
Abstract
Homozygous MTAP deletion occurs in ~15% of cancers, making them vulnerable to decreases in the concentration of S-adenosylmethionine (SAM). AG-270/S095033 is an oral, potent, reversible inhibitor of methionine adenosyltransferase 2 A (MAT2A), the enzyme primarily responsible for the synthesis of SAM. We report results from the first-in-human, phase 1 trial of AG-270/S095033 as monotherapy in patients with advanced malignancies (ClinicalTrials.gov Identifier: NCT03435250). Eligible patients had tumors with homozygous deletion of CDKN2A/MTAP and/or loss of MTAP protein by immunohistochemistry. Patients received AG-270/S095033 once daily (QD) or twice daily (BID) in 28-day cycles. The primary objective was to assess the maximum tolerated dose (MTD) of AG-270/S095033. Secondary objectives included safety, tolerability, pharmacokinetics (PK), pharmacodynamics (PD), and efficacy. Forty patients were treated with AG-270/S095033. Plasma concentrations of AG-270/S095033 increased with dose. Maximal reductions in plasma SAM concentrations ranged from 54% to 70%. Analysis of paired tumor biopsies showed decreases in levels of symmetrically di-methylated arginine (SDMA) residues. Reversible increases in liver function tests, thrombocytopenia, anemia and fatigue were common treatment-related toxicities. Two partial responses were observed; five additional patients achieved radiographically confirmed stable disease for ≥16 weeks. AG-270/S095033 has a manageable safety profile. Our data provide preliminary evidence of clinical activity and proof-of-mechanism for MAT2A inhibition.
Similar content being viewed by others


Introduction
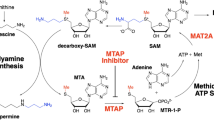
Methylthioadenosine phosphorylase (MTAP) is a key enzyme in the methionine salvage pathway. It metabolizes a byproduct of polyamine synthesis, 5′-methylthioadenosine (MTA), leading to the regeneration of methionine and adenine. Homozygous deletion of MTAP, the gene encoding this enzyme, occurs in a broad range of malignancies including pancreatic ductal adenocarcinoma, non-small cell lung cancer (NSCLC), gastric cancer, esophageal cancer, bladder cancer, diffuse large B-cell lymphoma, mesothelioma and glioblastoma multiforme (GBM)1,2,3,4,5,6,7,8. The frequency of homozygous MTAP deletion across all malignancies is ~15%8,9 and is associated with a poor prognosis10,11. Due to its proximity on the short arm of chromosome 9, MTAP is frequently (~75–80%) co-deleted with the tumor suppressor gene cyclin-dependent kinase inhibitor 2A (CDKN2A), which codes for the p16 and p14ARF proteins12,13,14,15; this supports the use of CDKN2A deletion as surrogate for MTAP deletion.
Synthetic lethal screens in multiple cancer cell lines have shown that cells with homozygous MTAP deletion are sensitive to decreases in the concentration of the methyl donor S-adenosylmethionine (SAM)16,17. This selective vulnerability is related to the fact that the catalytic activity of the SAM-utilizing enzyme protein arginine N-methyltransferase 5 (PRMT5) is inhibited by the accumulation of MTA that occurs when MTAP is deleted, since MTA competes with SAM for binding to PRMT518. Inhibition of PRMT5 activity reduces levels of symmetrically di-methylated arginine residues (SDMAs) on target proteins, many of which are involved in mRNA splicing19. As PRMT5 is highly sensitive to inhibition by MTA and requires high concentrations of SAM to maintain/catalyze SDMAs on proteins, cancer cells with homozygous deletion of MTAP are sensitive to modest reductions in SAM. The metabolic enzyme methionine adenosyltransferase 2A (MAT2A) is the primary producer of SAM and has been identified as a synthetic lethal target in MTAP-deleted cancers20.
AG-270/S095033 is a first-in-class, oral, potent, reversible small molecule inhibitor of MAT2A21. Preclinical studies in multiple MTAP-deleted cancer cell lines demonstrated that treatment with AG-270/S095033 led to a robust decrease in cell proliferation22. In vivo pharmacology studies with AG-270/S095033 in mice bearing MTAP-null tumors confirmed the ability of AG-270/S095033 to reduce the levels of SAM in both plasma and tumor tissue, and to inhibit tumor growth in a dose- and drug exposure-dependent manner. Cell cycle studies showed that AG-270/S095033-treated MTAP−/− cells had delayed progression through the S/G2/M phases and that these cells accumulated a variety of mitotic defects, such as multinucleated and micronucleated cells, indicating a functional impairment of mitosis and providing the rationale for synergistic combination with antimitotic taxanes22.
Together, these findings support testing MAT2A inhibitors as potential therapeutics in patients whose tumors have homozygous MTAP deletion. In this report, we present clinical data from the first-in-human phase 1 study of AG-270/S095033 as a single agent for the treatment of patients with advanced malignancies.
Results
Patient demographics/characteristics
As of the database lock date of May 26, 2023, 40 patients were treated with single agent AG-270/S095033 in this first-in-human phase 1 study (Fig. 1). Thirty-four patients received single agent AG-270/S095033 once daily (QD) at doses of 50 mg (N = 3), 100 mg (N = 7), 150 mg (N = 6), 200 mg (N = 12) and 400 mg (N = 6). Six patients received AG-270/S095033 at 200 mg twice daily (BID).

Phase 1 study schematic (ClinicalTrials.gov NCT03435250; EudraCT 2017-004384-13).
Patient demographics and baseline characteristics are shown in Table 1. Twenty-two patients (55%) were male and 18 patients (45%) were female. The median age was 64.5 years (range 32–87). All patients had advanced solid tumors. The most common primary tumor diagnoses were bile duct cancer (17.5%), pancreatic cancer (17.5%), non-small cell lung cancer (12.5%) and malignant mesothelioma (10%). Thirty-six patients (90%) had stage IV disease at screening. All patients had prior systemic antineoplastic therapy. Approximately half of the patients (N = 19; 47.5%) had ≥3 prior lines of systemic therapy. ECOG performance status at screening was 1 for 26 patients (65%) and 0 for 14 patients (35%). Thirty-five patients (87.5%) were enrolled based on homozygous CDKN2A deletion as estimated by next generation sequencing (NGS) or fluorescence in situ hybridization (FISH) assays performed at the study sites. Five patients were enrolled based on having ≤20% tumor cells staining positively for MTAP by the central IHC assay. Of the 35 patients enrolled based on homozygous CDKN2A deletion, the MTAP IHC assay was performed in 25 of them: 18 were found to have MTAP loss and 7 had MTAP that was intact. All patients discontinued treatment, 33 patients (82.5%) due to progressive disease or clinical suspicion of disease progression, 4 (10%) due to patient/guardian decision, 2 (5%) due to an adverse event, and 1 (2.5%) due to death.
Safety
All 40 patients treated with AG-270/S095033 monotherapy experienced at least one treatment-emergent adverse event (TEAE). The most common TEAEs included fatigue (N = 17; 42.5%), nausea (N = 13; 32.5%), blood bilirubin increase (N = 9; 22.5%), anemia, decreased appetite, diarrhea and dyspnea (N = 8 each; 20%) (Supplementary Table 1). Twenty-seven patients (67.5%) experienced a TEAE of Grade ≥3. The most common (≥10% of patients) Grade ≥3 TEAEs were blood bilirubin increased and anemia (N = 4 each; 10.0%) (Supplementary Table 2). There were no meaningful differences in the incidence of Grade ≥3 TEAEs between the dose level groups.
Treatment-related AEs (TRAEs) of any grade were reported in 28 (70%) patients who received AG-270/S095033 monotherapy (Table 2). TRAEs of Grade ≥3 were experienced by 7 patients (17.5%). The most commonly (≥10% of patients) reported TRAEs of any grade were fatigue (N = 10; 25%), blood bilirubin increase (N = 6; 15%), hyperbilirubinemia (N = 5; 12.5%), anemia (N = 4; 10%), and thrombocytopenia (N = 4; 10%). There were no meaningful differences in the incidence of TRAEs between the dose level groups.
Two patients (5.0%) who were treated with AG-270/S095033 experienced AEs leading to permanent treatment discontinuation. These AEs were nausea and vomiting in 1 patient who received 200 mg QD, and reversible liver injury suggestive of an immune-allergic hepatitis in 1 patient who received treatment at 200 mg BID. The AE of reversible liver injury was a DLT that occurred after Cycle 1.
Serious adverse events (SAEs) regardless of relationship to study treatment were experienced by 23 patients (57.5%). The most common (>1 patient) SAEs were dyspnea, pneumonia (N = 3 each; 7.5%), and abdominal pain, liver injury, pancreatitis, sepsis, and tumor pain (N = 2 each; 5.0%). Two patients (5.0%) experienced fatal SAEs: one fatal AE of pneumonia and one fatal AE of sepsis related to disease progression. None of the fatal AEs was related to treatment with AG-270/S095033.
Asymptomatic, exposure-dependent and reversible increases in unconjugated bilirubin were observed starting at 100 mg QD. In addition, treatment-related hyperbilirubinemia as an adverse event was reported in 10 patients (25%). One patient who received 150 mg QD experienced an SAE of Grade 3 hyperbilirubinemia during Cycle 1 which was considered related to AG-270/S095033 and considered to be a DLT. Low grade (Grade 1–2) treatment-related increases in transaminases were observed between the 100 mg and 400 mg dose levels, but two patients treated at 200 mg BID developed dose-limiting ≥grade 3 liver injury with increases in bilirubin and transaminases. One of the two cases was suggestive of an underlying immunologic mechanism, with rash preceding the increases in liver function tests and a liver biopsy showing eosinophilic inflammation. Both events resolved after treatment with corticosteroids.
Treatment with AG-270/S095033 was associated with sporadic erythematous rashes that resolved within 1 week of stopping treatment. Exposure-dependent and reversible thrombocytopenia was consistently observed and was largely Grade 1–2 in severity at doses between 100 and 200 mg QD. Higher grade thrombocytopenia was observed at 200 mg BID.
No differences were observed between the different dose level groups in ECOG performance status, ECG abnormalities and ophthalmological findings. All patients had an ECOG score of 0 or 1 at baseline, and the majority of post-baseline ECOG results were also 0 or 1. Eight (20.0%) patients had a worst post-baseline ECOG score of 2, and 1 (2.5%) patient had a worst post-baseline ECOG score of 3. There was no discernable effect of AG-270/S095033 on ECG parameters or an association with clinically significant cardiac events. The maximum post-baseline values for ECG parameters included QT/QTc interval increase from baseline >30 ms in 4 (10.0%) patients, QT/QTc interval >450 ms in 6 (15.0%) patients, PR interval >200 ms in 4 (10.0%) patients, and QRS duration >120 ms in 3 (7.5%) patients. TEAEs related to ECG findings included 1 case (2.5%) of serious electrocardiogram T wave inversion, 1 case (2.5%) of serious atrial fibrillation, 1 case (2.5%) of serious sinus tachycardia, 2 cases (5%) of non-serious tachycardia, and 1 case (2.5%) of non-serious supraventricular tachycardia.
As AG-270/S095033 was associated with non-reversible retinal atrophy at non-tolerated doses in a mouse GLP toxicology study, all patients had ophthalmologic examinations at screening and during treatment. No treatment-related events of clinically significant retinopathy were reported. Ophthalmological evaluations revealed TEAEs of non-serious blurred vision (N = 2; 5%), optic disc hemorrhage (N = 1; 2.5%) and retinal hemorrhage (N = 1; 2.5%). None of these events was considered related to treatment with AG-270/S095033.
Dose limiting toxicities (DLTs)
Four patients (10%) experienced at least one DLT during cycle 1. DLTs included: Grade 2 hypersensitivity reaction to AG-270/S095033 in 1 patient at 100 mg QD; Grade 3 hyperbilirubinemia and Grade 3 maculo-papular rash in 1 patient at 150 mg QD; Grade 3 neutrophil count decreased in 1 patient at 200 mg QD; and Grade 3 rash and Grade 3 drug-induced liver injury in 1 patient at 200 mg BID, associated with an eosinophilic inflammatory infiltrate on liver biopsy, which was suggestive of an immune-allergic hepatitis. Because of the hepatic toxicity observed at the 200 mg BID dose the sponsor and investigators felt that this dose should not be further evaluated. Insofar as the 400 mg QD dose failed to achieve significantly greater exposure and reductions in plasma SAM concentrations than the 200 mg QD dose, the 200 mg QD dose was considered to be the MTD. Moreover, the 200 mg QD dose satisfied the criteria required for the MTD in the statistical model.
Efficacy
Median duration of exposure to AG270/S095033 was 1.9 months (range 0.3–36.3). The median number of treatment cycles received was 2 (range 1–37). Best percentage change from baseline in target lesion size is presented in Fig. 2A. Investigator-assessed best overall response (BOR) was 2 (5%) partial response (PR), 7 (17.5%) stable disease (SD), 24 (60%) progressive disease (PD) and 7 (17.5%) not evaluable (NE) (Supplementary Table 3).Disease control rate (CR+PR+SD≥16 weeks) was 17.5% (95%–CI 7.3, 32.8).

a Waterfall plot of best percentage change in tumor size by RECIST. b Serial axial CT/PET images of a 54-year-old male patient with high-grade neuroendocrine carcinoma of the lung treated with single agent AG270/S095033 at a dose of 200 mg QD. White arrows indicate ___location of tumor. PR partial response. c Serial axial CT images of a 53-year-old male patient with a refractory sex cord stromal cell cancer treated with single agent AG270/S095033 at a dose of 100 mg QD. White arrows indicate ___location of tumor. PR partial response. d Swimmer’s plot of treatment duration and best response by RECIST. BDC bile duct cancer, MM malignant mesothelioma, NSCLC non-small cell lung cancer, PC pancreatic cancer.
One durable PR lasting 28 months was achieved in a 53-year-old male patient with a treatment-refractory and progressive sex cord stromal cell cancer who was treated at a dose of 100 mg QD. His tumor was reported to have homozygous CDKN2A deletion by NGS, but central IHC evaluation of the tissue showed more than 20% of tumor cells staining positively for MTAP. After 28 months of treatment the patient developed enlargement of a supraclavicular lymph node mass, without changes in other areas of disease. This prompted an increase in his AG270/S095033 dose to 200 mg/day, but the mass continued to enlarge and was associated with worsening anemia and fatigue. The isolated mass was therefore surgically resected, with improvement in his anemia and fatigue, and he continued treatment with AG270/S095033 until disease progression occurred in multiple other locations. The second PR was achieved in a 54-year-old male patient with NSCLC (considered likely to be a high-grade neuroendocrine carcinoma) treated at a dose of 200 mg QD, which lasted 48 weeks until he developed leptomeningeal disease. This patient was reported to have homozygous CDKN2A deletion by FISH, and central IHC analysis showed less than 20% of tumor cells staining positively for MTAP. CT images from these two patients are presented in Fig. 2B, C.
Analysis of the duration of exposure and response showed that 5 patients (12.5%) experienced SD for 4 months or longer (Fig. 2D). A 62-year-old woman with bile duct cancer treated at a dose of 150 mg QD achieved durable SD that lasted for 9 months. All five of these patients were enrolled based on homozygous CDKN2A deletion, but central IHC analysis was available for only three of them: two patients had ≤20% of tumor cells staining positively for MTAP, and one had >20% of tumor cells with positive staining.
Pharmacokinetics of AG-270/S095033 as single agent
After single and multiple oral dosing, AG-270/S095033 was well absorbed. Plasma concentrations increased in a dose-proportional manner from 50 mg to 200 mg. However, at 400 mg QD exposure was less than dose proportional. The plasma concentration-time profile of AG-270/S095033 after a single dose on C0D-3 at different doses is shown in Supplementary Fig. 1. In most of the cohorts the mean plasma trough concentrations of AG-270/S095033 demonstrated that steady-state was achieved by Day 15. The plasma concentration-time profile and exposure (AUC0–24) after multiple doses of AG-270/S095033 on C1D15 across different doses is shown in Fig. 3. The geometric mean of maximum concentration (Cmax) achieved on C1D15 ranged from 1712 to 13,581 ng/mL across the dosing cohorts and the median time to maximum plasma concentration (Tmax) ranged from 1 to 6 h. The geometric mean exposure (AUC0–tau where tau was 24 h) at steady-state (C1D15) in the 50 mg QD to 200 mg QD cohorts, ranged from 33375 to 107979 h*ng/mL and was 57285 h*ng/mL for the 400 mg QD cohort. The geometric mean exposure (AUC0–tau where tau was 12 h) in the 200 mg BID cohort was 126833 ng*h/mL, resulting in an AUC0–24 of 243715 h*ng/mL. The median terminal half-life (t½) of AG-270 ranged from 16 to 38 h across the tested doses. The median accumulation ratios based on exposure (Racc, AUC0–tau) in the 50, 100, 150, 200, 400 mg QD cohorts, were 4.92, 3.39, 4.95, 5.95, and 3.75 respectively and was 4.74 in the 200 mg BID cohort. The geometric mean accumulation-based half-life (T½eff) of AG-270 on C1D15 ranged from 43 to 99 h for the tested doses in the range of 50 to 200 mg QD and 200 mg BID. The PK parameters of AG-270/S095033 after a single dose and after multiple doses are presented in Supplementary Table 4.

a AG-270/S095033 concentration-time profile after multiple-dose administration at steady state (C1D15). Geometric mean ± SD is shown. b AG-270/S095033 AUC0-24h at steady state (C1D15). Box denotes 25th to 75th percentiles, horizontal bar the mean, and dotted bar the median, with whiskers extending to the minimum and maximum values. (a, b) AUC0-24 h AUC from 0 to 24 h, ss steady state. AUC0-24, ss = area under the concentration × time curve at steady state from time 0 to 24 h, BID twice daily, QD once daily, SD standard deviation, hr hour, n number of patients evaluated in each cohort, ng/mL nanogram per milliliter.
Pharmacodynamics
PD assessments included analysis of SAM and methionine concentrations in plasma as proximal target engagement biomarkers, and SDMA levels in tumor tissue as a marker of distal pathway inhibition. Plasma SAM concentrations at C1D15 decreased by 65–70% from baseline across doses of 50–200 mg QD and 200 mg BID, reaching a plateau from the first dose level of 50 mg QD, confirming MAT2A inhibition (Fig. 4A). A lower-than-expected reduction in plasma SAM concentrations (~54%) was observed at 400 mg QD, consistent with the lower-than-expected plasma concentrations of AG-270/S095033 at this dose. Plasma analysis revealed increases in methionine concentrations in a drug exposure-dependent manner up to 200 mg QD (Fig. 4B). Analysis of 9 paired tumor biopsies by IHC showed decreases in levels of tumor SDMA residues (p = 0.055), consistent with PRMT5 inhibition (Fig. 4C, D); the average H-score reduction compared to baseline was 36.4% [−98.8%, +21.4%] (Supplementary Table 5). In the 53-year-old patient with a sex cord stromal cell cancer who experienced a prolonged partial response, SMDA levels were increased ~2-fold in the tumor tissue at the time of disease progression relative to the baseline biopsy.

a Reductions in plasma SAM concentration at steady state (C1D15) in patients. Average reductions in plasma SAM concentration are within the range associated with maximum tumor growth inhibition in preclinical models (60–80%). b Increase in plasma methionine concentration at steady state (C1D15) in patients. In (a, b), box denotes 25th to 75th percentiles, horizontal bar the mean, and dotted bar the median, with whiskers extending to the minimum and maximum values. n indicates the number of patients evaluated in each cohort for each biomarker (SAM or methionine). c, d SDMA assessment by IHC in paired tumor biopsies collected before treatment and upon treatment of patients at C1D28. In c, H-scores found in 9 patients treated with AG-270/S095033 at the indicated doses, are shown (p-value = 0.055, two sided Wilcoxon matched-pairs test). In d, an example of SDMA staining is shown for a NSCLC patient with 39% SDMA reduction after 1 cycle of treatment with AG-270/S095033 at 50 mg QD.
Discussion
Dysregulation and reprogramming of energy metabolism are known hallmarks of cancer23, but successful therapeutic targeting of metabolic abnormalities in cancer remains limited. MTAP deletion occurs in approximately 15% of all cancers and creates vulnerabilities in tumor cells, as it promotes cellular dependence upon an exogenous supply of the essential amino acid methionine, affecting critical metabolic pathways that include nucleotide synthesis and folate metabolism24. Functional genomic screens of MTAP-deleted cells have identified MAT2A as a synthetic lethality target18, supporting efforts to develop MAT2A inhibitors. AG-270/S095033 was the first MAT2A inhibitor to enter clinical development (NCT03435250). Although other MAT2A inhibitors are in early clinical development25, this is to our knowledge the first trial to report the results of single agent MAT2A inhibition in patients with cancer.
AG-270/S095033 was generally well tolerated at doses up to 200 mg QD, and the toxicities were largely reversible and manageable. Treatment with AG-270/S095033 was associated with sporadic maculopapular erythematous rashes that resolved within 1 week of stopping treatment. Exposure-dependent and reversible thrombocytopenia was consistently observed and was largely Grade 1–2 in severity at doses between 100 and 200 mg QD. Higher grade thrombocytopenia was observed at 200 mg BID. Asymptomatic and exposure-dependent increases in unconjugated bilirubin were consistent with the known potential of AG-270/S095033 to inhibit UGT1A1 and the hepatocyte transporter OATP1B121. Other liver function tests were largely unaffected at doses below 200 mg BID. However, at the 200 mg BID dose, two patients experienced acute liver injury. One of the two cases was suggestive of an underlying immunologic mechanism, with rash preceding the increases in liver function tests and a liver biopsy showing eosinophilic inflammation. The hepatic toxicity seen with AG-270/S095033 may also been related to partial inhibition of MAT1A. MAT1A is mainly expressed in hepatocytes and liver injury has been associated with MAT1A deficiency26,27. Based on the need for identification of novel MAT2A inhibitors with increased specificity and improved properties, a strategic decision was made to pause further clinical development of AG-270/S095033.
AG-270/S095033 demonstrated approximately dose-proportional increases in systemic exposure within the range of 50 to 200 mg QD and 200 mg BID. The effective half-life of AG-270/S095033 was found to be in the range of 43 to 99 h, so that after 2 weeks of daily dosing most patients had achieved steady-state systemic exposure to the compound, steady-state reductions in plasma SAM concentrations and accumulation of plasma methionine. Average reductions in plasma SAM concentrations were similar between daily doses of 50 and 200 mg and were within the range associated with maximum tumor growth inhibition in preclinical models (60–80%)22. The modest differences in the reduction in plasma SAM concentration within this dose range are consistent with the predictions based on non-clinical data suggesting that 50 mg QD would be close to the dose associated with maximum pharmacologic activity. As anticipated from AG-270/S095033’s mechanism of action, marked increases in plasma methionine concentrations were also observed with increasing doses up to 200 mg QD. However, no high-grade treatment related toxicities were reported that could be directly attributed to the high plasma concentrations of methionine, such as drowsiness, irritability and cardiovascular AEs.
The 400 mg QD dose of AG-270/S095033 resulted in lower than predicted increases in steady-state exposure to the compound. This was most likely the result of solubility-limited absorption21. This observation prompted evaluation of a dose of 200 mg BID. While the 200 mg BID dose resulted in a 1.9-fold increase in exposure relative to the 200 mg QD dose, no further significant reduction in steady-state plasma concentrations of SAM or elevation in steady-state plasma concentration of methionine were achieved. Therefore, because of the lack of greater suppression of MAT2A activity, and because of the hepatotoxicity seen in two patients at the 200 mg BID dose, the MTD was declared to be 200 mg QD.
Analysis of nine paired tumor biopsy samples revealed decreases in levels of SDMA residues with AG-270 treatment, consistent with the predicted reduction in PRMT5 activity22,28. However, the strong trend toward reduction in SDMA levels did not reach statistical significance (p = 0.055), likely due to the limited sample size. As was the case for the plasma concentration of SAM, no clear dose-SDMA reduction relationship in tumor tissue was observed. Although the sample size for SDMA analysis in this study was limited, similar effects on plasma SAM and tumor SDMA levels have been reported for the MAT2A inhibitor IDE39729. The increase in tumor SDMA levels at the time of disease progression in the patient with a sex chord stromal cell cancer is of interest because it suggests a potential mechanism of resistance to MAT2A inhibition, and may be evaluated in future studies of MAT2A inhibitors.
AG-270/S095033 demonstrated modest signals of anti-tumor activity. Although tumor response was uncommon in this group of patients with treatment-refractory malignancies, durable responses, and disease stabilization were observed with a disease control rate at 16 weeks of 17.5%. Partial responses have also been reported for the MAT2A inhibitor IDE397. Preliminary findings from the Phase 2 expansion of IDE397 evaluating this MAT2A inhibitor in patients with selected indications (squamous NSCLC and bladder cancer) showed that two out of eight patients (one patient with NSCLC and one patient with bladder cancer) achieved a PR. The PR in the patient with bladder cancer later improved to become a complete response (CR)30. More recent reports of data from the monotherapy expansion study of IDE397 in patients with MTAP-deleted urothelial cancer and lung cancer showed an overall response rate of ~39%, with a disease control rate of ~94% and ctDNA molecular response rate of ~81%31. Together, these data highlight the potential of MAT2A inhibition as a therapeutic strategy for patients with advanced malignancies that have loss of MTAP.
While MAT2A inhibition appears to be promising, our study indicates a high rate of resistance in unselected patient populations. More investigation is warranted to unravel the full potential of MAT2A inhibition. In parallel to exploring monotherapy in larger selected patient populations there is a need for exploration of (i) rational combination strategies, and (ii) better predictive biomarkers of response. A leading hypothesis is that MAT2A inhibition may selectively enhance the activity of MTA-cooperative PRMT5 inhibitors in tumors with MTAP deletion22. MTA and SAM are structurally similar and compete for binding in the substrate pocket of PRMT5. PRMT5 inhibitors that depend on MTA for binding to the enzyme take advantage of the higher MTA concentrations found in MTAP-deleted tumors. A concomitant reduction in concentrations of SAM enhances the activity of such inhibitors32,33,34. Different MTA-cooperative PRMT5 inhibitors have already entered clinical development with preliminary data indicating marked reductions in levels of SDMA and early signs of clinical activity for single agent treatment across various tumor types35,36,37. A phase 1/2 study evaluating the safety and efficacy of dual MAT2A and PRMT5 inhibition is already underway (NCT05975073).
In addition to their combination with PRMT5 inhibitors, preclinical studies indicate that the antiproliferative effects of MAT2A inhibitors may have synergistic activity when combined with taxane-based chemotherapy22. This synergism has been reported to be mediated by effects of splicing dysregulation in Fanconi anemia (FA) pathway proteins leading to their reduction, which may sensitize cells to the antimitotic activity of taxanes38. In preclinical studies, MTAP−/− cells treated with AG-270/S095033 accumulated various mitotic defects, such as multinucleated and micronucleated cells, indicating a functional impairment of mitosis leading to greater in vitro and in vivo antitumor activity when combined with taxanes22. Based on these findings, our first-in-human clinical trial of AG-270/S095033 included two additional arms investigating (i) the combination of AG-270/S095033 with docetaxel in patients with MTAP-deleted NSCLC, and (ii) the combination of AG-270/S095033 with nab-paclitaxel and gemcitabine in patients with MTAP-deleted pancreatic cancer (NCT03435250). Early clinical trials of other MAT2A inhibitors also include testing combinations with taxane-based chemotherapy in selected patient populations (NCT04794699).
Targeting MAT2A in patients with MTAP-deleted cancers may also have an impact on T-cell function in the tumor microenvironment. MTA can activate adenosine signaling and promote pro-tumorigenic M2 macrophage polarization39,40, while both SAM and MTA can induce T-cell dysfunction41,42. Hence MAT2A inhibition may be of relevance for orthogonal combination strategies with immunotherapies in selected indications. CDKN2A genomic loss-of-function alterations have been associated with reduced benefit from immune checkpoint inhibitor (ICI) therapy in urothelial carcinoma43. A more recent analysis of >900 patients with 6 cancer types (NSCLC, melanoma, urothelial carcinoma, esophagogastric carcinoma, head and neck squamous cell carcinoma and renal cell carcinoma) demonstrated that co-occurrence of MTAP/CDKN2A deletions were associated with worse ICI outcomes in patients with urothelial carcinoma and melanoma44. Whether the inhibition of MAT2A may lead to beneficial changes in the composition of immune cells within the microenvironment of tumors with homozygous deletion of MTAP is a hypothesis that warrants further investigation.
A limitation of our study is that at the time it was conducted, most NGS panels did not include a direct evaluation of copy number changes in the MTAP gene. Considering that the MTAP gene is only 100 kb away from the CDKN2A locus, resulting in reported co-deletion of MTAP and CDKN2A in ~80–90% of the cases45,46,47,48, homozygous CDKN2A loss was used as a surrogate for MTAP loss. Since the correlation of MTAP deletion with CDKN2A deletion is imperfect, an immunohistochemistry (IHC) assay was developed to detect the loss of the MTAP protein. Our data confirm the discrepancy between homozygous CDKN2A deletion and MTAP deletion: only 72% of the patients enrolled based on homozygous CDKN2A deletion had MTAP expression in ≤20% of tumor cells. In addition, one patient with a partial response had expression of MTAP protein in >20% of tumor cells. This result suggests that the cut-off value for the percentage of tumor cells expressing MTAP protein, which was based on the performance of the MTAP IHC assay in known MTAP-null human tumor cell lines and xenografts, may need further evaluation and refinement in human malignancies. Therefore, patient preselection based on direct assessment of MTAP gene status by NGS is recommended for future studies.
In conclusion, AG-270/S095033 is a potent, reversible small molecule inhibitor of MAT2A. The first-in-human dose finding study of AG-270/S095033 monotherapy demonstrated modest signals of clinical antitumor activity, supporting the hypothesis that reducing intracellular concentrations of SAM in cancer cells with homozygous deletion of MTAP may result in a selective growth disadvantage. Consistent with this, our study is the first to demonstrate reductions in plasma SAM concentrations and in tumor SDMA levels at well-tolerated doses, providing pharmacodynamic evidence for proof-of-mechanism (Fig. 5). Although the overall safety profile was manageable, dose escalation was limited by off-target hepatobiliary toxicities. This risk may be mitigated through the development of a second generation MAT2A inhibitor (S095035), which is currently under clinical investigation (NCT06188702).

The MAT2A inhibitor AG-270/S095033 leads to a reduction in SAM concentrations and inhibition of PRMT5 activity in MTAP-deleted tumors. This results in a decrease in SDMA residues on target proteins involved in mRNA splicing and the induction of cell death.
Methods
Study oversight
This study was performed in accordance with the Declaration of Helsinki, the principles of Good Clinical Practice, and applicable local laws and regulations. The study was approved by an independent ethics committee or Institutional Review Board at each study center. All patients provided written informed consent before any study procedures. The study was sponsored by Agios Pharmaceuticals, Inc. until January 28, 2022, and subsequently by the Institut de Recherches Internationales Servier (I.R.I.S). The sponsor provided the study drug and worked with the investigators to design the study, collect, analyze, and interpret the data. The study was registered at ClinicalTrials.gov No. NCT03435250 and EudraCT No. 2017-004384-13.
Clinical study design
This phase 1 open-label, multicenter study investigated the safety and efficacy of AG-270/S095033 as a monotherapy or in combination with taxane-based chemotherapy in adult patients with advanced solid tumors or lymphoma with homozygous deletion of CDKN2A and/or MTAP, and/or loss of MTAP by IHC. The study had 3 dose-finding treatment arms: one arm evaluating AG-270/S095033 as single agent; a second arm evaluating AG-270/S095033 in combination with docetaxel; and a third arm evaluating AG-270/S095033 in combination with nab-paclitaxel and gemcitabine.
Study objectives
The primary objective was to determine the maximum tolerated dose (MTD) of single agent AG-270/S095033 and characterize its dose-limiting toxicities (DLTs). Key secondary objectives included characterization of the safety and tolerability of single agent AG-270/S095033, assessment of pharmacokinetics (PK) and pharmacodynamics (PD), and preliminary evaluation of antitumor activity. Analysis of biomarkers in tumor tissue was a key exploratory objective.
Patient population
Eligible patients were adults (≥18 years) with a histologically confirmed diagnosis of an advanced solid tumor or lymphoma that had progressed under at least one prior line of treatment, and for which additional effective standard therapy was not available or had been declined. Eligibility required homozygous loss of CDKN2A, which is frequently co-deleted with MTAP, or homozygous loss of MTAP by fluorescence in situ hybridization (FISH) or DNA sequencing of their tumor tissue. In case the status of both CDKN2A and MTAP was unknown, archival or fresh tumor tissue must have been shown to have loss of the MTAP protein by immunohistochemistry (IHC). Additional key inclusion criteria were Eastern Cooperative Oncology Group (ECOG) performance status ≤2 and adequate bone marrow, liver and kidney function. Key exclusion criteria included primary central nervous system (CNS) malignancy, symptomatic CNS metastases or CNS metastases requiring therapy with corticosteroids or anti-convulsant medication, degenerative retinal disease, clinically significant cardiac disease and impairment of gastrointestinal (GI) function or GI disease that could significantly alter the absorption of AG-270/S095033.
CDKN2A deletion and MTAP protein expression
CDKN2A and/or MTAP status was assessed locally by FISH or DNA sequencing of tumor tissue. Immunohistochemistry (IHC) was performed centrally for assessment of MTAP protein expression. A cutoff of ≤20% of tumor cells with positive immunostaining was used to identify patients whose tumors had likely lost both copies of the MTAP gene. A detailed description of this methodology is provided in the Supplementary Methods section.
Treatment plan and drug administration
AG-270/S095033 was administered by mouth (PO) on an empty stomach QD or BID. The starting dose was 50 mg QD. In the monotherapy arm, doses of 50 mg QD, 100 mg QD, 150 mg QD, 200 mg QD, 400 mg QD and 200 mg BID were tested. A single dose of AG-270/S095033 was administered on Cycle 0, Day-3 (C0D-3) with no further dosing over the subsequent 72 h. This initial single dose of AG-270/S095033 facilitated characterization of the drug’s half-life and pharmacokinetics after a single dose. Daily dosing with AG-270/S095033 then began on C1D1. One cycle of therapy was defined as 28 days of daily treatment with AG-270/S095033.
Treatment continued until disease progression, unacceptable toxicity, or patient/investigator decision. Dose escalation decisions were based on all available data, including dose-limiting toxicities, safety, PK, and PD data, and were guided by a two-parameter Bayesian logistic regression model (BLRM) following the escalation with overdose control (EWOC) principle. Dose escalation was designed to determine the MTD of AG-270/S095033 as single agent. However, if the MTD could not be determined, then the goal was to determine the dose with maximum pharmacologic activity (i.e., maximum suppression of circulating SAM concentrations) or the maximum feasible dose.
Safety assessments
Safety assessments included dose-limiting toxicities (DLTs), adverse events (AEs), serious adverse events (SAEs), changes in laboratory values, clinical examinations, vital signs, ECOG performance status, ophthalmological and ECG assessments. Adverse events were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) Version 4.03. The DLT evaluation period for dose escalation decisions with single agent AG-270/S095033 was one cycle (i.e., 28 days).
Efficacy assessments
Efficacy was evaluated by local investigator assessments using relevant response criteria, i.e., Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 for solid tumors49 or Lugano criteria for lymphoma50. Tumor assessments were performed during screening (up to 28 days prior to first dose) and after the completion of every 2 cycles (±7 days) of treatment until disease progression or withdrawal from the study. Response to treatment was also assessed at the end of treatment (EOT) if the patient discontinued treatment for reasons other than radiographically documented disease progression.
Pharmacokinetic and pharmacodynamic assessments
Both PK and PD evaluations of AG-270/S095033 in plasma were performed using LC-MS/MS analyses of blood samples collected at various timepoints pre-dose and post-dose. A detailed description of the bioanalytical methods is provided in the Supplementary Methods section. The PD markers of the pharmacologic activity of AG-270/S095033 included the plasma concentrations of SAM and methionine. Blood samples for PD were collected twice during screening and before administration of the first dose of AG-270/S095033 on C0D-3. A single dose of AG-270/S095033 was given on C0D-3 followed by no further dosing over the following 72 h. Blood samples were taken at 0.5, 1, 2, 3, 4, 6, 8, 24, 48 and 72 h after this single dose. Upon the conclusion of this 72-h sampling period (C1D1) daily dosing with AG-270/S098033 began, with samples taken at 0.5, 1, 2, 3, 4, 6, 8, and 24 h after the C1D1 dose. To characterize the PK and PD of AG-270/S095033 at steady-state, serial blood sampling occurred on C1D15, pre-dose and post-dose at 0.5, 1, 2, 3, 4, 6, 8, and 24 h. Additional blood samples were collected pre-dose on C1D8, C1D16, and on D1 of each cycle from C2.
Data analysis and processing were completed using Phoenix® WinNonlin® version 8.3.5 (Certara, Princeton, NJ) and Microsoft Excel for Office 365. Enrolled patients who received at least one dose of AG-270/S095033 and who had sufficient plasma sample data to assess PK or PD parameters comprised the PK or PD analysis populations. All plasma PK parameter calculations were performed using nominal times. PK parameters were determined using non-compartmental analysis (NCA) methods based on individual plasma concentration–time data for AG-270/S095033 and included, but were not limited to, maximum observed plasma concentration (Cmax), time of maximum plasma concentration (Tmax), area under the plasma concentration–time curve from time 0 to 24 h (AUC0–24), and area under the plasma concentration–time curve during a dosing interval (AUC0–tau).
Biomarker assessments in tumor tissue
Archival or fresh tumor biopsy samples were collected during screening/baseline and at the end of the first cycle of treatment. The tissue obtained by core needle biopsy was fixed in 10% normal buffered formalin and embedded in paraffin. Slides of this material were then made for standard hematoxylin and eosin staining (to confirm the presence and to estimate the percentage of tumor cells), for evaluation of MTAP staining in tumor cells by IHC, and for assessment of SDMA levels in tumor cells using IHC. A detailed description of the methodology is provided in the Supplementary Methods section.
Statistics and reproducibility
A two-parameter adaptive Bayesian logistic regression model (BLRM) using escalation with overdose control (EWOC) was employed to guide dose escalation and to estimate the MTD51,52.
Where (2) \({{\rm{logit}}}\left({{\rm{\pi }}}\left(d\right)\right)=\log \left(\frac{{{\rm{\pi }}}\left(d\right)}{1-{{\rm{\pi }}}\left(d\right)}\right),\) and π(d) is the probability of a DLT at total daily dose d, Doses are rescaled as d/d* with reference dose d* = 650 mg total daily dose of AG-270/S95033.
During the conduct of the actual trial, the BLRM with EWOC incorporates all observed DLT information at all dose levels together with pre-specified prior information to estimate the probabilities of DLT at each dose level. Three intervals are defined: [0, 0.16] under-dosing, [0.16, 0.33] targeted toxicity, and [0.33, 1.00] excessive toxicity.
The recommended next dose will, in general, have the highest chance that the DLT rate will fall in the target toxicity interval [0.16, 0.33] and will satisfy the following rules:
-
The dose for the next cohort will not exceed a 100% increase from the previous
dose, and
-
The EWOC principle will be satisfied, that is, <25% chance that the dose falls in the excessive toxicity interval (≥0.33).
Dose escalation will continue until identification of the MTD or RP2D. This will occur when the following conditions are met:
-
At least six evaluable patients have been treated at the recommended dose
-
This recommended dose satisfies one of the following conditions:
-
a.
The posterior probability of targeted toxicity [0.16, 0.33] at this dose exceeds 50% and is the highest among all potential doses OR
-
b.
A minimum of 21 patients have already been treated on the study
-
a.
-
It is the dose recommended for patients, either according to the model or by review of all clinical data by the clinical study team
The dose escalation uses a mixture prior consisting of three components (low, intermediate, and high toxicity). The assigned weights are 0.35, 0.6, and 0.05, respectively. Prior distributions of model parameters \(\log \left(\alpha \right)\) and \(\log \left(\beta \right)\) are described below.
Parameter | Means | Standard deviations | Correlation | Weight |
|---|---|---|---|---|
Intermediate toxicity | −1.279, 0.300 | 2.099,0.874 | −0.508 | 0.6 |
Low toxicity | −3.073, 0.090 | 2.574, 0.917 | −0.669 | 0.35 |
High toxicity | 0.003, 0.339 | 1.992, 0.857 | −0.151 | 0.05 |
Clinical data was analyzed using SAS 9.4. PK data analysis and processing were completed using Phoenix® WinNonlin® version 8.3.5 (Certara, Princeton, NJ) and Microsoft Excel for Office 365. Statistical analysis of SDMA IHC data was performed with GraphPad Prism using a two-tailed non-parametric Wilcoxon test for paired samples.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The study started enrolling patients before January 1st, 2019. The clinical study was registered at ClinicalTrials.gov No. NCT03435250 and EudraCT No. 2017-004384-13. Study-level clinical data from this study will be made available upon reasonable request from a qualified medical or scientific professional for the specific purpose laid out in that request and may include deidentified individual participant data. The data for this request will be available after a data access agreement has been signed. Please send your data-sharing request to https://clinicaltrials.servier.com/data-request-portal. Access to patient-level data depends on a number of constraints, such as the year the study was performed and an anonymization procedure. Requests are reviewed by a qualified panel of Servier experts and, if necessary, by an independent review board. Decisions will be communicated within 3 months, as detailed on the website. The clinical trial protocol may be available upon request. Source data are provided with this paper.
References
Hustinx, S. R. et al. Homozygous deletion of the MTAP gene in invasive adenocarcinoma of the pancreas and in periampullary cancer: a potential new target for therapy. Cancer Biol. Ther. 4, 83–86 (2005).
Schmid, M. et al. Homozygous deletions of methylthioadenosine phosphorylase (MTAP) are more frequent than p16INK4A (CDKN2) homozygous deletions in primary non-small cell lung cancers (NSCLC). Oncogene 17, 2669–2675 (1998).
Kim, J. et al. Downregulation of methylthioadenosin phosphorylase by homozygous deletion in gastric carcinoma. Genes Chromosomes Cancer 50, 421–433 (2011).
De Souza, A. L. et al. Clinical features of patients with MTAP-deleted bladder cancer. Am. J. Cancer Res. 13, 326–339 (2023).
Dreyling, M. H., Roulston, D., Bohlander, S. K., Vardiman, J. & Olopade, O. I. Codeletion of CDKN2 and MTAP genes in a subset of non-Hodgkin’s lymphoma may be associated with histologic transformation from low-grade to diffuse large-cell lymphoma. Genes Chromosomes Cancer 22, 72–78 (1998).
Barekatain, Y. et al. Homozygous MTAP deletion in primary human glioblastoma is not associated with elevation of methylthioadenosine. Nat. Commun. 12, 4228 (2021).
Hmeljak, J. et al. Integrative molecular characterization of malignant pleural mesothelioma. Cancer Discov. 8, 1548–1565 (2018).
ICGC/TCGA. Pan-cancer analysis of whole genomes consortium. Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020).
Beroukhim, R. et al. The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905 (2010).
Hansen, L. J. et al. MTAP loss promotes stemness in glioblastoma and confers unique susceptibility to purine starvation. Cancer Res. 79, 3383–3394 (2019).
Su, C.-Y. et al. MTAP is an independent prognosis marker and the concordant loss of MTAP and p16 expression predicts short survival in non-small cell lung cancer patients. Eur. J. Surg. Oncol. 40, 1143–1150 (2014).
Zhang, H., Chen, Z. H. & Savarese, T. M. Codeletion of the genes for p16INK4, methylthioadenosine phosphorylase, interferon-alpha1, interferon-beta1, and other 9p21 markers in human malignant cell lines. Cancer Genet. Cytogenet. 86, 22–28 (1996).
Vrugt, B. et al. Deletions of CDKN2A and MTAP detected by copy-number variation array are associated with loss of p16 and MTAP protein in pleural mesothelioma. Cancers 15, 4978 (2023).
Chapel, D. B., Dubuc, A. M., Hornick, J. L. & Sholl, L. M. Correlation of methylthioadenosine phosphorylase (MTAP) protein expression with MTAP and CDKN2A copy number in malignant pleural mesothelioma. Histopathology 78, 1032–1042 (2021).
Huang, H. Y. et al. Homozygous deletion of MTAP gene as a poor prognosticator in gastrointestinal stromal tumors. Clin. Cancer Res. 15, 6963–6972 (2009).
Kryukov, G. V. et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 351, 1214–1218 (2016).
Mavrakis, K. J. et al. Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 351, 1208–1213 (2016).
Marjon, K. et al. MTAP deletions in cancer create vulnerability to targeting of the MAT2A/PRMT5/RIOK1 axis. Cell Rep. 15, 574–587 (2016).
Fong, J. Y. et al. Therapeutic targeting of RNA splicing catalysis through inhibition of protein arginine methylation. Cancer Cell 36, 194–209.e9 (2019).
Bedard, G. T. et al. Combined inhibition of MTAP and MAT2a mimics synthetic lethality in tumor models via PRMT5 inhibition. J. Biol. Chem. 300, 105492 (2024).
Konteatis, Z. et al. Discovery of AG-270, a first-in-class oral MAT2A inhibitor for the treatment of tumors with homozygous MTAP deletion. J. Med. Chem. 64, 4430–4449 (2021).
Kalev, P. et al. MAT2A inhibition blocks the growth of MTAP-deleted cancer cells by reducing PRMT5-dependent mRNA splicing and inducing DNA damage. Cancer Cell 39, 209–224.e11 (2021).
Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Discov. 12, 31–46 (2022).
Bray, C., Balcells, C., McNeish, I. A. & Keun, H. C. The potential and challenges of targeting MTAP-negative cancers beyond synthetic lethality. Front. Oncol. 13, 1264785 (2023).
Johnson, M. L. et al. 492TiP a phase I study of synthetic lethal, IDE397 (MAT2A inhibitor) as a monotherapy and in combination with chemotherapy in advanced solid tumors harboring MTAP deletion. Ann. Oncol. 33, S766–S767 (2022).
Ramani, K. & Lu, S. C. Methionine adenosyltransferases in liver health and diseases. Liver Res. 1. https://doi.org/10.1016/j.livres.2017.07.002 (2017).
Lu, S. C. et al. Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. Proc. Natl. Acad. Sci. USA 98, 5560–5565 (2001).
Fischer, M. M. et al. Abstract 1278: MAT2A inhibitor, IDE397, displays broad anti-tumor activity across a panel of MTAP-deleted patient-derived xenografts. Cancer Res. 81, 1278–1278 (2021).
IDEAYA. IDEAYA Biosciences reports IDE397 interim phase 1 clinical data. https://media.ideayabio.com/2022-03-15-IDEAYA-Biosciences-Reports-IDE397-Interim-Phase-1-Clinical-Data (2023).
IDEAYA. IDEAYA announces top-line phase 2 results at ESMO 2023 of Darovasertib and Crizotinib combo in MUM, and clinical efficacy updates for neoadjuvant UM, GNAQ/11 cutaneous melanoma, and synthetic lethality pipeline. https://media.ideayabio.com/2023-10-23-IDEAYA-Announces-Top-Line-Phase-2-Results-at-ESMO-2023-of-Darovasertib-and-Crizotinib-Combo-in-MUM,-and-Clinical-Efficacy-Updates-for-Neoadjuvant-UM,-GNAQ-11-Cutaneous-Melanoma,-and-Synthetic-Lethality-Pipeline (2023).
IDEAYA. IDEAYA announces positive interim phase 2 monotherapy expansion data for IDE397 a potential first-in-class MAT2A inhibitor in MTAP-deletion urothelial and lung cancer. https://ir.ideayabio.com/2024-07-08-IDEAYA-Announces-Positive-Interim-Phase-2-Monotherapy-Expansion-Data-for-IDE397-a-Potential-First-in-Class-MAT2A-Inhibitor-in-MTAP-Deletion-Urothelial-and-Lung-Cancer (2023).
Fischer, M. M. et al. Abstract 1644: dual inhibition of MAT2A and PRMT5 delivers synergistic anti-tumor responses in preclinical models of MTAP-deleted cancer. Cancer Res. 83, 1644–1644 (2023).
Sun, L. et al. Structural insights into protein arginine symmetric dimethylation by PRMT5. Proc. Natl. Acad. Sci. USA 108, 20538–20543 (2011).
Antonysamy, S. et al. Crystal structure of the human PRMT5:MEP50 complex. Proc. Natl. Acad. Sci. USA 109, 17960–17965 (2012).
Engstrom, L. D. et al. MRTX1719 is an MTA-cooperative PRMT5 inhibitor that exhibits synthetic lethality in preclinical models and patients with MTAP-deleted cancer. Cancer Discov. 13, 2412–2431 (2023).
AMG 193 effective in multiple tumor types. Cancer Discov. 13, 2492 (2023).
Rodon, J. et al. First-in-human study of AMG 193, an MTA-cooperative PRMT5 inhibitor, in patients with MTAP-deleted solid tumors: results from phase I dose exploration. Ann. Oncol. 35, 1138–1147 (2024).
Abdul-Sater, Z. et al. FANCA safeguards interphase and mitosis during hematopoiesis in vivo. Exp. Hematol. 43, 1031–1046.e12 (2015).
Henrich, F. C. et al. Suppressive effects of tumor cell-derived 5′-deoxy-5′-methylthioadenosine on human T cells. Oncoimmunology 5, e1184802 (2016).
Hansen, L. J. et al. MTAP loss correlates with an immunosuppressive profile in GBM and its substrate MTA stimulates alternative macrophage polarization. Sci. Rep. 12, 4183 (2022).
Hung, M. H. et al. Tumor methionine metabolism drives T-cell exhaustion in hepatocellular carcinoma. Nat. Commun. 12, 1455 (2021).
Gjuka, D. et al. Enzyme-mediated depletion of methylthioadenosine restores T cell function in MTAP-deficient tumors and reverses immunotherapy resistance. Cancer Cell 41, 1774–1787.e9 (2023).
Adib, E. et al. CDKN2A alterations and response to immunotherapy in solid tumors. Clin. Cancer Res. 27, 4025–4035 (2021).
Adib, E. et al. Dual CDKN2A/MTAP loss compared to CDKN2A loss alone and response to immune-checkpoint inhibitors (ICI) in advanced solid tumors. J. Clin. Oncol. 40, 2622 (2022).
Carrera, C. J., Eddy, R. L., Shows, T. B. & Carson, D. A. Assignment of the gene for methylthioadenosine phosphorylase to human chromosome 9 by mouse-human somatic cell hybridization. Proc. Natl. Acad. Sci. USA 81, 2665–2668 (1984).
Han, G. et al. 9p21 loss confers a cold tumor immune microenvironment and primary resistance to immune checkpoint therapy. Nat. Commun. 12, 5606 (2021).
Illei, P. B., Rusch, V. W., Zakowski, M. F. & Ladanyi, M. Homozygous deletion of CDKN2A and codeletion of the methylthioadenosine phosphorylase gene in the majority of pleural mesotheliomas. Clin. Cancer Res. 9, 2108–2113 (2003).
National Cancer Institute. The Cancer Genome Atlas: TCGA Research Network.
Eisenhauer, E. A. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247 (2009).
Cheson, B. D. et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J. Clin. Oncol. 32, 3059–3068 (2014).
Neuenschwander, B., Branson, M. & Gsponer, T. Critical aspects of the Bayesian approach to phase I cancer trials. Stat. Med. 27, 2420–2439 (2008).
Babb, J., Rogatko, A. & Zacks, S. Cancer phase I clinical trials: efficient dose escalation with overdose control. Stat. Med. 17, 1103–1120 (1998).
Acknowledgements
This work was supported by Agios Pharmaceuticals, Inc. and by the Institut de Recherches Internationales Servier (I.R.I.S). The authors thank the patients who participated in the trial and their families, as well as the staff at each site who assisted with the study. The authors also acknowledge Perrine Soret, Charlotte Corbel, Natacha Moulharat, Cedric Vinson, Eric Baron, Dongwei (Emily) Zhu, Fanny Cazade, Dale Porter, Stefan Gross, Donald Simons and Annette Ervin-Haynes.
Author information
Authors and Affiliations
Contributions
J.T., H.B., F.W., E.G., C.A., S.N., M.H., and M.C. conceived and designed the trial. M.G., M.J., R.H., G.S., S.P., F.W., E.G., G.W., H.B., and J.T. identified and treated the patients enrolled into the study. M.H. and R.N. were primarily responsible for the analysis of the pharmacokinetic data. E.A. and M.R. were primarily responsible for the analysis of the pharmacodynamic data. P.H. was primarily responsible for the statistical analyses. All of the authors were involved in the writing and review of this manuscript, and in its final approval.
Corresponding author
Ethics declarations
Competing interests
P.H., M.R., M.H., R.N., A.S.-D., M.C., and V.A. are employed by Servier. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Dean Fennell, Stephanie Daignault, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gounder, M., Johnson, M., Heist, R.S. et al. MAT2A inhibitor AG-270/S095033 in patients with advanced malignancies: a phase I trial. Nat Commun 16, 423 (2025). https://doi.org/10.1038/s41467-024-55316-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55316-5
