
Abstract
One of the hallmarks of RNA viruses is highly structured untranslated regions (UTRs) which are often essential for viral replication, transcription, or translation. In this report, we discovered a series of coumarin derivatives that bind to a four-way RNA helix called SL5 in the 5’ UTR of the SARS-CoV-2 RNA genome. To locate the binding site, we developed a sequencing-based method namely cgSHAPE-seq, in which an acylating probe was directed to crosslink with the 2’-OH group of ribose at the binding site to create read-through mutations during reverse transcription. cgSHAPE-seq unambiguously determined a bulged G in SL5 as the primary binding site, which was validated through mutagenesis and in vitro binding experiments. The coumarin derivatives were further used as a warhead in designing RNA-degrading chimeras to reduce viral RNA expression levels. The optimized RNA-degrading chimera C64 inhibited live virus replication in lung epithelial carcinoma cells.
Similar content being viewed by others


Introduction
RNA viruses usually have highly structured 5’ and 3’ UTRs in their RNA genome, which can potentially serve as therapeutic targets1. In this report, we used SARS-CoV-2 as a specific test-case example to explore the use of RNA degraders to inhibit viral replication and to develop a method for identifying the binding sites of RNA binders. SARS-CoV-2 is an enveloped ssRNA(+) virus. The whole genome of SARS-CoV-2 ( ~ 30,000 nucleotides) is encoded in a single RNA molecule2. The viral RNA in transmitted virions is 5’ capped and 3’ polyadenylated, and, therefore, it is first recognized and treated as mRNA3. In this step, the 5’ UTR is used to hijack the host ribosome to translate viral proteins4. Furthermore, the 5’ UTR plays an essential role in RNA transcription for each coronavirus structural protein, which is accomplished through a “discontinuous” transcription mechanism. Specifically, the replication transcription complex binds to the 5’ UTR leader transcriptional regulatory sequences (TRS-L), and then “hops” onto the body TRS (TRS-B) sequence located at the 5’-end of each structural gene5,6. That said, all SARS-CoV-2 transcripts share the same 5’ UTR leader sequence. In addition, the SARS-CoV-2 5’ UTR was reported essential for viral RNA packaging7.
Given the importance of the UTRs in SARS-CoV-2, we and others elucidated the RNA structures in SARS-CoV-2 UTRs8,9,10,11,12. The 5’ UTR RNA structures in cell-free buffers, virus-infected cells, and our reporter cell model are highly consistent8,9,10,11,12,13, suggesting superior stability and suitability serving as drug targets. The 5’ UTR of SARS-CoV-2 contains five stem-loops, namely SL1–5. The start codon resides in SL5, a unique four-way helix8,9,10,11,12 (Fig. 1a). SL5 exists in all betacoronovirus species, including MERS and SARS-CoV, and the shapes of this RNA structure are similar9. We aligned the SARS-CoV-2 RefSeq and different lineages and demonstrated that the SL5 is highly conserved among all strains1. Although a predominant mutation was found in SL5B loop region from recent viral lineages (C241T), this mutation is unlikely to change the overall structure of SL51. In SARS-CoV-2, several structures in the 5’ UTR, including SL4, SL5A, and SL6, were found binding to amilorides14 (Fig. 1a). Amilorides demonstrated antiviral activity in SARS-CoV-2 infected cells.

a RNA secondary structures in SARS-CoV-2 5’ UTR and the pipeline in identification of the ligand binding site and the development of RNA-degrading chimeras. b Principle of cgSHAPE-seq for identifying small molecule binding sites in four steps. Step 1: Synthesis of FAI conjugated chemical probe. Step 2: Chemical-guided acylation at the 2’-OH of ribose at the binding site. Step 3: Reverse transcription in the presence of Mn2+ creates single-point mutations at the acylation site. Step 4: Mutational profiling and quantification identify the putative binding sites. c RNA-degrading chimeras (RIBOTACs) recruit RNase L at the target RNA to degrade viral RNAs. Created in BioRender. Tang, Z. (2024) https://BioRender.com/w93l410. Elements of this figure were provided by figdraw.com.
Here, we report a pipeline in antiviral discovery and optimization of RNA-degrading chimeras targeting SL5, highlighting a novel sequencing-based method namely chemical-guided (cg) selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) sequencing (seq), or cgSHAPE-seq, to rapidly locate the RNA ligand binding site (Fig. 1a). First, we screened a small coumarin derivative library in SL5 RNA binding assay and optimized the hit through structure-activity-relationship studies. To elucidate the RNA ligand binding site, we synthesized and applied a type of chemical probe that can selectively acylate the 2’-OH on the ribose at the ___location of binding (Fig. 1b)15,16. The 2’-OH acylation locations were “recorded” onto RNA molecules by reverse transcriptase as single-point mutations at the modification sites during primer extension. The mutation sites were then captured and deconvoluted by next-generation sequencing17,18. Mutational profiling analysis in cgSHAPE-seq unambiguously identified a bulged G in SL5 as the primary binding site in the SARS-CoV-2 5’ UTR. In the literature, other sequencing-based methods were reported using affinity probes bearing nitrogen mustard or diazirine moiety (e.g., ChemCLIP-seq19,20,21,22,23 and PEARL-seq24). However, a major limitation of these methods is a labeling bias toward guanosines25. Similar to the SHAPE, cgSHAPE reacts with the 2’-OH group of the ribose in all nucleotides, A, U, G, or C. This can potentially increase the scope and accuracy of proximity-induced chemical reactions on RNAs for mapping purposes. While we were preparing for the manuscript, the Kool lab also reported a proximity-induced acylation approach and determined RNA-binding sites for several FDA-approved drugs26.
We further developed RNA-degrading chimeras by replacing the 2’-OH acylating moiety with RNase L recruiter (RLR) moieties on the cgSHAPE probe, as well as by conjugating the RLR moieties on other putative solvent-accessible sites on the RNA ligand. RNA-degrading chimera utilizing endogenous ribonuclease (RNase) L was first reported by the Silverman group in 199327. Recently, the modality of RNA-degrading chimera was further developed by the Disney group and was demonstrated to be active using small-molecule RNA ligands15,16,17,18,28,29,30,31 (Fig. 1c). The Disney group also coined the name “ribonuclease targeting chimera” or RIBOTAC for this type of RNA degraders. RIBOTACs were shown efficacious to degrade microRNA in cells15,29 and mouse models16. Importantly, a small-molecule RIBOTAC was recently used to degrade the SARS-CoV-2 RNA genome by targeting an RNA structure named attenuator hairpin near the programmed frameshift (PFS) regulatory element28. The viral RNA transcript level was shown to be reduced in a model cell system by ~50% at 8 μM RIBOTAC28. Antisense-based RNA-degrading chimeras targeting spike-protein or envelope-protein encoding RNAs also demonstrated efficacy in SARS-CoV-2 infected cells18. Our optimized RIBOTAC robustly degraded SARS-CoV-2 RNA in cellular models at 1 μM and inhibited virus replication at 20 µM in lung epithelial cells. No significant toxicity was observed. Interestingly, we discovered the natural RNase L binding moiety is similar to or less active than the synthetic RLR in the RIBOTAC modality.
Results
Chemical optimization of the coumarin derivatives for SL5 RNA binding
We previously synthesized a collection of coumarin derivatives that are known to bind to RNAs32. Each of these coumarin derivatives is fluorescent (excitation/emission ~400/480 nm), enabling us to use fluorescence polarization (FP) to determine the in vitro binding affinity with the RNA receptors. We in vitro transcribed SL5 RNA (144–303, RefSeq NC_045512) and screened the compound library using the FP assay (Supplementary Fig. S1). C2 binds to SL5 at a dissociation constant (Kd) of 1.45 μM (Table 1). Elimination of the ethyl substituent on the A ring (C4) did not improve the binding affinity to SL5 RNA (Table 1). In contrast, a bulky substituent on the A ring (C6) impeded the interaction with SL5 (Table 1). This indicated that a non-sterically hindered linker might be suitable for conjugation on ring A without affecting the ligand binding affinity to SL5 RNA. We found C29 the best ligand in our compound collection, which binds to SL5 RNA at a Kd of 0.47 μM. Compared to C4, C29 has a different set of substituents on ring E, and therefore, we further investigated ring E while keeping ring A as unsubstituted piperazine (Table 1). In vitro FP assay showed that a fluorinated analog, C30, further improves the binding affinity (Kd = 0.22 µM). Changing the F group into Cl (C32) or CF3 (C36) groups or alternating the fluorinated site (C31) all reduced the binding affinities (Table 1). In contrast, replacing the fluorine group in C30 with methoxy group maintained the binding affinity (C34, Kd = 0.14 µM). To our knowledge, C30 and C34 are the strongest small-molecule ligand to the SARS-CoV-2 SL5 RNA.
cgSHAPE-seq uncovered the bulged G in SL5 as the C30 binding site
We were inspired by a commonly used method for RNA structure elucidation called SHAPE and sought to develop a chemical-guided sequencing-based method to identify the binding site of C30/C34 in SL5 RNA. We named this chemical probing method chemical-guided SHAPE sequencing, or cgSHAPE-seq. Conventional SHAPE uses electrophilic reagents that can form ester adducts on the 2’-OH of the ribose. The unpaired nucleotides have higher accessibility for acylation reactions, which is the basis of structure-based differential acylation activity. Therefore, identification of the acylation site would provide information on RNA base-pairing in the conventional SHAPE. Importantly, the electrophile-ribose adduct can create a mutation during reverse transcription (i.e., primer extension). As a result, conventional SHAPE coupled with quantitative mutational profiling has become a gold standard method to explore RNA topology in vitro and in vivo33,34,35,36,37. The key advantage of SHAPE is that the electrophile can usually react with all four nucleotides (A, U, G, or C). We wondered if the ribose acylation could be repurposed for identification of small-molecule binding sites by covalently linking electrophile moieties to RNA-binding chemical ligands.
First, we selected furoyl acylimidazole (FAI) as the acylating electrophile for synthesizing the chemical probe. Compared to other electrophile moieties such as anhydride and acyl cyanide, FAI is more resistant to hydrolysis with a reported half-life of ~73 min in aqueous solutions38,39,40. The hydrolysis-resistant probe design renders the synthesis and storage less demanding for anhydrous experimental facilities. An azide group on the furoyl moiety was used to provide a click-chemistry handle to conjugate with alkyne-modified C30 (Fig. 2a). We confirmed that this alkyne group does not eliminate the binding of C30 to SL5 RNA, even though the binding affinity decreased 7-fold (Supplementary Fig. S2). To obtain a pure acylation probe C30-FAI, we converted the carboxylic acid precursor (C30-FCA) to an acyl fluoride and then reacted this intermediate with equimolar imidazole to afford C30-FAI (Fig. 2a). We confirmed the final product was 91% pure and free of imidazole by NMR characterization (see Supplementary Information). We also compared this pure C30-FAI probe with a crude product synthesized by coupling of C30-FCA with carbonyl diimidazole (CDI), in which an excess amount of imidazole remained in the product. The downstream chemical probing results observed with this crude C30-FAI probe were almost identical to the pure probe (Supplementary Fig. S3), indicating that a low concentration of imidazole probably would not interfere with the chemical probing process.

a cgSHAPE-seq probe (C30-FAI) synthetic route. Reaction conditions: (i) 4 Å molecular sieves, Et3N, dry CH2Cl2, room temperature; ii) dry CDCl3, room temperature. b cgSHAPE-seq mutational profiling analysis of the SL5 sequence in total RNA extract treated with C30-FAI (0.1 mM). Δmutation rate (FAI-N3 – DMSO) indicates the background structure-based differential acylation. ΔΔmutation rate [(C30-FAI – DMSO) – (FAI-N3 – DMSO)] indicates the proximity-based differential acylation. The cgSHAPE-seq experiments were performed with three replicates (n = 3). c Scatter plot of –LogP vs ΔΔmutation rate. P values were calculated from one-sided t-test. d Comparison of the Δmutation rates of G174 and on average in RNAs treated with different concentrations of C30-FAI or FAI-N3. Experiments were performed with three replicates (n = 3). Sequencing data were analyzed by ShapeMapper 2 to obtain the mutation rate and SD of each nucleotide. Data are presented as mean values ± SD. Source data are provided in the Source Data file.
Next, we applied pure C30-FAI in SARS-CoV-2 5’ UTR RNA to elucidate the binding site of C30. It is known that conventional SHAPE experiments usually require high concentrations (10–100 mM) of acyl imidazole (e.g., FAI) for ribose acylation. To avoid obtaining structure-based SHAPE activity caused by FAI moiety alone, we chose to use a much lower dose of the chemical probe for cgSHAPE-seq. We reasoned that at low concentrations of the probe (0.02–1 mM), the differential acylation activity would be predominantly caused by ligand binding (i.e., proximity-promoted acylation). Briefly, the total RNA was extracted from SARS-CoV-2 5’ UTR expressing cells (see Method) and refolded in buffer. C30-FAI, FAI-N3, or DMSO was individually reacted with the folded RNA for 15 min at 37 °C. After the reaction, we used reverse transcriptase in the presence of MnCl2 (3 mM) for primer extension using a protocol modified from the literature report (see Method)33. In this step, we screened several commercially available reverse transcriptases and found ProtoScript II (New England Biolabs) one of the best enzymes that can tolerate Mn2+ in the reaction (Supplementary Fig. S4). The cDNA was then amplified by PCR in the SARS-CoV-2 5’ UTR region and the resulting amplicon was subsequently sequenced. We applied an existing software package ShapeMapper2 developed by the Weeks group for mutational profiling analysis33,39. We calculated the background Δmutation rate (FAI-N3 – DMSO) for each nucleotide and pleasingly observed a very low background signal at 0.02 and 0.1 mM of FAI-N3, indicating that the structure-based differential acylation activity is negligible in cgSHAPE-seq at these concentrations (Supplementary Fig. S5). In contrast, FAI-N3 at 1 mM significantly increased the Δmutation rate, implying the contribution of structure-based SHAPE activities started to emerge at high concentrations, confirming the suitable probe concentration for cgSHAPE is in the range 0.02−0.1 mM (Supplementary Fig. S5). We then calculated the RNA ligand-induced ΔΔmutation rate [(C30-FAI – DMSO) – (FAI-N3 – DMSO)] for each nucleotide and identified G174 as the only significantly mutated nucleotides in 0.02–0.1 mM probe-treated samples (Fig. 2b,c, Supplementary Fig. S6). In 1 mM probe-treated samples, the cgSHAPE signal of G174 is comparable with 0.1 mM probe-treated ones, but a high signal-to-noise ratio was observed due to the structure induced SHAPE background (Supplementary Fig. S3d). Mapping the nucleotide with previously identified secondary structures uncovered that G174 is a single-nucleotide bulge in the SL5 stem region8,9,10,11,12. It is worth noting that a previous study showed that G174 was highly reactive in a regular SHAPE experiment using another acylation agent, NAI, at 100 mM11. This concentration is at least 1,000 times higher than the optimal cgSHAPE probe concentration determined in our study (0.02−0.1 mM), and therefore, the cgSHAPE signal is unlikely attributed to the structure-induced SHAPE background at this position (Fig. 2d, Supplementary Fig. S3d).
We then validated the C30 binding site in SL5 by testing individual substructures of the SL5 RNA. The loop region of SL5A, SL5B, and a minimized four-helix junction, named SL5M (containing shorter stems) were synthesized chemically or enzymatically (Fig. 3a). The in vitro binding results demonstrated that only SL5M retained similar binding affinity to C30 (Fig. 3a, Supplementary Fig. S7). To further validate the putative binding site G174 in SL5M, we designed and synthesized SL5M RNAs with different mutations that disrupt the bulged G or other RNA structures (Fig. 3b). As expected, deletion of G174 or base-pairing G174 with an additional C both resulted in a 7-fold decrease in binding affinity to C30. Replacement of G174 with A, C, or U also significantly reduced C30 binding. We also expanded the bulged G by inserting different nucleotides between C173 and U175 or between A270 and G271, all these mutated RNAs demonstrated 5–6-fold reduced binding affinity to C30. These results suggested the importance of a single bulged G in accommodating C30’s binding. Changing the closing U-A base pair into C-G (3’-end of G174) or C-G base pair into U-A (5’-end of G174) also resulted in a 4-fold decreased binding. Mutations on other parts of the RNA have less impact (i.e., within 2-fold) in changing the binding affinity to C30 (Fig. 3b). Altogether, these observations validated that the bulged G region is the primary binding site in SL5 RNA for C30. We concluded that cgSHAPE-seq is a validated method for identifying the binding site of RNA-binding small molecules.

a Structural fragments of SL5 and their binding affinities to C30. b SL5M mutants and their binding affinities to C30. Mutated nucleotides are colored purple.
Comparison of cgSHAPE-seq and other sequencing-based RNA ligand localization methods
We compared cgSHAPE-seq with two existing sequencing-based methods for determining RNA ligand binding sites, PEARL-seq24 and Chem-CLIP19,20,21,22,23. PEARL-seq used photocrosslinking moieties such as diazirine to covalently link the bound RNA nucleotides. On the other hand, Chem-CLIP uses a nitrogen mustard moiety to alkylate nucleobases by nucleophilic reactions. To compare these two approaches with cgSHAPE-seq, we synthesized three new C30-based probes: both C30-D and C30-BD contain a diazirine group for photocrosslinking, and C30-NM contains a nitrogen mustard group for alkylation. We also confirmed that these probes have similar binding affinity to SL5 RNA compared to the cgSHAPE probe (Supplementary Fig. S2 and S8). These three chemical probes were used to treat the total RNA extracted from SARS-CoV-2 5’ UTR expressing cells at 0.1 mM under appropriate crosslinking conditions (see Supplementary Information). We used the same reverse transcription and analysis pipeline as those used in our cgSHAPE-seq experiments. We observed that C30-D in PEARL-seq can also identify G174 as the binding site albeit with a higher background than cgSHAPE-seq, while the other two probes didn’t significantly differentiate G174 from other modified nucleotides (Supplementary Fig. S9). Taken together, we concluded that the FAI-based acylation probe is a more sensitive chemical probe to identify RNA binding sites of small molecules.
Comparison of two RNAse L recruiting moieties in RIBOTACs
We then conjugated C30/C34 with RLRs to synthesize SL5-targeting RNA degraders (RIBOTACs). For RLR conjugation on the A ring, the acylating moiety in the above cgSHAPE probe (C30-FAI) was replaced with RLR moieties. We noticed that in most reported co-NMR structures of RNA bulges and small molecules that are similar to C30/C34, both termini of the small molecules are solvent accessible41,42,43, and, therefore, ring E was also explored for conjugation to RLRs, where the methoxy group of C34 was substituted for RLR attachment (Fig. 4a).

a Synthesis of C30-based RIBOTACs using conjugation sites on rings A or E of C30. b Comparison of two RLR moieties in the RIBOTAC modality using the in vitro RNase L degradation assay with purified SL5 RNA. RNase L and compounds were preincubated at 4 °C for 12 h to induce dimerization of RNase L. Cyanine 5 (Cy5)-labeled SL5 RNA was then added, and the mixture was incubated at 22 °C for an additional 2 h. RNA degradation products were analyzed by polyacrylamide gel electrophoresis (PAGE) using the red fluorescence channel (right), while the RNA ladder was visualized with SYBR Safe stain and imaged using the green fluorescence channel (left). Experiments were performed three times independently, yielding similar results. c Cellular activity of RIBOTACs in SARS-CoV-2 5’ UTR expressing cells. d Inhibitory effect of RIBOTAC C64 in SARS-CoV-2 infected A549 cells. The cytotoxicity of the compound was also evaluated. The dose-response curves are representative of three independent measurements (n = 3). Data are presented as mean values ± SD. Source data are provided in the Source Data file.
For RLR moieties, the natural RNase L ligand 2’-5’-linked oligoadenylate (2-5A) and its synthetic mimic D1 were both previously reported to be used in RNA-degrading chimeras (Supplementary Fig. S10)28,29. Combining the two conjugation sites and two RLR structures, we obtained four RIBOTAC candidates, C47, C48, C64, and C65, for SL5 RNA degradation (Fig. 4a). We validated that the polyethylene glycol (PEG) linker on 2-5A does not affect the activity in a reported RNase L degradation assay with a 5’ 6-fluorescein-tagged model RNA containing multiple RNase L cleavage sites44,45 (Supplementary Fig. S10a). It was demonstrated that the binding affinity between RNase L and D1 (Kd ≈ 18 μM) is 80,000-fold weaker than that observed for 2-5A45. Consistent with this reported in vitro binding data, the synthetic D1 alone is > 10,000 times weaker than 2-5A in the in vitro RNase L degradation assay (Supplementary Fig. S10b,c).
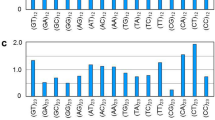
Next, we tested the four RIBOTACs in the in vitro RNase L degradation assay with purified SL5 RNA and observed their activities in order: C64 > C47 ≈ C48 > C65 (Fig. 4b, Supplementary Fig. S11a). To our surprise, the RIBOTAC C64 with D1 as the RLR moiety is much stronger than C65 with 2-5A at 50 μM (Fig. 4b). This result is contrary to what we would have predicted based on the activities of the RLR moieties per se. We validated these in vitro findings in SARS-CoV-2 5’ UTR expressing 293 T cells. In this cell model, the SARS-CoV-2 5’ UTR sequence was fused to a CMV promoter-controlled Gaussia luciferase expression cassette (Fig. 4c; for sequences, see Method). Consistent with the RNase L degradation assay result, the maximum potency of C64 (i.e., RNA reduction level) was significantly better than C65 (Fig. 4c). The activities of C47 and C48 in this cell model are similar to those of C64 and C65 (Supplementary Fig. S11b).
Efficacy of RIBOTAC in live virus infection assay
Finally, we tested the activity of C64 in SARS-CoV-2 infected cells. The SARS-CoV-2 virus was engineered to include a Nano Luciferase (NLuc) reporter by fusing NLuc onto ORF7 of the SARS-CoV-2 genome46. In this way, the NLuc signal is proportional to the viral protein copy number in cells. We applied a human lung epithelial carcinoma cell line A549 that expresses high level of the angiotensin converting enzyme 2 (ACE2) gene as the host cell46. The cells were infected with the SARS-CoV-2-NLuc virus at a multiplicity of infection (MOI) of 2.0 at 1 h before the treatment with RIBOTACs C64 for 3 d. To our satisfaction, C64 showed > 95% inhibition at 20 μM (Fig. 4d). At the same concentration, no major toxicity is observed in A549 cells (Fig. 4d).
Discussion
In cgSHAPE-seq, it is critical to employ a concentration at which the predominant source of SHAPE activity is the proximity-enhanced activity. Specifically, a standard SHAPE probing should be performed parallelly with an identical probe concentration as that of the cgSHAPE probe to determine the full set of SHAPE reactivity in the folded RNA (Fig. 2d). At the optimal cgSHAPE probe concentration (i.e., 0.02−0.1 mM in this study), the standard SHAPE activity should be minimal (typically <0.1% as demonstrated in Fig. 2d). In addition, the putative binding sites determined by cgSHAPE-seq require extensive validation. In the current study, we used mutagenesis and in vitro binding assay to validate the binding site (Fig. 3). We envision that reverse genetics with a mutated binding site coupled with relevant functional assays can also be used for validation purposes if the RNA binder has well-defined biological consequences.
Multiple crucial factors merit attention when designing cgSHAPE probes: (1) the cgSHAPE probe should be considerably stable in water solution to allow sufficient target engagement and to avoid hydrolysis by moisture in the air during the experiment preparation steps. In this current study, we chose FAI moiety with a reported half-life of ~73 min. We attempted to use C30-NAI conjugate but observed that the probe is considerably less stable than C30-FAI (data not shown). (2) The conjugation site of acylation moiety should be solvent accessible and not impede ligand binding. In this current study, the C30-FAI precursor for cgSHAPE and C30-D probe for PEARL-seq both showed significant binding to the RNA target, albeit 3−7 fold less binding affinity (Supplementary Figs. S2 and S8). (3) The functional group should be compatible with acylating moiety. For example, the FAI-based probes used in this report would not be compatible with nucleophilic RNA ligands14 due to self-reaction. cgSHAPE-seq potentially has a limitation with bias towards certain types of small molecule-RNA interactions. As shown in conventional SHAPE, FAI moiety has a higher reactivity towards unpaired RNA nucleotides. Although most of the reported RNA ligands target the unpaired region47,48, cgSHAPE-seq may be less reactive for ligands that bind to the double-stranded RNA grooves.
In our cgSHAPE-seq result, apart from G174, we also observed a cluster of nucleotides from A131 to G149 showing slightly higher mutation rates than others (Fig. 2b,c). This can be potentially caused by the nonspecific binding of C30 with flexible sequence32. RNA targeting strategies are known to have off-target effects due to shallow binding sites on RNAs and relatively weak binding affinity for small molecules. C64 at 3 μM can cause >100 genes up- or down-regulated (|Log2FoldChange | > 2) in the transcriptome (Supplementary Fig. S12, Supplementary Table S1). The activity of the RIBOTAC might be improved if a more potent and selective RNase L recruiter is used49. Specifically, we showed that the natural RNase L recruiter/activator 2-5A, which is negatively charged, is sometimes not compatible with the positively charged RNA binder C30 (Fig. 4b). For this reason, new synthetic RNase L recruiter should probably be considered to be neutral or positively charged as most of the reported RNA ligands are also positively charged50,51,52.
In summary, we developed a generalizable chemical probing method called cgSHAPE-seq for quickly identifying small molecule-RNA binding sites by sequencing. cgSHAPE probes react with the 2’-OH groups on the ribose close to the binding sites with a mitigated dependency of the nucleobase identity observed in other reported methods. We used cgSHAPE-seq to identify a bulged G on SL5 as the primary binding site on the SARS-CoV-2 5’ UTR targeted by the newly discovered coumarin derivatives, such as C30 and C34. Finally, we developed a novel C30/C34-based RNA degrader (RIBOTAC) capable of degrading viral RNA transcripts in cells and inhibiting virus replication in SARS-CoV-2 infected cells, offering crucial insights into RNA degrading chimeras’ design.
Methods
Synthesis of C30-FAI (cgSHAPE probe)
Compound C30-alkyne (50 mg, 0.08 mmol) in DMSO (1 mL) was added 2-(azidomethyl)furan-3-carboxylic acid (15 mg, 0.09 mmol), THPTA (9 mg, 0.02 mmol), sodium ascorbate (8 mg, 0.04 mmol) and CuSO4 (3 mg, 0.02 mmol). The reaction vial was sealed, evacuated, and refilled with N2 three times and stirred at room temperature overnight. DMSO was removed under vacuum and the residue was purified by silica gel column chromatography (0–10% CH3OH in CH2Cl2) to afford C30-FCA as a yellow solid (45 mg, 70%). MS-ESI (m/z) [M + 1]+ 746.28.
1H NMR (500 MHz, DMSO-d6) δ 8.71–8.68 (m, 2H), 8.52 (s, 1H), 8.09 (s, 1H), 7.71–7.68 (m, 2H), 7.38 (dd, J = 10.1, 2.6 Hz, 1H), 7.02 (dd, J = 9.0, 2.3 Hz, 1H), 6.97 (td, J = 7.6, 2.6 Hz, 1H), 6.88 (d, J = 2.4 Hz, 1H), 6.72 (d, J = 1.9 Hz, 1H), 5.92 (s, 2H), 4.52 (s, 2H), 3.58 – 3.51 (m, 14H), 3.38 (t, J = 5.1 Hz, 4H), 2.59 (t, J = 5.1 Hz, 4H), 2.56 (t, J = 5.8 Hz, 2H).
13C NMR (126 MHz, DMSO-d6) δ 160.2 (d, J = 252 Hz), 159.5, 154.9, 153.3, 152.2, 144.4, 144.3, 143.4, 139.4, 138.6, 129.6, 129.2 (d, J = 11.8 Hz), 124.2, 114.5, 112.1, 111.7, 111.4, 110.2, 104.2 (d, J = 29.8 Hz), 99.6, 99.4, 69.8, 69.7, 69.1, 63.4, 57.0, 54.9, 52.6, 46.7, 44.8.
To a solution of C30-FCA (40 mg, 53.6 μmol) and 4 Å molecular sieves (10 mg) in dry CH2Cl2 (1 mL) was added 2,4,6-trifluoro-1,3,5-triazine (21.7 mg, 161 μmol) and Et3N (27.1 mg, 268 μmol, 37.3 μL). The mixture was stirred at 25 °C for 1 h. The reaction mixture was diluted with CDCl3 (10 ml) and quenched with ice-cold NaHCO3 aqueous solution (10 mL). The organic layer was washed with ice-cold water (10 ml × 2), dried over Na2SO4, and filtered. The filtrate was concentrated and redissolved in dry CDCl3 to afford C30-FCF (23 mg/0.5 ml) which was kept on dry-ice before use.
1H NMR (400 MHz, CDCl3) δ 8.62 (s, 1H), 8.43 (s, 1H), 8.22 - 7.99 (m, 1H), 7.73 (s, 1H), 7.54–7.41 (m, 2H), 7.19 (dd, J = 2.0, 9.6 Hz, 1H), 6.84 (dd, J = 2.0, 8.8 Hz, 1H), 6.75 (dd, J = 1.6, 7.6 Hz, 2H), 6.67 (dt, J = 2.4, 7.2 Hz, 1H), 5.86 (s, 2H), 4.68 (s, 2H), 3.72–3.68 (m, 2H), 3.68–3.62 (m, 12H), 3.42–3.32 (m, 4H), 2.72–2.59 (m, 6H).
19F NMR (376 MHz, CDCl3) δ 32.70, −112.07.
To a solution of C30-FCF (23 mg/0.5 ml, 30 µmol) in dry CDCl3 was added 1H-imidazole in CDCl3 (36 mg/ml, 60 µl, 30 µmol). The mixture was stirred at 20 °C for 5 min. Complete reaction was confirmed by 1H NMR. C30-FAI was obtained as HF salt. CDCl3 was removed under nitrogen and the product was redissolved in 0.5 mL dry DMSO-d6 for 1H NMR reverification and RNA modification. 1H NMR in DMSO-d6 shows a purity of 90%.
1H NMR (400 MHz, CDCl3) δ 8.62 (s, 1H), 8.42 (s, 1H), 8.20 (s, 1H), 8.09 (dd, J = 5.6, 7.2 Hz, 1H), 7.87 (s, 1H), 7.59–7.51 (m, 2H), 7.45 (d, J = 8.8 Hz, 1H), 7.22–7.15 (m, 2H), 6.84 (dd, J = 2.4, 8.8 Hz, 1H), 6.74 (d, J = 2.0 Hz, 2H), 6.67 (dt, J = 2.4, 7.2 Hz, 1H), 5.83 (s, 2H), 4.68 (s, 2H), 3.71–3.68 (m, 2H), 3.67–3.61 (m, 12H), 3.41 - 3.35 (m, 4H), 2.71 - 2.62 (m, 6H).
13C NMR (100 MHz, CDCl3) δ 159.6, 159.5 (d, J = 250 Hz), 159.1, 154.4, 154.3, 152.5, 144.6, 144.2 (d, J = 14 Hz), 143.0, 139.2, 137.7, 137.8, 136.2, 130.4, 128.1, 126.3 (d, J = 11 Hz), 122.3, 116.3, 114.4, 111.3, 111.0, 110.2, 109.4, 103.8 (d, J = 29 Hz), 99.4 (d, J = 24 Hz), 99.4, 69.6, 69.5, 69.4, 68.8, 67.8, 63.6, 56.7, 52.1, 46.3, 44.1.
1H NMR (400 MHz, DMSO-d6) δ 8.70 - 8.67 (m, 2H), 8.51 (s, 1H), 8.33 (s, 1H), 8.16 (s, 1H), 7.93 (d, J = 2.1 Hz, 1H), 7.74 (s, 1H), 7.69 (d, J = 8.9 Hz, 1H), 7.38 (dd, J = 10.1, 2.6 Hz, 1H), 7.17 (s, 1H), 7.05 (d, J = 2.1 Hz, 1H), 7.02 (dd, J = 8.9, 2.3 Hz, 1H), 6.97 (td, J = 7.6, 2.6 Hz, 1H), 6.88 (d, J = 2.3 Hz, 1H), 5.88 (s, 2H), 4.52 (s, 2H), 3.59–3.51 (m, 14H), 3.40 (t, J = 5.1 Hz, 4H), 2.69–2.63 (m, 6H).
cgSHAPE-seq using total RNA extract from cells
SARS-CoV-2 5’ UTR expressing cells were harvested and pelleted. Total RNA was extracted using TRIzol Reagent (Invitrogen) per the user’s manual. An on-column DNA digestion was performed to remove the residual genomic DNA in total RNA using DNase I (10 U/μL, Roche) and RDD buffer (Qiagen). Purified total RNA was dissolved in water and stored at –80 °C before use. For RNA modification, 5 μg total RNA was used for each reaction. C30-FAI and FAI-N3 were prepared at 20 mM, 2 mM, and 0.4 mM in DMSO as 20× working solution. Briefly, total RNA was added water and 5× folding/reaction buffer (500 mM HEPES pH 7.4, 500 mM KCl, 30 mM MgCl2) to make a 47.5 μL solution. The solution was incubated at 37 °C for 30 min to refold. 2.5 μL C30-FAI (cgSHAPE probe), FAI-N3 (background control) or DMSO was added to the total RNA and mixed well by pipetting. The mixture was incubated at 37 °C for 15 min and then quenched by adding RLT buffer (Qiagen). The RNA was then extracted using RNeasy kit (Qiagen). 500 ng total RNA was used for reverse transcription and then PCR as described below. All reactions were performed in triplicates.
For reverse transcription (10 μL reaction), probe or DMSO treated RNA and reverse transcription primer (0.5 μM in final reaction buffer) were heated at 70 °C for 5 min and snap-cooled on ice for 1 min. 5× reaction buffer (375 mM Tris-HCl, 500 mM KCl, 15 mM MnCl2, pH 7.4, 2 μL), DTT (100 mM, 1 μL), dNTP (10 mM, 0.5 μL), ProtoScript II (0.5 μL, New England Biolabs, M0368L) and RNase inhibitor (0.2 μL, ApexBio, K1046) were added. The reaction was incubated at 42 °C for 1 h and deactivated at 70 °C for 15 min. In each PCR reaction (50 μL), cDNA (2.5 μL) was mixed with Phire Hot Start II DNA Polymerase (Thermo Fisher, 1 μL), dNTP (10 mM, 1 μL), 5× Phire Green reaction buffer (10 μL), primers (0.5 μM in final reaction buffer) and water (35.5 μL). After reaction, the amplicon was purified using a DNA Clean & Concentrator kit (Zymo Research) following the user’s manual. The purified DNA was submitted for next-generation sequencing (Amplicon-EZ, Azenta Life Sciences).
An integrated software package developed by Busan and Weeks, ShapeMapper2 was used to analyze the fastq files for mutational profiling and the result was used to generate Fig. 2b, c, and Supplementary Data Fig. S3, S5, S6 and S939. The reference sequence (SARS-CoV-2_5_UTR.fa) required for ShapeMapper2 is listed below.
>SARS-CoV-2_5_UTR
aggtttataccttcccaggtaacAAACCAACCAACTTTCGATCTCTTGTAGATCTGTTCTCTAAACGAACTTTAAAATCTGTGTGGCTGTCACTCGGCTGCGTGCTTAGTGCACTCACGCAGTATAATTAATAACTAATTACTGTCGTTGACAGGACACGAGTAACTCGTCTATCTTCTGCAGGCTGCTTACGGTTTCGTCCGTGTTGCAGCCGATCATCAGCACATCTAGGTTTCGTCCGGGTGTGACCGAAAGGTAAGATGGAGAGCCTTGTCCCTGGTTTCAACGAGGGAGTCAAAGTTCTGTTTGCCCTGATCTGCATCGCTGTGGCCGAGGCCAAGCCCACCGAGAACAACGAagacttcaacatcgtggccg. (lowercase = primer binding sequences).
In vitro RNAse L degradation assay
Purified recombinant GST-tagged RNase L was purchased from MyBioSource (MBS1041064). The buffer of RNase L was exchanged into a buffer containing 50 mM Tris-HCl (pH 7.4) and 100 mM NaCl using Zeba Desalting Column (Thermo Fisher, 8766) using the manufacturer’s protocol. T7 transcribed SL5 RNA was first labeled with Cy5 using T4 RNA Ligase (Thermo Fisher, EL0021) and pCp-Cy5 (Jena Bioscience, NU-1706-CY5) using manufacturer’s protocol and then purified by polyacrylamide gel electrophoresis (PAGE) and recovered using small-RNA PAGE Recovery Kit (Zymo Research, R1070). RNase L (1.3 µg in 5 µL) was incubated in the presence of C47, C48, C64, C65, or DMSO control in the cleavage buffer (final reaction volume is 8 µL) at 4 °C for 12 h. The 1× cleavage buffer contains 25 mM Tris-HCl (pH 7.4), 10 mM MgCl2, 100 mM KCl, 50 µM ATP, and 7 mM β-mercaptoethanol. The SL5 RNA (120 ng in 2 µl H2O) was then added into the reaction mixture and incubated for another 2 h at 22 °C. The reaction was stopped by adding RNA Gel Loading Dye (Thermo Fisher, R0641) at 1:1 ratio. The samples (4 µl) were then loaded on TBE-urea polyacrylamide gel (20%) for electrophoresis (180 V for 85 min). The gel was stained with SYBR safe (1/50,000, ApexBio, A8743) in TBE buffer for 1 min and visualized on gel imager (Thermo Fisher, iBright FL1500).
The RNA sequence of SL5 used in this assay is: 5’-UCGUUGACAG GACACGAGUAACUCGUCU AUCUUCUGCAGGCUGCUUACGGUUUCGUCCGUGUUGCAGCCGAUCAUCAGCACAUCUAGGUUUCGUCCGGGUGUGACCGAAAGGUAAGAUGGAGAGCCUUGUCCCUGGUUUCAACGA.
For RNase L degradation of a model 6-fluorescein (FAM)-tagged RNA, RIBOTACs in the above protocol were replaced with D1 (0.37 µg in 0.75 µl DMSO) or 2-5A-N3 (0.12 ng in 0.75 µl H2O)44,45. After electrophoresis, the gel was not stained and was directly visualized on the gel imager at the green fluorescence channel. The 6-FAM-tagged RNA (5’-6-FAM-UUAUCAAAUUCUUAUUUGCCCCAUUUUUUUGGUUUA-BHQ) was purchased from IDT.
SARS-CoV-2 5’ UTR expressing stable cell line
293 T cells (Thermo Fisher, R70007) were cultured in DMEM growth medium (Gibco, 11995040) supplemented with 10% FBS (Cytiva, SH30910.03) and 1% Antibiotic-Antimycotic (Gibco, 15240062) at 37 °C in 5% CO2 atmosphere. For producing the lentivirus, 293 T cells were seeded in a 6-well plate (Fisher, FBO12927) at 3 ×105 cells per well and transfected with 1 µg of SARS-CoV-2 5’ UTR expressing lentivirus vector (pLV-SARS-CoV-2-5’UTR-GLuc) along with the packaging plasmids pMD2.G (0.4 µg) and psPAX2 (0.6 µg) using Lipofectamine 2000 (Invitrogen, 11668019). At 24 h post-transfection, the cell medium was replaced with fresh growth medium. 48 h after the change of media, the supernatant containing the lentivirus particles was siphoned and centrifuged at 500 g for 10 min at 4 °C to remove the cell debris. The virus particles were further concentrated at 10X in volume using Lenti-X-concentrator (Clontech, PT4421-2) according to the manufacturer’s protocol. The lentivirus can be quantified using literature method53. Usually, 107–108 plaque forming units (pfu)/mL lentivirus was obtained after the concentrator treatment. For lentiviral transduction, 293 T cells were inoculated with the concentrated viral suspension (multiplicity of infection ~10) using polybrene (Sigma-Aldrich, TR-1003-G) at a final concentration of 8 μg/mL. At 24 h post-transduction, the culture medium was replaced with fresh growth media. After recovery for 24 h, the transduced cells were then selected in blasticidin (10 μg/mL, Invivogen, ant-bl) for 2 weeks. For stable single clone selection, the cells were diluted in the growth medium containing blasticidin (10 μg/mL) to a final density of 1 cell per 100 µL. The diluted cell suspension was then dispensed to a 96-well plate (100 µL per well). The plate was incubated at 37 °C for 4 weeks. A single cell colony from one of the wells was then selected for experiments.
pLV-SARS-CoV-2-5UTR-Luc was constructed by inserting the SARS-CoV-2 5’ UTR and Gaussian luciferase into the pLV vector, under the control of the CMV promoter.
Quantitative reverse transcription PCR (RT-qPCR) assay
The SARS-CoV-2 5’ UTR expressing cells were seeded at 3 × 105 cells per well in 12-well plates in 1 mL growth medium at 37 °C for 3 h. The cells were then treated with the compounds (C47, C48, C64, or C65) at various concentrations (1.3 nM–3 µM) for 48 h. After treatment, the supernatant was aspirated from each well and the total RNA was then extracted from the cells using RNeasy mini kit (Qiagen, 74104). The total RNAs were quantified by ultraviolet absorption at 260 nm (Thermo Fisher, NanoDrop 1000). Usually 10–20 μg total RNA was obtained from each well. cDNAs were synthesized from 500 ng of total RNA for each sample using M-MLV reverse transcriptase (Promega, M1701) and (dT)25 according to the manufacturer’s protocol. 1 µl of cDNA mixture was used in a 15 µl RT-qPCR reaction (Apex-Bio, K1070). The human GAPDH RNA level was used as the reference for normalization. The RT-qPCR primer sequences used for the PCR are shown below:
SL5-SYBR-FW: 5’-CGTTGACAGGACACGAGTAA
SL5-SYBR-RV: 5’-TTGAAACCAGGGACAAGGCTC
GAPDH-FW: 5’-GACAAGGCTGGGGCTCATTT
GAPDH-RV: 5’-CAGGACGCATTGCTGATGAT
SARS-CoV-2 inhibition assay
Vero-E6 cells (ATCC® CRL-1586™) and A549 cells (ATCC® CCL-185) were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Cytiva Life Science, SH30022) with addition of 10% fetal bovine serum (FBS, Millipore Sigma, F0926) at 37 °C under 5% CO2 atmosphere. A549 cells were transduced with a human ACE2-expressing lentivirus vector, and the transduced were cultured in the DMEM plus 2 µg/µL puromycin46.
Virus and titration
SARS-CoV-2-Nluc was created by engineering the nanoluciferase (Nluc) gene into the OFR7 of the SARS-CoV-2 genome. The insertion site of Nluc at ORF7 was based on previous mNeonGreen reporter SARS-CoV-254. The virus was propagated in Vero-E6 cells once, aliquoted in DMEM, and stored at –80˚C. A biosafety protocol to work on SARS-CoV-2 in the BSL3 Lab was approved by the Institutional Biosafety Committee of the University of Kansas Medical Center.
Plaque assay 55,56
Vero-E6 cells were seeded in 24-well plates at a density of 0.5 ×106 cells per well. A virus stock was serially diluted at 10-fold in Dulbecco’s phosphate-buffered saline, pH7.4 (DPBS). 200 µL of the diluent were added to each well and incubated for 1 h on a rocking rotator. After removing the virus diluent, 0.5 mL of overlay media (1% methylcellulose in DMEM with 5% FBS) were added to each well. The plates were incubated at 37 °C under 5% CO2 for 4 days. The methylcellulose overlays were aspirated, and the cells were fixed with 10% formaldehyde solution for 30 min and stained with 1% crystal violet solution followed by extensive washing. Plaques in each well were counted and multiplied by the dilution factor to determine the virus titer at pfu/mL.
Determination of half-maximal inhibitory concentration (IC 50 ) 46,57
ACE2-A549 cells were seeded into 96 well plates. When the cells were confluent, SARS-CoV-2-NLuc viruses were diluted with cold PBS and added into each well at a multiplicity of infection (MOI) of 2 (2 pfu/cell). The plates were kept in the CO2 incubator for 1 hour. Compound C64 was diluted at 2 x serials from 20 µM to 0.002 µM. The virus-PBS solution was aspirated. Each well was washed with cold PBS three times and was loaded with the diluted compounds. Each concentration was loaded in triple wells in the plates, and the total volume of each well was 0.2 mL. The plates were kept in the incubator. After 3 days post-infection, the culture media were aspirated from each well and the wells were washed with PBS three times. The nano-luciferase activity assay (Promega, N1110) was carried out following the manufacturer’s instructions. Briefly, 100 ul of cell lysis buffer was added to each well for 10 minutes to completely lyse the cells. Then 100 ul of nano-luciferase reaction reagent were added to each well and the luminescent signal was determined at A490 absorbance on a plate reader (Bio-Tek, Synergy). The IC50 was calculated using GraphPad Prism 8.0 software.
Statistical analysis
All data are shown as means ± SD with sample size (n) listed for each experiment. Statistical analysis was performed with R version 4.2.1. The P values were calculated by R using one-sided t- test. For data generated from ShapeMapper2, the standard error (stderr) associated with the mutation rate at a given nucleotide in the S (probe treated) or U (DMSO treated) samples was calculated as: stderr = \(\sqrt{{mutation\; rate}}/\sqrt{{reads}}\). The standard error of the Δmutation rate at a given nucleotide is: \(\sqrt{{{{stderr}}_{S}}^{2}+{{{stderr}}_{U}}^{2}}\).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are provided with this paper. The cgSHAPE-seq data for C30-FAI, FAI-N3, and DMSO-treated RNAs, and RNA-seq data for C64-treated cells were deposited in NCBI SRA with accession numbers PRJNA1029650 and PRJNA947619, respectively. Source data are provided with this paper.
References
Hegde, S., Tang, Z., Zhao, J. & Wang, J. Inhibition of SARS-CoV-2 by targeting conserved viral RNA structures and sequences. Front. Chem. 9, 802766 (2021).
Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020).
Malone, B., Urakova, N., Snijder, E. J. & Campbell, E. A. Structures and functions of coronavirus replication-transcription complexes and their relevance for SARS-CoV-2 drug design. Nat. Rev. Mol. Cell Biol. 23, 21–39 (2022).
de Breyne, S. et al. Translational control of coronaviruses. Nucleic Acids Res. 48, 12502–12522 (2020).
Zúñiga, S., Sola, I., Alonso, S. & Enjuanes, L. Sequence motifs involved in the regulation of discontinuous coronavirus subgenomic RNA synthesis. J. Virol. 78, 980–994 (2004).
Sola, I., Almazán, F., Zúñiga, S. & Enjuanes, L. Continuous and discontinuous RNA synthesis in coronaviruses. Annu. Rev. Virol. 2, 265–288 (2015).
Escors, D., Izeta, A., Capiscol, C. & Enjuanes, L. Transmissible gastroenteritis coronavirus packaging signal is located at the 5′ end of the virus genome. J. Virol. 77, 7890–7902 (2003).
Zhao, J., Qiu, J., Aryal, S., Hackett, J. L. & Wang, J. The RNA architecture of the SARS-CoV-2 3′-untranslated region. Viruses 12, 1473 (2020).
Sun, L. et al. In vivo structural characterization of the SARS-CoV-2 RNA genome identifies host proteins vulnerable to repurposed drugs. Cell 184, 1865–1883.e20 (2021).
Huston, N. C. et al. Comprehensive in vivo secondary structure of the SARS-CoV-2 genome reveals novel regulatory motifs and mechanisms. Mol. Cell 81, 584–598.e5 (2021).
Manfredonia, I. et al. Genome-wide mapping of SARS-CoV-2 RNA structures identifies therapeutically-relevant elements. Nucleic Acids Res. (2020) https://doi.org/10.1093/nar/gkaa1053.
Sanders, W. et al. Comparative analysis of coronavirus genomic RNA structure reveals conservation in SARS-like coronaviruses. bioRxiv Prepr. Serv. Biol. (2020) https://doi.org/10.1101/2020.06.15.153197.
Lan, T. C. T. et al. Structure of the full SARS-CoV-2 RNA genome in infected cells. bioRxiv (2020) https://doi.org/10.1101/2020.06.29.178343.
Zafferani, M. et al. Amilorides inhibit SARS-CoV-2 replication in vitro by targeting RNA structures. Sci. Adv. 7, eabl6096 (2021).
Costales, M. G., Suresh, B., Vishnu, K. & Disney, M. D. Targeted degradation of a hypoxia-associated non-coding RNA enhances the selectivity of a small molecule interacting with RNA. Cell Chem. Biol. 26, 1180–1186.e5 (2019).
Costales, M. G. et al. Small-molecule targeted recruitment of a nuclease to cleave an oncogenic RNA in a mouse model of metastatic cancer. Proc. Natl. Acad. Sci. 117, 2406–2411 (2020).
Liu, X. et al. Targeted degradation of the oncogenic MicroRNA 17-92 cluster by structure-targeting ligands. J. Am. Chem. Soc. 142, 6970–6982 (2020).
Su, X. et al. Efficient inhibition of SARS-CoV-2 using chimeric antisense oligonucleotides through RNase L activation. Angew. Chem. Int. Ed. Engl. 60, 21662–21667 (2021).
Velagapudi, S. P. et al. Defining RNA-small molecule affinity landscapes enables design of a small molecule inhibitor of an oncogenic noncoding RNA. ACS Cent. Sci. 3, 205–216 (2017).
Suresh, B. M. et al. A general fragment-based approach to identify and optimize bioactive ligands targeting RNA. Proc. Natl. Acad. Sci. USA. 117, 33197–33203 (2020).
Velagapudi, S. P. et al. Design of a small molecule against an oncogenic noncoding RNA. Proc. Natl. Acad. Sci. USA. 113, 5898–5903 (2016).
Velagapudi, S. P., Li, Y. & Disney, M. D. A cross-linking approach to map small molecule-RNA binding sites in cells. Bioorganic Med. Chem. Lett. 29, 1532–1536 (2019).
Guan, L. & Disney, M. D. Covalent small-molecule-RNA complex formation enables cellular profiling of small-molecule-RNA interactions. Angew. Chemie Int. Ed. 52, 10010–10013 (2013).
Mukherjee, H. et al. PEARL-seq: a photoaffinity platform for the analysis of small molecule-RNA interactions. ACS Chem. Biol. 15, 2374–2381 (2020).
Balaratnam, S. et al. A chemical probe based on the PreQ1 metabolite enables transcriptome-wide mapping of binding sites. Nat. Commun. 12, 5856 (2021).
Fang, L. et al. Pervasive transcriptome interactions of protein-targeted drugs. Nat. Chem. 15, 1374–1383 (2023). 2023 1510.
Torrence, P. F. et al. Targeting RNA for degradation with a (2’-5’)oligoadenylate-antisense chimera. Proc. Natl. Acad. Sci. USA. 90, 1300–1304 (1993).
Haniff, H. S. et al. Targeting the SARS-CoV-2 RNA genome with small molecule binders and ribonuclease targeting chimera (RIBOTAC) degraders. ACS Cent. Sci. 6, 1713–1721 (2020).
Costales, M. G., Matsumoto, Y., Velagapudi, S. P. & Disney, M. D. Small molecule targeted recruitment of a nuclease to RNA. J. Am. Chem. Soc. 140, 6741–6744 (2018).
Zhang, P. et al. Reprogramming of protein-targeted small-molecule medicines to RNA by ribonuclease recruitment. J. Am. Chem. Soc. 143, 13044–13055 (2021).
Bush, J. A. et al. Ribonuclease recruitment using a small molecule reduced c9ALS/FTD r(G4C2) repeat expansion in vitro and in vivo ALS models. Sci. Transl. Med. 13, eabd5991 (2021).
Tang, Z. et al. Recognition of single-stranded nucleic acids by small-molecule splicing modulators. Nucleic Acids Res. 49, 7870–7883 (2021). PMC8373063.
Smola, M. J., Rice, G. M., Busan, S., Siegfried, N. A. & Weeks, K. M. Selective 2 ′ -hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nat. Protoc. 10, 1643–1669 (2015).
Weeks, K. M. & Mauger, D. M. Exploring RNA structural codes with SHAPE chemistry. Acc. Chem. Res. 44, 1280–1291 (2011).
Siegfried, N. A., Busan, S., Rice, G. M., Nelson, J. A. E. & Weeks, K. M. RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nat. Methods 11, 959 (2014).
Spitale, R. C. et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature 519, 486–490 (2015).
Flynn, R. A. et al. Transcriptome-wide interrogation of RNA secondary structure in living cells with icSHAPE. Nat. Protoc. 11, 273–290 (2016).
Chan, D., Beasley, S., Zhen, Y. & Spitale, R. C. Facile synthesis and evaluation of a dual-functioning furoyl probe for in-cell SHAPE. Bioorg. Med. Chem. Lett. 28, 601–605 (2018).
Busan, S. & Weeks, K. M. Accurate detection of chemical modifications in RNA by mutational profiling (MaP) with ShapeMapper 2. RNA 24, 143–148 (2018).
Chan, D., Feng, C., Zhen, Y., Flynn, R. A. & Spitale, R. C. Comparative analysis reveals furoyl in vivo selective hydroxyl acylation analyzed by primer extension reagents form stable ribosyl ester adducts. Biochemistry 56, 1811–1814 (2017).
Campagne, S. et al. Structural basis of a small molecule targeting RNA for a specific splicing correction. Nat. Chem. Biol. 15, 1191–1198 (2019). 2019 1512.
Nagano, K., Kamimura, T. & Kawai, G. Interaction between a fluoroquinolone derivative and RNAs with a single bulge. J. Biochem. 171, 239–244 (2022).
Ichijo, R., Kamimura, T. & Kawai, G. Interaction between a fluoroquinolone derivative KG022 and RNAs: Effect of base pairs 3′ adjacent to the bulged residues. Front. Mol. Biosci. 10, 168 (2023).
Thakur, C. S., Xu, Z., Wang, Z., Novince, Z. & Silverman, R. H. A convenient and sensitive fluorescence resonance energy transfer assay for RNase L and 2’,5’ oligoadenylates. Methods Mol. Med. 116, 103–113 (2005).
Thakur, C. S. et al. Small-molecule activators of RNase L with broad-spectrum antiviral activity. Proc. Natl. Acad. Sci. USA. 104, 9585–9590 (2007).
Adhikary, P. et al. Discovery of small anti-ACE2 peptides to inhibit SARS-CoV-2 infectivity. Adv. Ther. 4, 2100087 (2021).
Hargrove, A. E. Small molecule–RNA targeting: starting with the fundamentals. Chem. Commun. 56, 14744–14756 (2020).
Childs-Disney, J. L. et al. Targeting RNA structures with small molecules. Nat. Rev. Drug Discov. 21, 736–762 (2022).
Meyer, S. M. et al. DNA-encoded library screening to inform design of a ribonuclease targeting chimera (RiboTAC). J. Am. Chem. Soc. 144, 21096–21102 (2022).
Morgan, B. S., Forte, J. E., Culver, R. N., Zhang, Y. & Hargrove, A. E. Discovery of key physicochemical, structural, and spatial properties of RNA-targeted bioactive ligands. Angew. Chemie - Int. Ed. 56, 13498–13502 (2017).
Velagapudi, S. P., Gallo, S. M. & Disney, M. D. Sequence-based design of bioactive small molecules that target precursor microRNAs. Nat. Chem. Biol. 10, 291–297 (2014).
Disney, M. D. et al. Inforna 2.0: a platform for the sequence-based design of small molecules targeting structured RNAs. ACS Chem. Biol. 11, 1720–1728 (2016).
Barczak, W., Suchorska, W., Rubiś, B. & Kulcenty, K. Universal real-time PCR-based assay for lentiviral titration. Mol. Biotechnol. 57, 195–200 (2015).
Xie, X. et al. A nanoluciferase SARS-CoV-2 for rapid neutralization testing and screening of anti-infective drugs for COVID-19. Nat. Commun. 11, 5214 (2020).
Hao, S. et al. Long-term modeling of SARS-CoV-2 infection of in vitro cultured polarized human airway epithelium. MBio 11, 1–17 (2020).
Zou, W. et al. The sars-cov-2 transcriptome and the dynamics of the s gene furin cleavage site in primary human airway epithelia. MBio 12, (2021).
Hao, S. et al. Establishment of a replicon reporter of the emerging tick-borne bourbon virus and use it for evaluation of antivirals. Front. Microbiol. 11, 2229 (2020).
Acknowledgements
Research reported in this article was supported by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) under award numbers R35GM147498 and P20GM113117, W. M. Keck Foundation, and the University of Kansas General Research Funds. We also thank Dr. Robert Silverman at Cleveland Clinic for providing the authentic 2-5 A samples for comparison. We have obtained SARS-CoV-2-Nluc from Drs. Shi and Menachery through The University of Texas Medical Branch (UTMB)’s World Reference Center for Emerging Viruses and Arboviruses. We thank the University of Kansas Medical Center Genomics Core for performing RNA-seq experiments. The KUMC Genomics Core was supported by Kansas Intellectual and Developmental Disabilities Research Center (NIH U54 HD 090216), the Molecular Regulation of Cell Development and Differentiation COBRE (P30 GM122731-03), the NIH S10 High-End Instrumentation Grant (NIH S10OD021743) and the Frontiers CTSA grant (UL1TR002366).
Author information
Authors and Affiliations
Contributions
J.W. conceived the work. J.W., Z.T., S. Hegde and J.Q. wrote the paper. Z.T. and M.S. performed chemical synthesis. Z.T. and J.W. performed the next-generation sequencing analysis. S. Hegde collected RNA degradation data. S. Hao performed the live virus assay.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tang, Z., Hegde, S., Hao, S. et al. Chemical-guided SHAPE sequencing (cgSHAPE-seq) informs the binding site of RNA-degrading chimeras targeting SARS-CoV-2 5’ untranslated region. Nat Commun 16, 483 (2025). https://doi.org/10.1038/s41467-024-55608-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55608-w
