for three times. The deposition of Pd on the NF surface is a spontaneous oxidation-reduction process50. In a 20 mL vial, a piece of NF (10 × 20 × 1.7 mm) was placed into 10 mL H2PdCl4 solution with different concentrations. After stirring (300 rpm) at room temperature for 15 min, the solution changed from yellow to colorless. The prepared Pd/NF was cleaned three times with ethanol and ultrapure water and then dried overnight under vacuum at 60 °C. Depending on the concentration of the H2PdCl4 solution used, the electrodes were named Pd/NF-1 (i.e., 1 mM H2PdCl4), Pd/NF-2, Pd/NF-4, Pd/NF-7, Pd/NF-8.5, Pd/NF-10. The preparation of Pt/NF follows the procedure described above and named as Pt/NF-1 (i.e., 1 mM H2PtCl6).
Prepared of Pd/Ti and Au/Ti
Similar to NF, Ti foil needs to be cleaned before preparing Pd/Ti and Au/Ti. The cleaning process was the same as NF cleaning. The preparation of Pd/Ti was carried out by ion sputtering (Hitachi MC1000). At 100 V and 15 mA, Pd/Ti was obtained by continuous sputtering Pd on Ti foil (10 × 20 × 0.1 mm) for 5 min. The preparation of Au/Ti follows the same process.
Synthesis of 55 nm Au Nanoparticles
The preparation of 55 nm diameter gold nanoparticles follows a typical method64. Add 100 mL of HAuCl4 · 3H2O solution (0.01 wt%) to a round-bottom flask and heat until boiling with stirring. 0.7 mL of sodium citrate solution was rapidly injected into the boiling solution. After continuous boiling for 30 min, cool it to room temperature in naturally.
Characterization
A Hitachi HF5000 microscope equipped with a cold field emission gun operating at 200 kV was used to obtain scanning transmission electron microscopy images65. Using the Kratos Axis Ultra DLD with Mg Kα as the excitation source, X-ray photoelectron spectroscopy (XPS) analyzes were conducted on the sample. The resulting spectra were processed using CasaXPS software (version 2.3.24), with calibration based on the C 1 s peak at 284.8 eV. The scanning electron microscope (SEM) images and corresponding energy-dispersive X-ray spectroscopy (EDS) mapping were acquired from Hitachi SU3800 (hot field emission gun) at 15 kV and Hitachi SU8240 (cold field emission gun) at 5 kV. Using an inductively coupled plasma atomic emission spectrometer (ICP-AES, Avio 200, PerkinElmer), the metal content in the electrode and electrolyte was determined. A Bruker D8 Advance diffractometer with Cu Kα as radiation was used to acquire the XRD patterns. The electrochemical oxidation products of ethylene glycol were quantified by high-performance liquid chromatography (HPLC, LC-20A with a UV-vis detector (210 nm), Shanghai Chromatography Instrument Co., Ltd., China) with a Bio-Rad Aminex HPX-87H column. Sulfuric acid (5 mM) was used as the mobile phase.
In situ surface enhanced Raman spectroscopy (SERS) measurements were performed on a confocal Raman system (Renishaw inVia Qontor) with ×50 microscope objectives. A 633 nm laser was used as the excitation light. To enhance the Raman spectral signal, gold nanoparticles (2 μL, 55 nm) were dropped on the Au/Ti electrode surface and vacuum dried overnight. Raman spectrum was calibrated by the 520 cm−1 peak of Si. Unless otherwise specified, all Raman measurements were performed in an air atmosphere.
At the BL14W and BL11B beamlines of the Shanghai Synchrotron Radiation Facility (SSRF), operating at 3.5 GeV and 200 mA current, we collected X-ray absorption fine structure (XAFS) spectra at the Au L3-edge (E0 = 11919 eV) and Pd K-edge (E0 = 24350 eV). In situ measurements in fluorescence mode were conducted with an Ar-filled Lytle detector, and the X-ray energy calibration was carried out using the white line peak of Au and Pd foils. The data were analyzed using the Athena software65.
Electrochemical measurements
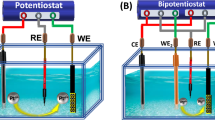
The electrochemical tests for the ethylene glycol oxidation reaction were carried out using a DH7002A electrochemical workstation from Jiangsu Donghua Analytical Instrument Co., Ltd. (China)65. A standard three-electrode system was employed for these tests, utilizing the pre-prepared Pd/NF as the working electrode, an Ag/AgCl (Saturated KCl) electrode as the reference electrode and a platinum mesh as the counter electrode. It is worth noting that the electrochemical data obtained were used directly without any further processing. Before use, Pd/NF was connected with Cu wire and sealed with hot glue. The geometric area exposed of the working electrode was 1*1 cm2. All working electrodes were treated identically. Before the electrochemical test, all working electrodes were cleaned by continuous cyclic voltammetry scanning in 1 M KOH in the potential range of −1.0 ~ +0.2 V vs. Ag/AgCl (Saturated KCl). Stably overlapping cyclic voltammetry curves were considered to indicate that the working electrodes had reached a stable state.
The performance of EG oxidation reaction was evaluated in an H-cell system separated by Nafion 117 membrane. Before use, Nafion 117 membrane was soaked in 5% H2O2 aqueous solution and 0.05 M H2SO4 aqueous solution at 80 °C for 1 h, and finally soaked in ultrapure water for 3 h. EG oxidation reaction was carried out in freshly prepared 1 M KOH solution, which was kept saturated with N2 during the electrochemical test. KOH solution was stored in a polytetrafluoroethylene container. According to the equation E (RHE) = E (Ag/AgCl) + 0.1989 V + 0.0591 × pH, all potentials are converted to values relative to the reversible hydrogen electrode (E vs. RHE)65. The frequency measurement range of electrochemical impedance spectroscopy at specific potentials was all from 0.1 Hz to 10 kHz, with an applied amplitude of 5 mV. Unless otherwise stated, all reported potentials are not corrected for iR drop. In the calculation of EG conversion rate, the running time of the potential cycle is given by multiplying the number of cycles by the time per cycle.
Electrochemical in situ SERS spectra were investigated in flow alkaline solution (0.3 M EG + 1 M KOH) at 30 rpm with a standard three electrodes system (GaossUnion, C031). Au/Ti deposited with gold nanoparticles was used as working electrode. Ag/AgCl electrode (Saturated KCl) and platinum wire were employed as the reference electrode and counter electrode, respectively. The cathode chamber and anode chamber of the electrolytic cell were separated by a Nafion 117 membrane.
The flow cell system was performed in a custom-made electrolytic cell with N2-saturated 1 M KOH solution as the electrolyte, and a Nafion 117 membrane separated the two electrode chambers. Commercial Pt/C supported on carbon paper was used as the counter electrode, and an Ag/AgCl (saturated KCl) electrode was used as the reference electrode. Pre-prepared Pd/NF (geometric area: 2 × 2 cm2) was used as the working electrode. The anolyte (0.3 M EG + 1 M KOH) was fixed at 200 mL, accompanied by stirring at 300 rpm, and a peristaltic pump was used to close-circulate the electrolyte at a flow rate of 20 mL min−1. Potentials in the flow cell system are not iR-corrected.
Hydrolysis of PET
To obtain monomeric ethylene glycol from PET, 12.6 g of dry powder (PET CB-102, 300 mesh, DuPont) was mixed with 200 mL of 2 M KOH aqueous solution in a round-bottom flask. The mixture was heated and stirred (900 rpm) in an oil bath at 60 °C for 24 h, resulting in a clear and transparent PET hydrolysate. The concentration of KOH in the hydrolysate was then diluted to 1 M and used directly as the electrolyte.
Product analysis
The products obtained under alkaline conditions are all present as salts, but for simplicity and comparison, they are labeled as acids in all figures of this work. The calibration curves were obtained by measuring different concentrations of standard solutions at first. The quantification of the products was calculated according to the calibration curve. The conversion rate of ethylene glycol, selectivity, and the Faradaic efficiency (FE) for EGOR were calculated with the following equations.
where the mole of formic acid needs to be divided by 2 for the calculation of formic acid selectivity.
where n is the number of electron transfer for each product, 3 for formic acid, 4 for glycolic acid, and 8 for oxalic acid; 96485 C mol−1 is the Faraday constant.
Data availability
All data for this study are available within the article and supplementary files or from the corresponding author upon reasonable request. Supplementary data generated for this study are provided in the Supplementary Information file. Source data are provided with this paper.
References
Rahimi, A. & García, J. M. Chemical recycling of waste plastics for new materials production. Nat. Rev. Chem. 1, 0046 (2017).
Tournier, V. et al. An engineered PET depolymerase to break down and recycle plastic bottles. Nature 580, 216–219 (2020).
Stubbins, A., Law, K. L., Muñoz, S. E., Bianchi, T. S. & Zhu, L. Plastics in the earth system. Science 373, 51–55 (2021).
Chen, Z. et al. Biodegradation of highly crystallized poly(ethylene terephthalate) through cell surface codisplay of bacterial PETase and hydrophobin. Nat. Commun. 13, 7138 (2022).
Kratish, Y., Li, J., Liu, S., Gao, Y. & Marks, T. J. Polyethylene terephthalate deconstruction catalyzed by a carbon-supported single-site molybdenum-dioxo complex. Angew. Chem. Int. Ed. 59, 19857–19861 (2020).
Korley, L. T. J., Epps, T. H., Helms, B. A. & Ryan, A. J. Toward polymer upcycling—adding value and tackling circularity. Science 373, 66–69 (2021).
Li, Y. et al. Catalytic transformation of PET and CO2 into high-value. Chem. Angew. Chem. Int. Ed. 61, e202117205 (2022).
Zhang, M.-Q. et al. Catalytic strategies for upvaluing plastic wastes. Chem 8, 2912–2923 (2022).
Cao, R. et al. Co-upcycling of polyvinyl chloride and polyesters. Nat. Sustainability 6, 1685–1692 (2023).
Griffin, M. B. et al. The selective oxidation of ethylene glycol and 1,2-propanediol on Au, Pd, and Au–Pd bimetallic catalysts. J. Catal. 307, 111–120 (2013).
Yan, H. et al. Engineering Pt-Mn2O3 interface to boost selective oxidation of ethylene glycol to glycolic acid. Appl. Catal. B Environ. 284, 119803 (2021).
Uekert, T., Kasap, H. & Reisner, E. Photoreforming of nonrecyclable plastic waste over a carbon nitride/nickel phosphide catalyst. J. Am. Chem. Soc. 141, 15201–15210 (2019).
Zhou, H. et al. Electrocatalytic upcycling of polyethylene terephthalate to commodity chemicals and H2 fuel. Nat. Commun. 12, 4679 (2021).
Qin, Y. C. et al. Extraordinary p–d hybridization interaction in heterostructural Pd-PdSe nanosheets boosts C−C bond cleavage of ethylene glycol electrooxidation. Angew. Chem. Int. Ed. 61, e202200899 (2022).
Wang, Y. et al. Implanting Mo atoms into surface lattice of pt3mn alloys enclosed by high-indexed facets: promoting highly active sites for ethylene glycol oxidation. ACS Catal. 9, 442–455 (2019).
Wang, J. et al. Electro-reforming polyethylene terephthalate plastic to co-produce valued chemicals and green hydrogen. J. Phys. Chem. Lett. 13, 622–627 (2022).
Ren, T. L. et al. Electrochemical co-production of ammonia and biodegradable polymer monomer glycolic acid via the co-electrolysis of nitrate wastewater and waste plastic. ACS Catal. 13, 10394–10404 (2023).
Budak, K., Sogut, O. & Aydemir Sezer, U. A review on synthesis and biomedical applications of polyglycolic acid. J. Polym. Res. 27, 208 (2020).
Gomes, J. F. & Tremiliosi-Filho, G. Spectroscopic studies of the glycerol electro-oxidation on polycrystalline Au and Pt surfaces in acidic and alkaline media. Electrocatalysis 2, 96–105 (2011).
Li, T. & Harrington, D. A. An overview of glycerol electrooxidation mechanisms on Pt, Pd and Au. ChemSusChem 14, 1472–1495 (2021).
Tang, C. et al. Ethanol electrooxidation on an island-like nanoporous gold/palladium electrocatalyst in alkaline media: electrocatalytic properties and an in situ surface-enhanced raman spectroscopy study. Inorg. Chem. 61, 19388–19398 (2022).
Si, D., Xiong, B. Y., Chen, L. S. & Shi, J. L. Highly selective and efficient electrocatalytic synthesis of glycolic acid in coupling with hydrogen evolution. Chem. Catal. 1, 941–955 (2021).
Chatterjee, M., Chatterjee, A., Ghosh, S. & Basumallick, I. Electro-oxidation of ethanol and ethylene glycol on carbon-supported nano-Pt and -PtRu catalyst in acid solution. Electrochim. Acta 54, 7299–7304 (2009).
Yongprapat, S., Therdthianwong, A. & Therdthianwong, S. Au/C catalysts promoted with metal oxides for ethylene glycol electro-oxidation in alkaline solution. J. Electroanal. Chem. 697, 46–52 (2013).
Xu, H. et al. Facile construction of fascinating trimetallic PdAuAg nanocages with exceptional ethylene glycol and glycerol oxidation activity. Nanoscale 9, 17004–17012 (2017).
Chen, Y. et al. Trimetallic PtRhCo petal-assembled alloyed nanoflowers as efficient and stable bifunctional electrocatalyst for ethylene glycol oxidation and hydrogen evolution reactions. J. Colloid Interface Sci. 559, 206–214 (2020).
Chang, S. C., Ho, Y. & Weaver, M. J. Applications of real-time FTIR spectroscopy to the elucidation of complex electroorganic pathways: electrooxidation of ethylene glycol on gold, platinum, and nickel in alkaline solution. J. Am. Chem. Soc. 113, 9506–9513 (1991).
Morallón, E., Rodes, A., Vázquez, J. L. & Pérez, J. M. Voltammetric and in-situ FTIR spectroscopic study of the oxidation of methanol on Pt(hkl) in alkaline media. J. Electroanal. Chem. 391, 149–157 (1995).
Miyazaki, K. et al. Electrochemical oxidation of ethylene glycol on Pt-based catalysts in alkaline solutions and quantitative analysis of intermediate products. Electrochim. Acta 56, 7610–7614 (2011).
Wang, L. et al. In situ FTIR spectroelectrochemical study on the mechanism of ethylene glycol electrocatalytic oxidation at a Pd electrode. Phys. Chem. Chem. Phys. 13, 2667–2673 (2011).
Lin, J. L., Ren, J., Tian, N., Zhou, Z. Y. & Sun, S. G. In situ FTIR spectroscopic studies of ethylene glycol electrooxidation on Pd electrode in alkaline solution: the effects of concentration. J. Electroanal. Chem. 688, 165–171 (2013).
Wang, H. et al. Electrocatalysis of ethylene glycol oxidation on bare and bi-modified Pd concave nanocubes in alkaline solution: an interfacial infrared spectroscopic investigation. ACS Catal. 7, 2033–2041 (2017).
Li, Z. H. et al. Alcohols electrooxidation coupled with H2 production at high current densities promoted by a cooperative catalyst. Nat. Commun. 13, 147 (2022).
Lai, J. P. et al. Efficient bifunctional polyalcohol oxidation and oxygen reduction electrocatalysts enabled by ultrathin PtPdM (M = Ni, Fe, Co) nanosheets. Adv. Energy Mater. 9, 1800684 (2019).
Gao, F., Zhang, Y. P., Ren, F. F., Shiraishi, Y. & Du, Y. K. Universal surfactant-free strategy for self-standing 3D tremella-like Pd–M (M = Ag, Pb, and Au) nanosheets for superior alcohols electrocatalysis. Adv. Funct. Mater. 30, 2000255 (2020).
Zhao, M. et al. Pd-Ru alloy nanocages with a face-centered cubic structure and their enhanced activity toward the oxidation of ethylene glycol and glycerol. Small Methods 4, 1900843 (2020).
Tang, J. X. et al. Tetrahexahedral PdRh nanocrystals with tunable composition as a highly efficient electrocatalyst for ethylene glycol oxidation. J. Mater. Chem. A 9, 11049–11055 (2021).
Hong, W., Shang, C. S., Wang, J. & Wang, E. Bimetallic PdPt nanowire networks with enhanced electrocatalytic activity for ethylene glycol and glycerol oxidation. Energy Environ. Sci. 8, 2910–2915 (2015).
Li, S. P., Lai, J. P., Luque, R. & Xu, G. B. Designed multimetallic Pd nanosponges with enhanced electrocatalytic activity for ethylene glycol and glycerol oxidation. Energy Environ. Sci. 9, 3097–3102 (2016).
Wang, Y. X. et al. Selective electro-reforming of waste polyethylene terephthalate-derived ethylene glycol into C2 chemicals with long-term stability. Green. Chem. 25, 5872–5877 (2023).
Li, T. T. et al. Galvanic replacement mediated 3D porous PtCu nano-frames for enhanced ethylene glycol oxidation. Chem. Commun. 55, 14526–14529 (2019).
Liu, F. L. et al. Concerted and selective electrooxidation of polyethylene-terephthalate-derived alcohol to glycolic acid at an industry-level current density over a Pd−Ni(OH)2 catalyst. Angew. Chem. Int. Ed. 62, e202300094 (2023).
Yan, Y. F. et al. Electrocatalytic upcycling of biomass and plastic wastes to biodegradable polymer monomers and hydrogen fuel at high current densities. J. Am. Chem. Soc. 145, 6144–6155 (2023).
Li, S. et al. Interfacial synergistic effect in SnO2/PtNi nanocrystals enclosed by high-index facets for high-efficiency ethylene glycol electrooxidation. Nano Res. 15, 7877–7886 (2022).
Wattanakit, C., Yutthalekha, T., Asssavapanumat, S., Lapeyre, V. & Kuhn, A. Pulsed electroconversion for highly selective enantiomer synthesis. Nat. Commun. 8, 2087 (2017).
Timoshenko, J. et al. Steering the structure and selectivity of CO2 electroreduction catalysts by potential pulses. Nat. Catal. 5, 259–267 (2022).
Arán-Ais, R. M., Scholten, F., Kunze, S., Rizo, R. & Roldan Cuenya, B. The role of in situ generated morphological motifs and Cu(i) species in C2+ product selectivity during CO2 pulsed electroreduction. Nat. Energy 5, 317–325 (2020).
Pei, S. Z., You, S. J. & Zhang, J. N. Application of pulsed electrochemistry to enhanced water decontamination. ACS EST Eng. 1, 1502–1508 (2021).
Wang, D. X. et al. Alternating current electrolysis enabled formal C−O/O−H cross-metathesis of 4-alkoxy anilines with alcohols. Angew. Chem. Int. Ed. 61, e202201543 (2022).
Shi, R. et al. Electrocatalytic reforming of waste plastics into high value-added chemicals and hydrogen fuel. Chem. Commun. 57, 12595–12598 (2021).
Chen, L. et al. Improved ethanol electrooxidation performance by shortening Pd–Ni active site distance in Pd–Ni–P nanocatalysts. Nat. Commun. 8, 14136 (2017).
Zhang, Z. et al. Single-atom catalyst for high-performance methanol oxidation. Nat. Commun. 12, 5235 (2021).
Wang, J. Y. et al. Electrocatalytic valorization of poly(ethylene terephthalate) plastic and CO2 for simultaneous production of formic acid. ACS Catal. 12, 6722–6728 (2022).
Goetz, M. K., Bender, M. T. & Choi, K. S. Predictive control of selective secondary alcohol oxidation of glycerol on NiOOH. Nat. Commun. 13, 5848 (2022).
Liu, Y. F., Hu, P. F., Wei, M. J. & Wang, C. Electrocatalytic study of ethylene glycol oxidation on Pt3Sn alloy nanoparticles. ChemElectroChem 6, 1004–1008 (2019).
Li, C. Y. et al. In Situ monitoring of electrooxidation processes at gold single crystal surfaces using shell-isolated nanoparticle-enhanced Raman spectroscopy. J. Am. Chem. Soc. 137, 7648–7651 (2015).
Ye, L. et al. Self- regeneration of Au/CeO2 based catalysts with enhanced activity and ultra-stability for acetylene hydrochlorination. Nat. Commun. 10, 914 (2019).
Lai, S. C. S. & Koper, M. T. M. Ethanol electro-oxidation on platinum in alkaline media. Phys. Chem. Chem. Phys. 11, 10446–10456 (2009).
Liu, Q. et al. Converting CO2 to ethanol on Ag nanowires with high selectivity investigated by operando Raman spectroscopy. Sci. China Chem. 66, 259–265 (2023).
Sun, X. et al. Facile synthesis of precious-metal single-site catalysts using organic solvents. Nat. Chem. 12, 560–567 (2020).
Kim, D., Zhou, C. S., Zhang, M. & Cargnello, M. Voltage cycling process for the electroconversion of biomass-derived polyols. Proc. Natl Acad. Sci. 118, e2113382118 (2021).
Pourbaix M. et al. Atlas Of Electrochemical Equilibria In Aqueous Solutions, 2nd English edn. (National Association of Corrosion Engineers, 1974).
Kim, D. et al. Enhancing glycerol conversion and selectivity toward glycolic acid via precise nanostructuring of electrocatalysts. ACS Catal. 11, 14926–14931 (2021).
Li, J. F. et al. Surface analysis using shell-isolated nanoparticle-enhanced Raman spectroscopy. Nat. Protoc. 8, 52–65 (2013).
Zhao, G. et al. An efficient and stable high-entropy alloy electrocatalyst for hydrogen evolution reaction. Chin. J. Catal. 62, 156–165 (2024).
Acknowledgements
This work was supported by the National Key R&D Program of China (2021YFA1500300, 2022YFA1500146 and 2021YFB3800300), and the National Natural Science Foundation of China (Grant No. 22072090, 22272106, 21991153, 21991150, and 22372099). P. X. acknowledges the support of the Xiaomi Young Talents Program from Xiaomi Foundation. This work was supported by “Photon Science Research Center for Carbon Dioxide”, User Experiment Assist System of SSRF and beamline BL13SSW and BL06B for experiments supports. The authors also acknowledge the generous support from Renishaw (Shanghai) Trading Co., Ltd., Jiangsu Donghua Analytical Instrument Co., Ltd. (China) and Hitachi High-Tech Scientific Solutions (Beijing) Co., Ltd.
Author information
Authors and Affiliations
Contributions
P.X. and G.Z. conceived the idea. G.Z. performed this work including protocol design and execution, characterization, data analysis, and manuscript writing. G.Z., J.L. and M.L. co-participated in the measurement of in situ XAS and Raman. L.L. provided equipment support and protocol discussion for in situ XAS experiments. L.C., X.L. and P.X. supervised the research and wrote the manuscript. All authors commented on the manuscript and agreed to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Dequan Xiao and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, G., Lin, J., Lu, M. et al. Potential cycling boosts the electrochemical conversion of polyethylene terephthalate-derived alcohol into valuable chemicals. Nat Commun 15, 8463 (2024). https://doi.org/10.1038/s41467-024-52789-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52789-2
