
Abstract
The electrostatic effects of separators under the internal electric field are often overlooked, leading to the unreliability of traditional theoretical models. Here we introduce the dielectric constant as a descriptor and develop a high dielectric constant fiber separator primarily composed of phosphorylated cellulose. Under the internal electric field, the intense electron displacement polarization within the high dielectric constant separator enhances the charge transfer kinetics and optimizes the solvation structure, thus mitigating the formation of amorphous organic oligomers at the solid-electrolyte interphase. Furthermore, the separator induces the formation of LiF, thereby forming a robust and low-resistance solid-electrolyte interphase. The separator exhibits high ionic conductivity (0.76 mS cm−1 at 25 °C) and Li+ transference number (0.68). Consequently, the Li||LiFePO4 pouch cell with the prepared separator achieve high specific energy exceeding 350 Wh kg−1 (relative to the mass of pouch cells) under practical quantities of active materials and electrolyte.
Similar content being viewed by others

Introduction
Alkali metal batteries, characterized by their high theoretical specific capacity and low reduction potentials, are highly promising next-generation energy storage systems1,2,3. However, the unstable solid-electrolyte interphase (SEI) on metal negative electrodes and the vigorous growth of dendrites poses significant challenges to the practical application of alkali metal batteries4,5. The separator, which directly interfaces with the critical components such as the negative electrode and electrolyte, plays a pivotal dual role as both bridging and barrier, and has the potential to address these challenges6,7. Separator engineering strategies such as adjusting the potential profile to enhance ion transport and incorporating functionalized cluster to inhibit reduction have been successively reported8,9. These strategies aim to provide uniform ion transport pathways and reduce parasitic reactions between metal electrode and electrolytes. In addition, introducing strong dipole moments monolayers, establishing sacrificial layer on the separator, and coating fluoride onto separator to establish artificial SEI have also been proposed10,11. These strategies have successfully improved charge transport kinetics and established a stable negative electrode interface, thereby facilitating the practical application of alkali metal batteries.
Previous studies have primarily focused on modulating electrostatic interactions, van der Waals forces, and dipole-dipole interactions between the separator and electrolyte components (alkali metal ions, solvent molecules, and anions)12. However, their electrical properties have been overlooked, particularly under the influence of significant electric fields present within batteries. During the discharge of alkali metal batteries, internal electric fields exceed 1 × 105 V/m13,14. Such electric field intensities are sufficient to affect separator-negative electrode interfaces and interactions between the separator and electrolyte15. Under these intense electric fields, properties such as molecular polarizability and dipole moments are notably altered, with interaction energy changes potentially exceeding 50% before and after electric field application16,17. This oversight results in inaccurate descriptions of separator-negative electrode interfaces and interactions between the separator and electrolyte. Based on the facts that the electric field within the battery affects both the transport kinetics of solvent molecules and alkali metal ions, as well as significantly changing the electron distribution in the separator, a new descriptor was introduced for characterizing separator properties, the dielectric constant18. According to the Clausius-Mossotti equation, the dielectric constant is proportional to molecular polarizability and dipole moment, making it a valuable descriptor for assessing interactions between the separator and other components19. Moreover, the dielectric constant effectively exhibits the response capability of separators to electric fields.
Herein, we explore the response behavior of fiber separators with different dielectric constants under electric fields. We study the effect of a high-dielectric constant separator on the negative electrode surface of the alkali metal batteries under the premise of a uniform internal electric field through theoretical and experimental analysis. The internal electric field of the battery enhances the electron displacement polarization of the separator, optimizing the transfer kinetics and solvation structure of alkali metal ions. This also induces the formation of a robust SEI rich in LiF and low in oligomers. In addition, the high-dielectric constant separator regulates the electric field density distribution and mitigates the tip effect in the metal electrode. Through the application of the separator with high dielectric constant, alkali metal batteries exhibit greatly improved the electrochemical performance and cycling stability. We have extended the strategy into the wet process and achieved large-scale production of high-dielectric constant separators using economically viable biomaterials as raw materials. This strategy contributes to the fresh insight into the separator design for the long-life alkali metal batteries and provides an engineering solution.
Results
Design and characteristics of high-dielectric constant fiber separators
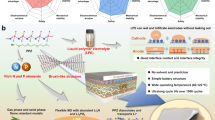
The electric field inside the battery affects the distribution of electrons, but this aspect has not received enough attention in previous research20. By combining the electric field inside the battery with the dielectric constant properties of the separator, we designed a high-dielectric constant separator for alkaline metal batteries. The electric field inside the battery could cause the electron cloud in the molecular chain of the separator to move to one side, thereby increasing the overall molecular dipole moment21. Accordingly, the separator with high-dielectric constant demonstrates a heightened polarization effect attributed to the alteration in electric dipoles. The resulting high dielectric ___domain affect the ion diffusion efficiency in the electrolyte phase and the solvation structure (Fig. 1a). In a typical lithium metal battery system, high dielectric domains within the outer Helmholtz plane (OHP) have the capacity to initiate Li+ desolvation from the solvation sheath, thereby decreasing the desolvation energy and promoting uniform Li+ flux22. In addition, according to the Gauss’s law, the relationship of electric displacement and the area density of the free charge between lithium electrode and separator with high-dielectric constant induced interfaces could be expressed as Eq. (1):
where DInterfaces and DLithium are the electric displacement in separator and lithium electrode, respectively; σ is the surface charge density23. Due to its high electrical conductivity, free electrons swiftly respond to neutralize the accumulated charge within the metal upon the applied electric current. Consequently, the magnitude of the net charge within is typically in the range of picocoulombs, markedly smaller than that at the surface (microcoulombs)24. Hence, the DLithium can be reasonably approximated as 0. In this situation, the relationship between electric field and dielectric constant can be described as Eq. (2), which can also be expressed as Eq. (3):
where E and ε are the electric field strength and dielectric constant, respectively. Hence, it can be inferred that the electric field applied to the electrolyte is inversely proportional to the dielectric constant of the medium. Separators with high-dielectric constants are essential for regulating the distribution of electric field and preventing the tip effect of metal negative electrodes, consequently improving the longevity of batteries.

a Schematic illustration of the promotion of lithium ion desolvation by high-dielectric constant ___domain and its impact on the tip effect of the metal negative electrode. The inset at the bottom right corresponds to the electric field intensity in the high dielectric constant ___domain (blue line) and the low dielectric constant ___domain (red line). b Dielectric constants and costs of various commercial separators and ANF, CS and PCS. The cost is expressed in US dollars per square meter ($ m−2), and the details of the calculations are listed in the Supplementary Table 1. c The energy gaps of PC and cellulose (calculate two repeating units of cellulose molecule chain). Li-DEC and Li-EC RDF (d) and integrated coordination numbers (e) with the existence of CS and PCS. f The ratio of Li+ based on the distribution of molecules within 4 Å of CS and PCS; Inset: The main distribution form of Li+ when PCS chains (the blue wireframe) and CS chains (the red wireframe) exist. The effect of PCS and CS on the desolvation energy during the step-by-step desolvation process of [Li(EC)3(DEC)]+ under an electric field (g) and without electric field (h). Source data are provided as a Source Data file.
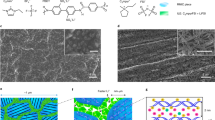
Large-scale production of separators was obtained using traditional wet process. Polar phosphate groups were chosen to introduce into cellulose to increase its dielectric constant (Supplementary Fig. 1, Supplementary Note 1 and Supplementary Data 1–5). The raw cellulose was obtained by a traditional pulping process, which was further phosphorylated by a simple impregnation and curing process (Supplementary Fig. 2). The phosphate groups were grafted into cellulose skeleton by treating cellulose pulp into the (NH4)2HPO4 solution. Additionally, the introduction of urea in the reaction system was employed to avoid the degradation of cellulose by suppressing the proton release from (NH4)2HPO425. The successful introduction of the phosphate group is discussed in Supplementary Fig. 3 and Supplementary Note 2. Notably, the impregnation and curing processes in the phosphorylation reaction of cellulose were carried out under the standard atmospheric pressure, presenting a reliable, environmentally friendly, and low-cost process for large-scale production. The realization of kilogram-scale preparation of phosphorylated cellulose (PC) is depicted in Supplementary Fig. 4, confirming the scale-up potential of the modification process. The optimal reaction degree and its relationship with dielectric constant and ion transport were explored (Supplementary Fig. 5 and Supplementary Note 3). Subsequently, a wet processing was implemented using PC as the main raw material to simulate large-scale production of high-dielectric constant separators, designated as phosphorylated cellulose separator (PCS). For comparison, the separators made from unmodified cellulose were referred to as CS. The mechanical properties and electrolyte compatibility of PCS are discussed in Supplementary Figs. 6–12 and Supplementary Note 4. Compared to traditional separators including polypropylene (PP), polyethylene (PE), polyimide (PI) and glass fiber (GF), PCS displays a highest dielectric constants and lower cost, demonstrating its commercial competitiveness (Fig. 1b). Finite element analysis-based multiphysics simulations further demonstrate that variations in the dielectric constant of the separator can modulate the tip effect caused by inevitable protrusions on the lithium metal negative electrode (Supplementary Fig. 13)26. At a dielectric constant of 2.8, the local electric field concentrates at the tips, leading to preferential Li+ deposition in these regions. This ultimately enhances the tip effect, resulting in dendritic growth and an unstable negative electrode interface. In contrast, when the dielectric constant reaches 29, the uneven local electric field is effectively mitigated. Thus, introducing PCS with a high dielectric constant can effectively reduce the local electric field, thereby slowing dendrite growth and promoting uniform, dense Li+ deposition.
Density functional theory (DFT) computation on the electronic structure was conducted to elucidate the structural basis of the high-dielectric constants of PCS (Fig. 1c and Supplementary Data 6–10). There is significant wavefunction amplitude localized on the phosphate groups for the highest occupied molecular orbital (HOMO) and moves to the glucose unit for the lowest unoccupied molecular orbital (LUMO)27. The charge transfer like transition reduces the energy gap (to 7.22 eV) compared to untreated cellulose (8.99 eV), which facilitates electronic excitation under electric fields thus leading to a higher polarizability and an increased dielectric constant28. This ultimately resulted in significant charge transfer occurs between Li+ and phosphate groups under an applied electric field, whereas the charge transfer with raw cellulose chains is considerably weaker (Supplementary Fig. 14a, b). Additionally, the total dipole moment of PCS and CS increases with the increase of the applied electric field (Supplementary Fig. 14c). Notably, PCS with higher dielectric constant exhibits more pronounced electron transfer and dipole moment increments compared to CS. This suggests that PCS may experience a stronger interaction with Li+ under the influence of an electric field16. Classical molecular dynamics (MD) simulations were conducted to evaluate the impact of phosphorylated cellulose chains on the solvation structure of Li+ (Supplementary Fig. 15 and Supplementary Data 11–20). The radial distribution function of Li+-solvent molecules significantly decreased with the existence of PCS chains (Fig. 1d). In the presence of CS, the coordination numbers of Li+ with EC and DEC are 4.09 and 0.92, respectively. When PCS is present, these coordination numbers decrease to 3.02 and 0.33, respectively (Fig. 1e). In addition, statistical analysis of the molecular distribution within 4 Å around the cellulose chains reveals that 32.7% of Li+ have no interactions with CS chains, significantly higher than the occurrence around PCS chains, which stands at 17.3% (Fig. 1f)29. The frequency of Li+ present around the PCS chains is consistently higher than that around the CS chains. It suggests that Li+ is more likely to interact with solvent molecules in the presence of CS chains, driven by solvation structure as the dominant state. While in the presence of PCS, Li exhibits a tendency to desolvate from solvent molecules and move along the PCS chain. This confirms that the PCS chains with high dielectric constant optimize the Li+ solvation structure, aiding in overcome the energetically unfavorable desolvation process and facilitating rapid Li+ transfer30. These findings further demonstrate the advantages of PCS in promoting charge transfer31. The stepwise dissolution energy barrier of [Li(EC)3(DEC)]+ under the influence of an electric field was also investigated to further reveal the effect of PCS on the desolvation process. As shown in Fig. 1g, h, the energy barrier for [Li(EC)3(DEC)]+ to remove EC and DEC molecules from its solvation sheath is lower in the presence of a PCS chain (5.86 eV) than in the presence of a pure cellulose chain (6.86 eV). After applied an electric field, the desolvation energy was further decreased to 3.12 eV in the presence of a PCS chain. In contrast, the change in desolvation energy in the presence of CS chain is negligible (6.47 eV). This can be attributed to the intense electronic transfer of PCS with high-dielectric constant under an electric field, thereby inducing the Li+ desolvation32. The results demonstrated that high-dielectric constant separators accelerated the dissolution and diffusion of Li+ under an internal electric field of batteries, a crucial yet underexplored factor for battery performance.
Alkali metal negative electrode stability and SEI chemistry with PCS
As shown in Supplementary Fig. 16 and Supplementary Note 5, to investigate the effects of high-dielectric constant PCS to metal ion transport kinetics, the electrochemical performance of typical Li||Cu asymmetric cells were evaluated with commercial ester-based electrolyte (1 M LiPF6 in EC/DEC) and ether-based electrolyte (1 M LiTFSI in DME/DOL + LiNO3 additive). The average Li plating/stripping were evaluated by Aurbach method (Fig. 2a and Supplementary Fig. 17a). When assembling PCS, the CEs of the Li||Cu cells using ether-based and ester-based electrolytes were 99.68% and 97.96%, respectively. These values were markedly higher than those of cells assembled with CS, which exhibited CE of 96.26% and 94.08% for ether-based and ester-based electrolytes, respectively. Similarly, the cells equipped with PCS maintained an average CE of 99.59% with ether-based electrolytes and 95.27% with ester-based electrolytes along 100 cycles (Supplementary Fig. 17b–d). This performance surpasses most previous reports and reaches advanced levels (Supplementary Table S2). The steady CE of cell with PCS can be well explained by the voltage-capacity curves during Li nucleation shown in Supplementary Fig. 17c. The nucleation overpotential of Li||Cu cells with PCS, CS were 59.0, and 181.7 mV, respectively. The low nucleation overpotential confirmed the enhance of the Li+ transport kinetics through the with high-dielectric constant PCS. We evaluated the critical current density (CCD) of Li||Cu cells with CS and PCS (Fig. 2b, c). The polarization voltage of the cells increased progressively with the current density. The Li|CS|Cu cell short circuited at a current density of 1.8 mA cm−2, whereas the CCD for Li|PCS|Cu cell reached 7.4 mA cm−2. High current densities promote dendrite formation, which ultimately causes short circuits. The high CCD of Li|PCS|Cu revealed that the presence of PCS enhanced the charge transfer kinetics, finally allowing for a more uniform distribution of the applied current. In addition, the symmetric Li||Li cells with PCSs exhibited stable voltage hysteresis values of 0.022, 0.026, 0.045, 0.071 and 0.081 V when the current density increased from 0.2 mA cm−2 to 0.5, 1, 5 and return to 0.2 mA cm−2, respectively, whereas the voltage-time curve of the cells with CS revealed higher overpotential at the corresponding current densities (Fig. 2d). The voltage-time curves of Li||Li cell with PCS at 1 mA cm−2, 1 mAh cm−2, exhibits overpotential of 51.3 mV, and no apparent voltage polarization or cell short circuiting was observed during a long plating/stripping time of over 1600 h (Fig. 2e). In a sharp contrast, the Li||Li cells with CS displayed a gradual increase in voltage hysteresis is observed in the cells with PCS after plating/stripping for 500 h. The enhanced CE, plating/stripping stability and rate capability suggest that high-dielectric constant PCS enable alkali metal batteries a rapid reaction kinetics and high reversibility of the plating/stripping process. We conducted Sand’s time test to verify the impact of PCS on ion transport kinetics (Supplementary Fig. 18)33. The voltage changes of Li||Cu cells with PCS, CS, and PP occurred at 18.31, 14.67, and 10.84 h, respectively. The extended Sand’s time with PCS indicates enhanced ion transport capability, suggesting that PCS effectively improves Li+ flux and mitigates dendrite growth.

a CEs of Li||Cu cells with PCS and CS by the Aurbach method using 1 M LiPF6 in EC/DEC. Time-controlled CCD of Li||Cu cells with CS (b) and PCS (c). The plating/stripping protocol was initiated at a current density of 0.2 mA cm−2, with the current density incremented by 0.2 mA cm−2 following each cycle. Each platting step was maintained for 1 h. Rate performance (d) and long-term plating/stripping performance (e) of Li||Li symmetric cells with PCS and CS. CLSM images and surface roughness curves of Li negative electrodes in Li||Li symmetric cell with CS (f) and PCS (g) after 20 cycles of plating/stripping (1 mA cm−2, 1 mAh cm−2). Top-view SEM images of the Li deposition in Li||Li symmetric cells with CS (h) and PCS (1 mA cm−2, 1 mAh cm−2) (i). j The calculated dissociation energy and F–P atom distance of LiPF6 with the existence of CS and PCS. k The ESP comparison of PCS, CS and PP. Source data are provided as a Source Data file.
The surface morphologies of deposited Li were investigated to reveal the effect of PCS on the Li+ transport. The confocal laser scanning microscopy (CLSM) images (Fig. 2f, g) showed that the roughness of deposited Li in a Li||Li symmetric cell with PCS (3.6 μm) is much lower than that with CS (57.2 μm). Scanning electron microscopy (SEM) images reveals that Li||Li symmetric cells and Li||Cu cells with PCS maintained a relatively uniform and flat structure after Li deposition (Fig. 2i, and Supplementary Fig. 19a). In contrast, deposited Li exhibited loosely stacked moss-like structure in the cells with CS (Fig. 2h, and Supplementary Fig. 19b). Additionally, the Li deposition thickness with PCS is closer to the theoretical density of Li compared to CS (Supplementary Fig. 19c). This uniform and dense deposition can be attributed to the high dielectric constant of PCS, which polarizes under the applied electric field to generate a directional electric field that regulates the electric field density distribution and prevents the tip effect in the metal negative electrode34. The above results demonstrate the advantage of PCS with high-dielectric constant for extending the cycle life of cells.
DFT calculations were conducted to further investigate the effect of high-dielectric constant PCS on the dissociation of electrolyte and metal deposition. Typical LiPF6 electrolyte was selected to investigate the effect of separators with various molecular chains on its dissociation. LiPF6 can decompose to LiF and LixPFy during the discharge progress, the energy required for P-F bond dissociation in this process is 3.580 eV and the distance of the F-P bond is 1.854 Å35. However, the dissociation energy and the F-P distance are changed to 3.615 eV and 1.854 Å with the existence of PP chain, which could be attributed to the slightly positive electrostatic surface potential (0.03 eV) of PP hinders the dissociation of LiPF6 (Fig. 2k and Supplementary Data 20–24)36. In contrast, the dissociation energy and the F-P distance are 3.372 eV and 1.874 Å when CS is employed, while the dissociation energy and the F-P distance are 2.327 eV and 1.960 Å after the cellulose is phosphorylated. The results confirm that the abundant phosphate groups in the cellulose skeleton significantly enhance charge transfer, facilitating the dissociation of LiPF6 to form LiF. Furthermore, the charge displacement curves between PCS and various electrolytes components (LiPF6, EC and DEC) under an electric field indicate that PCS has no significant effect on promoting the decomposition of EC and DEC (Supplementary Fig. 20). This is favorable for forming an SEI that is rich in LiF and low in organic components. The XPS analyses of the Li deposited layer after discharging confirmed LiF formation with the effect of PCS (Fig. 3a, and Supplementary Fig. 21a). The peaks in C 1s spectra deconvoluted into C‒C, C‒O, C=O, and CF3 that originate from the decomposed electrolyte and generate amorphous phase in SEI10. A high percentage of Li2O (37.5%) was detected in the O 1s spectrum in the presence of the CS, while it was only 9.8% in the cells with PCS. High resolution F 1s and Li 1s XPS spectra of Li deposit also higher content of LiF in the cells with PCS than those with CS.

a F 1s and Li 1s XPS spectra of the SEIs in Li||Li symmetric cells with PCS and CS, evaluated at a current density of 1 mA cm−2 and a capacity of 1 mAh cm−2 after 20 cycles of platting/stripping. b Magnified view of the Cryo-TEM image of SEI in the presence of PCS, with Li deposited on a Cu mesh at a current density of 0.5 mA cm−2 and a capacity of 0.5 mA cm−2. LiF, Li2CO3, LiO2, and inlay structure are outlined in blue, orange, white, and red lines, respectively. c Low-magnification Cryo-TEM image and corresponding elemental distributions of C, O, and F. 3D rendering images of several typical second ion fragments on the cycled Li surface from the Li||Li symmetric cells with PCS (d) and CS (e) after 20 cycles at 1 mA cm−2 and 1 mA h cm−2. Normalized sputtering depth distribution profiles of secondary ion fragments on the cycled Li surface from the Li||Li symmetric cells with PCS (f) and CS (g) from the TOF-SIMS measurements. Source data are provided as a Source Data file.
Currently, research on the composition of the SEI of alkali metal batteries mainly focused on its inorganic components, while the analysis of organic components is often overlooked. The organic SEI components were analyzed through nuclear magnetic resonance (NMR) technology to further elucidate the influence of high-dielectric constant ___domain on the SEI composition (Supplementary Fig. 21b). After completing 10 cycles of Li||Cu cells with CS and PCS, the cells were disassembled and organic solvents were allowed to evaporate in a glovebox, followed by extracting the organic components of the SEI in DMSO-d6 solvent and conducting NMR analysis. The organic SEI components with the existence of PCS consisted of lithium ethyl carbonate (LEC) and lithium ethylene mono carbonate (LEMC), which mainly generated from the EC electrochemical single-electron reduction and lithiation37. In sharp contrast, the organic SEI component signals under the influence of CS are more pronounced, and new signals corresponding to EG (3.29 ppm) and MeOH (3.19 ppm) also appeared38. The presence of EG and MeOH could be ascribed to the heightened reactivity of the produced alkyl carbonates with the proton solvent (DMSO), without implying their actual existence within SEI39. In addition, obvious signals of amorphous oligomers (3.37 ppm) were also observed, covering the signals of LEMC, as illustrated in Supplementary Fig. 21b, depicting the generation mechanism. EC undergone a nucleophilic reaction during the electrochemical reactions, producing intermediates that subsequently react with DEC to produce oligomers38. These oligomers can undergo extension reactions through further nucleophilic attacks. This amorphous oligomer type includes a substantial quantity of ether bonds, facilitating coordination with Li+ and enhancing its diffusion barrier. This finding elucidates the low SEI charge transfer barrier of PCS for Li+ (Supplementary Fig. 22 and Supplementary Note 6). Due to its high-dielectric constant, PCS interacts more strongly with Li+ under the influence of electric field in the battery, prompting its desolvation. Solvent molecules encounter difficulties in migrating with Li, hindering their accumulation at the SEI and leading to a reduced presence of amorphous organic compounds. Conversely, in the presence of CS, Li+ tends to coordinate with solvent molecules (e.g. [Li(EC)3(DEC)]+) and reach the negative electrode interface propelled by the electric field, forming low-conductivity amorphous oligomers through electrochemical reduction. Thus, the SEI induced by PCS is dense, uniform, and low in amorphous oligomers, exhibiting electronic insulation and low interfacial impedance. This structure minimizes parasitic reactions and prevents continuous consumption of the electrolyte. Amorphous oligomers readily dissolve in solvents, leading to an unstable SEI. This instability causes continuous SEI accumulation, resulting in poor stability and high SEI impedance. This explains why the plating/stripping stability and polarization of Li||Li and Li||Cu half cells with CS are inferior to those with PCS.
The structure of SEI after the electrochemical deposition in the presence of separator with high and low dielectric constant at subangstrom resolution using cryo-TEM was investigated, respectively (Fig. 3b and Supplementary Fig. 23). An amorphous phase with embedded Li2O, Li2CO3, and LiF nanocrystals is found in the classical mosaic SEI. This structure is consistent with the above XPS and NMR analysis. The measured lattice spacings of 2.33, 2.48, 2.66, and 2.80 Å correspond to the (111) plane of LiF, (110) plane of metallic Li, (111) plane of Li2O, and (002) plane of Li2CO3, respectively10,40. When a high-dielectric constant PCS is present, an embedded structure of SEI is observed, with a large amount of LiF being found embedded in Li2CO3. The Li2CO3/LiF structure has been reported to possess a low Li diffusion energy barrier (<40 kJ·mol−1) and a high diffusion coefficient (7.93 × 10−6 m2 s−1)41. This inlay structure could promote the transport of Li while ensuring the low electron conductivity of LiF42. Conversely, LiF is hardly observed when low dielectric constant separators are installed, and the structure of SEI is predominantly amorphous. The cryo-TEM mappings show distributions of C, O, and F elements (Fig. 3c) and confirm the richness of LiF at the interface of SEI. The above results confirm that high-dielectric constant PCS could assist in the formation of a low energy barrier and robust SEI under the driving of electric field. In this SEI, Li primarily diffusion along grain boundaries, which is significantly faster than the conventional lattice diffusion43. We conducted TOF-SIMS depth profiling after galvanostatic plating/stripping of Li||Li symmetric cells to further investigate the various chemical components of the SEI. The 3D rendering images further visualize the specific distribution of various components in SEI (Fig. 3d, e). The Li surface from the Li||Li symmetric cells with PCS showed strong LiF2– signals, and the signal intensity of LiO2−, LiCO3−, and organic components were lower. In contrast, the Li surface from the Li||Li symmetric cells with CS showed strong signal of organic components and weak signal of LiF2−. The sputtering depth distribution profiles reveal that the SEI signal intensity drops to ~0 with PCS after sputtering for 100 s (Fig. 3f). In contrast, the SEI signal intensity in cells with CS remains significant even after 600 s of sputtering (Fig. 3g). The initially low signal intensity of organic components in the SEI with CS can be attributed to its high solubility, leading to dissolution in the electrolyte. As sputtering time increases, the organic component signal intensity rises markedly, confirming that the SEI is predominantly organic. These results demonstrate that PCS induces the formation of a LiF-rich, thin, and robust SEI.
Electrochemical performance of cells with PCS
The effect of high-dielectric constant PCS to the kinetics of Li+ migration was further demonstrated by in situ distribution of relaxation times (DRT) analyses, activation energy (Ea) and Li+ transference numbers (Fig. 4a, b, and Supplementary Fig. 24). Specifically, the DRT mapping of Li||LFP cell with PCS exhibits weaker interface phase transport impedance (RSEI) and charge transfer impedance (Rct) than that with CS within an electrochemical cycle, confirming the faster charge transfer and higher conductive and robust SEI derived with the existence of PCS than that with CS. In addition, the high Li+ transference number (0.68) and the lower activation energy (54.46 KJ·mol−1) of Li | |Li cell with PCS indicating the lower energy barrier for the Li+ desolvation with the existence of PCS. The mechanism confirmed by the above results is illustrated in Fig. 4c, the introduction of a high-dielectric constant separator reduces the desolvation energy of alkali metals, thereby decreasing the Rct. Simultaneously, the SEI rich in LiF and low in oligomers reduces the RSEI.

In situ DRT spectra representing a cycle of Li||LFP cells with CS (a) and PCS (b) at 25 °C, operating under a current density of 1 C. c Schematic diagram of decreased Rct and RSEI in a high dielectric field. d Rate capacity of Li||LFP cells with PCS, and PP (1 C = 170 mA g−1). Long-term cycling performances of Li||LFP cells with PP and PCS at a current density of 0.5 C (e) and 1 C (f). g Cycling performance of Li||LFP pouch cell with PCS at 0.5 C. The specific energy is calculated in relation to the mass of the Li||LFP pouch cell (2.6 g). Rate capacity (h) and cycling performance (i) of Na||NVP cells with PCS and GF separators (1 C = 117.6 mA g−1). Source data are provided as a Source Data file.
PCS were loaded into Li metal cells for cyclic testing to evaluate its practical feasibility. As shown in Fig. 4d, the Li||LFP cell with PCS exhibited enhanced rate capability compared to that with CS and PP separator. The discharge capacities of Li||LFP cell with PCS were 164.4, 161.6, 154.3, 142.3, 133.3 and 123.8 mAh g−1 at 0.2, 0.5, 1, 2, 3 and 5C, respectively, higher than those of cell with PP separator (164.2, 158.6, 150.7, 136.0, 126.2, and 114.8 mAh g−1) and CS (160.4, 156.4, 148.8, 136.3, 127.2, and 114.3 mAh g−1), reflecting fast kinetics of Li+ transport. The comparison of cycling stability of Li||LFP cells with PCS, CS and commercial PP separators at 0.5 and 1 C were conducted (Fig. 4e, f). The Li||LFP cell with PCS demonstrated stable cycling performance with 93.1% retention of the initial capacity over 800 cycles at 0.5 C, which surpassed that of Li||LFP cell with PP separator (decaying rapidly after 400 cycles and failed after 600 cycles) and CS (decaying rapidly after 400 cycles and failed after 450 cycles). Moreover, the Li||LFP cell with PCS had an initial capacity of 151.4 mAh g−1 at 1 C and a reversible capacity of 141.2 mAh g−1 after 1000 stable cycles could be attained. In contrast, the Li||LFP cell with PP separator and CS presented a quick capacity decay within 1000 cycles and the efficiency exhibited obvious fluctuations during the cycles. This could be ascribed to the irreversible formation and growth of the SEI and side reactions44. Furthermore, the enhanced negative electrode interface stability provided by the PCS with high dielectric constant significantly improves Li reversibility. Even with a low negative electrode to the positive electrode ratio (N/P ratio of ~3), Li||LFP cell maintained a capacity of 129 mAh g−1 after 300 cycles, exceeding 80% of the initial capacity. The Li||LFP cell with PCS also delivers high and stable discharge specific capacities (163–116 mAh g−1) at current densities ranging from 0.1 to 5 C (Supplementary Fig. 25). This performance is substantially better that of Li||LFP cells using CS and PP. Due to the stable SEI induced by PCS, the electrolyte weight to capacity ratio (E/C ratio) was further reduced to about 5 g Ah−1, and the Li|PCS|LFP cell also showed significant cycle stability, remaining stable after 300 cycles. Even with the active material mass loading further increased to ~22 mg cm−2, the cell with PCS maintains an initial capacity of 150 mAh g−1, operates stably for over 300 cycles, and exhibit better rate performance of 105 mAh g−1 at 5 C. In stark contrast, the specific capacities of Li||LFP cells with CS and PP decrease to 9 and 12 mAh g−1 at 5 C, respectively. Under conditions including low N/P ratio, lean electrolyte, and high active material mass loading, batteries equipped with PCS still maintain stable cycling. This demonstrates the practical potential of PCS in the development of high specific energy batteries.
The potential for large-scale production of PCS is primary confirmed in Supplementary Fig. 26. The size of PCS prepared on the wetting process machine after rolling reached 31 × 62 cm2. In addition, the exhibited rollable, twistable, foldable, and stretchable properties of PCS confirm its possibility in using different processes (e.g. winding or stacking) to meet the commercial demands45. Moreover, after folding, cutting, and pricking, the pouch cell could still light up a light-emitting diode display (Supplementary Fig. 27). To demonstrate the universality of PCS, the less stable commercial active material NCM811 was also utilized. Similarly, the Li||NCM811 cells with PCS exhibited enhanced cyclability and rate performance compared to those with CS and PP (Supplementary Fig. 28). In addition, the prepared Li||LFP and Li||NCM811 pouch cell demonstrated significant cycle stability, remaining stable after 200 cycles. Even under conditions of high active material loading (~22 mg cm−2), lean electrolyte (~5 g Ah−1), and limited negative electrode excess (~3 or 2.8), pouch cells using LFP and NCM811 as active materials can both operate stably for over 200 cycles (Fig. 4g and Supplementary Fig. 28d). Both the cycling stability and specific energy surpass the advanced values reported in recent years, confirming the practical application potential of PCS as a separator (Supplementary Fig. 29). To ascertain the universality of PCS in alkali metal batteries, its application was extended to sodium metal batteries. The prevalent employment of glass fiber (GF) separators in sodium metal batteries, characterized by their substantial thickness as well as inadequate mechanical strength, constrains battery specific energy and impedes its practical application. The Na||NVP cell with PCS exhibited better rate capability than that with GF separator (Fig. 4h). The discharge capacities of Na||NVP cell with PCS were 160.9, 156.6, 149.1, 136.8, 128.3 and 116.1 mAh g−1 at 0.2, 0.5, 1, 2, 3 and 5 C, respectively, higher than those of cell with GF separator (157.5, 152.6, 145.7, 134.9, 126.2, and 114.8 mAh g−1). Moreover, the Na | |NVP cell with PCS had an initial capacity of 111.7 mAh g−1 at 1 C and a reversible capacity of 92.9 mAh g−1 after 1000 stable cycles could be attained (Fig. 4i). In stark contrast, the Na | |NVP cell with GF experienced a quick capacity decay (from 110.2 mAh g−1 to 74.8 mAh g−1) with in the 1000 cycles. These results confirm that PCS enable cells with significantly enhanced CE, cycling stability and rate performance, and has broad application prospects in the commercialization of alkali metal batteries.
Discussion
With the coordination of the electric field inside the battery with high-dielectric constant separator, we demonstrated a strategy that facilitated the reaction at the separator-negative electrode interface to construct a stable alkaline metal battery. Comprehensive simulations and characterizations demonstrate the critical role of separators with high-dielectric constant in promoting the regular distribution of electric field density. In addition, the high-dielectric constant PCS undergoes intense electronic transfer under an electric field, promoting the generation of LiF and reducing the energy barrier for Li transfer in SEI. Benefiting from the electric field-driven electron displacement polarization, the interaction between the high dielectric constant separator and alkali metal ions is enhanced, optimizing the solvation structure and dynamics of alkali metal ions and consequently mitigating the reaction of organic solvent molecules at SEI. With the prepared high-dielectric constant separator, Li and Na metal cells both exhibited improved cycling stability compared to cells with commercial PP or GF separators. These advancements hold significance in elucidating the reactions at the separator-negative electrode interface within intricate environment of batteries. Furthermore, this simple high-dielectric constant separator design strategy has the potential for widespread application in alkali metal batteries.
Methods
Materials
Bamboo fibers were provided by Babo Co., Ltd. (Chengdu, China). Diammonium hydrogen phosphate (99%), urea (99%), N-Methyl-2-pyrrolidone (NMP, 99.5%), and diethyl carbonate (99%) were purchased from Aladdin Reagent Co., Ltd. (Shanghai, China). Aramid nanofibers (ANF) was obtained from Jufang new materials Co., Ltd. (Bingzhou, China). DMSO-d6 (99.8%) was purchased from Deutero. All the electrolytes were obtained from DodoChem. LiNi0.8Co0.1Mn0.1O2 (NCM811), LiFePO4 (LFP), Na3V2(PO4)3 (NVP), polyvinylidene difluoride (PVDF, Mw = 1000000) and Super-p were purchased from Shenzhen Kejing STAR Technology Co., Ltd. (Shenzhen, China). Celgard2500 PP separator (thickness: 25 μm; lateral dimension: 60 mm; porosity: 55%; average pore size: 1.6 μm) was offered by Canrd New Energy Technology Co., Ltd. (Dongguan, China). GF/A separator (thickness: 260 μm; porosity: 55%) was provided by WhatmanTM. All the materials were used without any further purification.
Phosphorylation of cellulose
The bamboo fiber was beaten for 30 min and vacuum-dried at 50 °C for 48 h to obtain cellulose pulp. The prepared cellulose pulp was then phosphorylated46,47. Briefly, 10 g dried cellulose pulp was immersed in 150 mL aqueous solution containing 12 g urea and 4.5 g (NH4)2HPO4 for 1 h, followed by vigorous stirring for 30 min. The resultant mixture was dried in a ventilation oven at 50 °C for 48 h to remove water, and then cured at 150 °C for 30 min. Finally, the obtained products were washed 3 times with deionized water to remove unreacted reagents, and then vacuum dried at 50 °C for 48 h to obtain dried PC.
Preparation of cellulose-based separators
A traditional wet process was employed to prepare a series of separators48. Typically, 5.76 g small pieces of dried PC pulp was soaked in water for 12 h, followed by defibering for 5 min. Subsequently, 0.35 g ANF was added to the defibered PC pulp suspension and stirred evenly. Finally, the separators were obtained by transferring the above mixed pulp and vacuum-dried at 70 °C for 5 min.
Characterization
TGA was performed on a TA Instrument Q500 with a heating rate of 5 °C/min. HRR curves were measured using an ASTM D7309 micro calorimeter. The specific surface area and pore size distribution (PSD) were performed on a BSD-660M N2 adsorption analyzer. Fourier transform infrared (FTIR) spectroscopies were carried out on a Nicolet 8700 spectrometer. Li||Li and Li||Cu Cells were disassembled in a glove box after plating/stripping. The Cu foils or lithium metal electrodes were washed with dimethoxyethane to remove residual lithium salts, and after the dimethoxyethane evaporated, XPS and SEM analyses were conducted. Vacuum transfer chambers were utilized to prevent the influence of water and oxygen on the samples during transfer. XPS spectra were recorded an Escalab 250Xi X-ray Photoelectron Spectrometer. Morphological images were taken on Hitachi S-3000N scanning electron microscope. Gatan 698 cryo-transfer holder and Talos F200X G2 TEM were employed to study the morphology Li deposits. A 200 mesh Cu grid was lined against a Cu foil to assemble a Li||Cu cell for Li deposition, with the areal capacity of 0.5 mAh cm−2 at the current density of 0.5 mA cm−2. Shake the Cu mesh loaded on the Cu foil in the dimethoxyethane to make it fall off naturally to prevent damage to the carbon film. All the TEM images were taken at −170 °C.
Electrochemical measurement
Positive electrodes were prepared by coating slurry mixtures onto single-side of aluminum (Al) foils (99.8% purity, 14-μm thickness, untreated) using an MKS-AFA-ES200 automatic coating machine. The slurry was prepared with NMP as the solvent, maintaining a weight ratio of active materials (LFP, NCM811 or NVP), PVDF, Super-p at 8:1:1. Unless otherwise specified, the mass loading of the active material was ~3.0 mg cm−2. 2032-type coin cells, with casings and springs constructed from stainless steel, were utilized for assembly. The coated Al foils were subsequently cut into circular pieces with a 10 mm diameter using an MKS-T10 precision cutter. Li foil (500 μm) was employed as the negative electrode for lithium metal batteries and the electrolyte used for cells was 40 µL. Na foil (500 μm) was employed as the negative electrode for sodium metal batteries and the electrolyte used for cells was 100 µL. The mass loading of the LFP was adjusted to approximately 22 mg cm−2 to enable electrochemical measurements under a high active material mass loading condition. To evaluate Li||LFP cells with a low N/P ratio, an N/P ratio of 3 was maintained by depositing Li on Cu foils at a current density of 0.5 mA cm−2. Additionally, the E/C ratio of Li||LFP cells was controlled at approximately 5 g Ah−1 to conduct electrochemical measurements under a lean electrolyte condition. For the pouch cell, a single-sided coated Al foil (mass loading of ~22 mg cm−2) was employed as the positive electrode, and 60 μm lithium loaded on Cu foil (99.8% purity, 12-μm thickness, untreated) was used as the negative electrode. The stack pressure applied to pouch cells was ~200 kPa. All cells were assembled in an argon-filled glovebox with less than 0.1 ppm H2O and less than 0.1 ppm O2. Unless otherwise specified, all cycling experiments of batteries were carried out at 25 °C utilizing a BENTING chamber. The charge/discharge behaviors of batteries were performed on a LAND battery test system. The Li||LFP and Na||NVP cells were charged and discharged between the voltage ranges of 2.5–4.2 V, and 2.5–4 V, respectively. A cut-off voltage of 1 V was applied in symmetrical cell and Li||Cu half-cell test. The CV, EIS, and CA measurements were performed on a CHI 760E electrochemical workstation. EIS measurements were performed within a frequency range of 0.1 Hz to 105 Hz. A sinusoidal perturbation amplitude of 5 mV was applied, with 12 data points collected per frequency decade. The measurements were conducted under potentiostatic control, with the system stabilized at open-circuit potential for 2 s prior to data acquisition.
Computational simulation
The ORCA 6.0.1 software package was employed to realize the DFT calculations49. Molecules were built using Avogadro50. The geometries were optimized using the B3LYP functional along with all-electron def2-SVP basis sets without symmetry constraints. We utilized Grimme’s latest additive dispersion correction D4 model throughout the optimization to ensure accurate consideration of weak intermolecular interactions and atomic charge effects51. The optimized structures exhibit no imaginary frequencies. The subsequently HOMO-LUMO calculations were performed using B3LYP-D4/def2-TZVP. For ESP and weak interactions energy, precise calculations were conducted using ωB97M-V function along with def2-TZVP basis set. Counterpoise correction was applied and diffuse basis sets were not considered when calculating interaction energies as this study does not involve anionic systems. The restrained electrostatic potential (RESP) and ESP were calculated and visualized using Multiwfn and VMD, respectively52,53. Molecule structures along with their charge density were drawn using VMD and VESTA software54.
Classical MD simulations were carried out using the GROMACS package55. The molecular mechanics force field was parametrized from the general AMBER force field56. The RESP atomic charges were calculated employing Multiwfn program to describe electrostatic interactions precisely52. Atomic charges of Li+ and PF6− were multiplied by scale factor 0.8 to correct the polarization effect57. Each simulation box was created by randomly packing all molecules using Packmol package in a cubic box of size 50 × 50 × 50 Å3 with periodicity in all three directions58. Simulation boxes of the classical MD system contain 60 Li+, 60 PF6−, 450 EC molecules, 246 DEC molecules, and 6 fixed cellulose or PC chains corresponding to 1 M LiPF6 in EC/DEC (1:1 volume) electrolyte. The atom charges parameters were fitted specifically for this work (Supplementary Fig. 15c, d). The cutoff distance of 1.0 nm was selected for Lennard Jones interactions. Long range electrostatic interactions were calculated with the particle-mesh Ewald (PME) method with grid spacing of 0.12 nm and cutoff distance of 1.2 nm. An energy minimization was performed to relax the strained contacts in the initial configuration. Afterward, cellulose chains were frozen, and simulations were performed in the canonical ensemble (NVT) for 100 ps at 298.15 K to reach a stable energy state. Finally, 10 ns of NVT was carried out to sample adequately for further analysis. RDF, coordination number and atomic distribution were analyzed using VMD53.
Data availability
All data are available within the main text, the Supplementary Information file, and the Source Data files. Source data are provided with this paper.
References
Popovic, J. The importance of electrode interfaces and interphases for rechargeable metal batteries. Nat. Commun. 12, 6240 (2021).
Xing, X. et al. Graphite-based lithium-free 3D hybrid anodes for high energy density all-solid-state batteries. ACS Energy Lett. 6, 1831–1838 (2021).
Tang, Z. et al. Revealing the closed pore formation of waste wood-derived hard carbon for advanced sodium-ion battery. Nat. Commun. 14, 6024 (2023).
Jurng, S., Brown, Z. L., Kim, J. & Lucht, B. L. Effect of electrolyte on the nanostructure of the solid electrolyte interphase (SEI) and performance of lithium metal anodes. Energ. Environ. Sci. 11, 2600–2608 (2018).
Adenusi, H. et al. Lithium batteries and the solid electrolyte interphase (SEI)-progress and outlook. Adv. Energy Mater. 13, 2203307 (2023).
Francis, C. F. J., Kyratzis, I. L. & Best, A. S. Lithium-ion battery separators for ionic-liquid electrolytes: a review. Adv. Mater. 32, 1904205 (2020).
Zhu, X. et al. Ultra-stable zinc anodes facilitated by hydrophilic polypropylene separators with large scale production capacity. Adv. Funct. Mater. 34, 2407262 (2024).
Qin, J. et al. Achieving stable Na metal cycling via polydopamine/multilayer graphene coating of a polypropylene separator. Nat. Commun. 12, 5786 (2021).
Sheng, L. et al. Suppressing electrolyte-lithium metal reactivity via Li+-desolvation in uniform nano-porous separator. Nat. Commun. 13, 172 (2022).
Liu, Y. et al. Self-assembled monolayers direct a LiF-rich interphase toward long-life lithium metal batteries. Science 375, 739–745 (2022).
Liu, P. et al. Multifunctional separator allows stable cycling of potassium metal anodes and of potassium metal batteries. Adv. Mater. 34, 2105855 (2022).
Yao, N., Chen, X., Fu, Z.-H. & Zhang, Q. Applying classical, Ab Initio, and machine-learning moleculardynamics simulations to the liquid electrolyte for rechargeable batteries. Chem. Rev. 122, 10970–11021 (2022).
Li, Q. et al. Understanding the molecular mechanism of pulse current charging for stable lithium-metal batteries. Sci. Adv. 3, e1701246 (2017).
Simons, R. Electric field effects on proton transfer between ionizable groups and water in ion exchange membranes. Electrochim. Acta 29, 151–158 (1984).
Yang, B. et al. Strong internal electric field enhanced polysulfide trapping and ameliorates redox kinetics for lithium-sulfur battery. J. Energy Chem. 77, 376–383 (2023).
Bardak, C., Atac, A. & Bardak, F. Effect of the external electric field on the electronic structure, spectroscopic features, NLO properties, and interionic interactions in ionic liquids: a DFT approach. J. Mol. Liq. 273, 314–325 (2019).
Tran, T. N. et al. Electric field enhances the electronic and diffusion properties of penta-graphene nanoribbon anodes in lithium-ion batteries. RSC Adv. 14, 33524–33535 (2024).
Xu, J. et al. Understanding the electrical mechanisms in aqueous zinc metal batteries: from electrostatic interactions to electric field regulation. Adv. Mater. 36, 2309726 (2024).
Allen, P. B. Dipole interactions and electrical polarity in nanosystems: The Clausius–Mossotti and related models. J. Chem. Phys. 120, 2951–2962 (2004).
Huang, Y. et al. A relaxor ferroelectric polymer with an ultrahigh dielectric constant largely promotes the dissociation of lithium salts to achieve high ionic conductivity. Energ. Environ. Sci. 14, 6021–6029 (2021).
Sugahara, A. et al. Negative dielectric constant of water confined in nanosheets. Nat. Commun. 10, 850 (2019).
Holoubek, J. et al. Predicting the ion desolvation pathway of lithium electrolytes and their dependence on chemistry and temperature. J. Phys. Chem. Lett. 13, 4426–4433 (2022).
Andrews, M. H., Marshak, A. H. & Shrivastava, R. The effect of position-dependent dielectric constant on the electric field and charge density in a p-n junction. J. Appl. Phys. 52, 6783–6787 (1981).
Kim, J. Y. et al. Effect of the dielectric constant of a liquid electrolyte on lithium metal anodes. Electrochim. Acta 300, 299–305 (2019).
Zhang, Q. Y., Liu, X. H., Ren, Y. L. & Li, Y. S. Phosphorated cellulose as a cellulose-based filler for developing continuous fire resistant lyocell fibers. J. Clean. Prod. 368, 133242 (2022).
Lu, G. et al. High-voltage electrosynthesis of organic-inorganic hybrid with ultrahigh fluorine content toward fast Li-ion transport. Sci. Adv. 10, eado7348 (2024).
Lei, H. et al. DFT and molecular dynamic simulation for the dielectric property analysis of polyimides. Chem. Phys. Lett. 786, 139131 (2022).
Kim, K. et al. Phosphorescent dye-based supramolecules for high-efficiency organic light-emitting diodes. Nat. Commun. 5, 4769 (2014).
Yang, Y. et al. Diminishing space-charge layer effect of Zinc anodes by an anion-immobilized electrolyte membrane. ACS Energy Lett. 8, 1959–1968 (2023).
Ma, B. et al. Molecular-docking electrolytes enable high-voltage lithium battery chemistries. Nat. Chem. 16, 1427–1435 (2024).
Zhang, C. et al. Single-crystalline van der Waals layered dielectric with high dielectric constant. Nat. Matter 22, 832–837 (2023).
Zhang, M. et al. Polymer dielectrics with simultaneous ultrahigh energy density and low loss. Adv. Mater. 33, 2008198 (2021).
Yi, X. et al. Safe electrolyte for long-cycling alkali-ion batteries. Nat. Sustain. 7, 326–337 (2024).
Ning, Z. et al. Dendrite initiation and propagation in lithium metal solid-state batteries. Nature 618, 287–293 (2023).
Zhao, Q., Stalin, S. & Archer, L. A. Stabilizing metal battery anodes through the design of solid electrolyte interphases. Joule 5, 1119–1142 (2021).
Zheng, S. et al. Precise control of Li+ directed transport via electronegative polymer brushes on polyolefin separators for dendrite-free lithium deposition. Adv. Funct. Mater. 32, 2201430 (2022).
Wang, L. et al. Identifying the components of the solid–electrolyte interphase in Li-ion batteries. Nat. Chem. 11, 789–796 (2019).
Hu, J. et al. Deciphering the formation and accumulation of solid-electrolyte interphases in Na and K carbonate-based batteries. Nano Lett. 24, 1673–1678 (2024).
Michan, A. L., Leskes, M. & Grey, C. P. Voltage dependent solid electrolyte interphase formation in silicon electrodes: monitoring the formation of organic decomposition products. Chem. Mater. 28, 385–398 (2016).
Sheng, O. et al. In situ construction of a LiF-enriched interface for stable all-solid-state batteries and its origin revealed by Cryo-TEM. Adv. Mater. 32, 2000223 (2020).
Das Goswami, B. R. et al. Unraveling ion diffusion pathways and energetics in polycrystalline sei of lithium-based batteries: combined Cryo-HRTEM and DFT study. J. Phys. Chem. C. 127, 21971–21979 (2023).
Hu, T. et al. Impact of the local environment on Li ion transport in inorganic components of solid electrolyte interphases. J. Am. Chem. Soc. 145, 1327–1333 (2023).
Svirinovsky-Arbeli, A. et al. Using NMR spectroscopy to link structure to function at the Li solid electrolyte interphase. Joule 8, 1919–1935 (2024).
Zhang, W. et al. A “cation-anion regulation” synergistic anode host for dendrite-free lithium metal batteries. Sci. Adv. 4, eaar4410 (2018).
Dai, F. & Cai, M. Best practices in lithium battery cell preparation and evaluation. Commun. Mater. 3, 64 (2022).
Ghanadpour, M., Carosio, F., Larsson, P. T. & Wågberg, L. Phosphorylated cellulose nanofibrils: a renewable nanomaterial for the preparation of intrinsically flame-retardant materials. Biomacromolecules 16, 3399–3410 (2015).
Noguchi, Y., Homma, I. & Matsubara, Y. Complete nanofibrillation of cellulose prepared by phosphorylation. Cellulose 24, 1295–1305 (2017).
Liu, H. et al. Flexible, degradable, and cost-effective strain sensor fabricated by a scalable papermaking procedure. ACS Sustain. Chem. Eng. 6, 15749–15755 (2018).
Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2, 73–78 (2012).
Hanwell, M. D. et al. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics 4, 17 (2012).
Caldeweyher, E. et al. A generally applicable atomic-charge dependent London dispersion correction. J. Chem. Phys. 150, 154122 (2019).
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Humphrey, W., Dalke, A. & Schulten, K. V. M. D. Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Momma, K. & Izumi, F. VESTA 3for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272–1276 (2011).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25 (2015).
Duan, Y. et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 24, 1999–2012 (2003).
Yu, Z. et al. Rational solvent molecule tuning for high-performance lithium metal battery electrolytes. Nat. Energy 7, 94–106 (2022).
Martínez, L., Andrade, R., Birgin, E. G. & Martínez, J. M. PACKMOL: a package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 30, 2157–2164 (2009).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2022YFB3803502, G.X.), Fundamental Research Funds for the Central Universities (2232025A-05, G.X.), National Natural Science Foundation of China (52103076, G.X.), International Cooperation Fund of the Science and Technology Commission of Shanghai Municipality (24520713300, G.X.), and Shanghai Rising-Star Program (24QA2700100, G.X.).
Author information
Authors and Affiliations
Contributions
G.X. conceived and directed the project. T.Z. and X.Z. conducted most of the experiments with the assistance of the other authors. Z.X., and J.X. participated in parts of the experiments. T.Z. drafted the initial version of this manuscript. G.X., Y.C., K.C.G. and M.Z. contributed to the revision of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ge Li, Bingan Lu and Xiao Zhao for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, T., Zhu, X., Xiong, J. et al. Electron displacement polarization of high-dielectric constant fiber separators enhances interface stability. Nat Commun 16, 4867 (2025). https://doi.org/10.1038/s41467-025-60256-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60256-9
