
Abstract
Approximately 20% of paediatric and adolescent/young adult patients with renal tumours are diagnosed with non-Wilms tumour, a broad heterogeneous group of tumours that includes clear-cell sarcoma of the kidney, congenital mesoblastic nephroma, malignant rhabdoid tumour of the kidney, renal-cell carcinoma, renal medullary carcinoma and other rare histologies. The differential diagnosis of these tumours dates back many decades, when these pathologies were identified initially through clinicopathological observation of entities with outcomes that diverged from Wilms tumour, corroborated with immunohistochemistry and molecular cytogenetics and, subsequently, through next-generation sequencing. These advances enabled near-definitive recognition of different tumours and risk stratification of patients. In parallel, the generation of new renal-tumour models of some of these pathologies including cell lines, organoids, xenografts and genetically engineered mouse models improved our understanding of the development of these tumours and have facilitated the identification of new therapeutic targets. Despite these many achievements, paediatric and adolescent/young adult patients continue to die from such rare cancers at higher rates than patients with Wilms tumour. Thus, international coordinated efforts are needed to answer unresolved questions and improve outcomes.
Key points
-
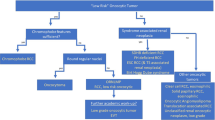
Non-Wilms tumours occur in ~20% of paediatric and adolescent/young adult patients with kidney cancers, and advances in next-generation sequencing have brought clarity to this group of rare and hard-to-treat cancers.
-
Translocation renal-cell carcinomas are primarily driven by translocations involving the MiT family members TFE3, TFEB and ELOC. A growing number of cell lines and patient-derived xenograft (PDX) models are available for these tumours, but genetically modified mouse models remain a gap in the field.
-
Renal medullary carcinomas are characterized by a biallelic loss of SMARCB1 and generally include a disruptive balanced translocation of one SMARCB1 allele. Robust models of renal medullary carcinomas (such as cell lines, organoids and PDXs) are now available or actively being developed.
-
Malignant rhabdoid tumours of the kidney are driven by biallelic loss of SMARCB1 and, to a lesser extent, SMARCA4. Here, there are a relatively large number of cell lines, organoids, PDX and genetically modified mouse models that are available for these tumours, which led to identification of several potential therapeutic targets.
-
Clear-cell sarcoma of the kidney, congenital mesoblastic nephromas and most of the other paediatric and adolescent and young adult non-Wilms renal tumours are driven by alterations in BCOR, EGFR, DICER1, BRAF and TSC1 or TSC2, along with gene fusions involving EWSR and NTRK. This group of tumours is in need of preclinical models. International collaborations will lead to the establishment of these models and the development of functional studies to facilitate the identification of therapeutic targets, ultimately resulting in informed clinical trials and improved outcomes.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
27,99 € / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
209,00 € per year
only 17,42 € per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others

References
Wilms, M. Die Mischgeschwülste der Niere. (Verlag von Arthur Georgi, 1899).
Knudson, A. G. Mutation and cancer: statistical study of retinoblastoma. Proc. Natl Acad. Sci. USA 68, 820–823 (1971).
Knudson, A. G. & Strong, L. C. Mutation and cancer: a model for Wilms’ tumor of the kidney. J. Natl Cancer Inst. 48, 313–324 (1972).
Perotti, D. et al. Hallmark discoveries in the biology of Wilms tumour. Nat. Rev. Urol. 21, 158–180 (2024).
46th Congress of The International Society of Paediatric Oncology (SIOP) 2014 Toronto, Canada, 22nd –25th October, 2014 SIOP Abstracts. Pediatr. Blood Cancer 61, S105–S433 (2014).
WHO Classification of Tumours Editorial Board. Urinary and Male Genital Tumours. (International Agency for Research on Cancer, 2022).
Cajaiba, M. M. et al. The classification of pediatric and young adult renal cell carcinomas registered on the Children’s Oncology Group (COG) protocol AREN03B2 after focused genetic testing. Cancer 124, 3381–3389 (2018).
Spreafico, F. Paediatrics: towards evidence-based management of paediatric RCC. Nat. Rev. Urol. 12, 426–428 (2015).
Denize, T. et al. Renal cell carcinoma in children and adolescents: a retrospective study of a French–Italian series of 93 cases. Histopathology 80, 928–945 (2022).
Argani, P. Translocation carcinomas of the kidney. Genes. Chromosomes Cancer 61, 219–227 (2022).
Cimadamore, A. et al. Towards a new WHO classification of renal cell tumor: what the clinician needs to know-a narrative review. Transl. Androl. Urol. 10, 1506–1520 (2021).
Sun, G. et al. Integrated exome and RNA sequencing of TFE3-translocation renal cell carcinoma. Nat. Commun. 12, 5262 (2021).
Linehan, W. M. & Ricketts, C. J. The Cancer Genome Atlas of renal cell carcinoma: findings and clinical implications. Nat. Rev. Urol. 16, 539–552 (2019).
Bakouny, Z. et al. Integrative clinical and molecular characterization of translocation renal cell carcinoma. Cell Rep. 38, 110190 (2022).
Argani, P. MiT family translocation renal cell carcinoma. Semin. Diagn. Pathol. 32, 103–113 (2015).
Gandhi, J. S., Malik, F., Amin, M. B., Argani, P. & Bahrami, A. MiT family translocation renal cell carcinomas: a 15th anniversary update. Histol. Histopathol. 35, 125–136 (2020).
Tretiakova, M. S. Chameleon TFE3-translocation RCC and how gene partners can change morphology: accurate diagnosis using contemporary modalities. Adv. Anat. Pathol. 29, 131–140 (2022).
Marcon, J. et al. Comprehensive genomic analysis of translocation renal cell carcinoma reveals copy-number variations as drivers of disease progression. Clin. Cancer Res. 26, 3629–3640 (2020).
Argani, P. et al. A distinctive pediatric renal neoplasm characterized by epithelioid morphology, basement membrane production, focal HMB45 immunoreactivity, and t(6;11)(p21.1;q12) chromosome translocation. Am. J. Pathol. 158, 2089–2096 (2001).
Moch, H. et al. The 2022 World Health Organization classification of tumours of the urinary system and male genital organs-part A: renal, penile, and testicular tumours. Eur. Urol. 82, 458–468 (2022).
Wei, S., Testa, J. R. & Argani, P. A review of neoplasms with MITF/MiT family translocations. Histol. Histopathol. 37, 311–321 (2022).
Argani, P. et al. TFEB-amplified renal cell carcinomas: an aggressive molecular subset demonstrating variable melanocytic marker expression and morphologic heterogeneity. Am. J. Surg. Pathol. 40, 1484–1495 (2016).
Alhalabi, O. et al. Immune checkpoint therapy combinations in adult advanced MiT family translocation renal cell carcinomas. Oncologist 28, 433–439 (2023).
Kauffman, E. C. et al. Preclinical efficacy of dual mTORC1/2 inhibitor AZD8055 in renal cell carcinoma harboring a TFE3 gene fusion. BMC Cancer 19, 917 (2019).
Ishiguro, M., Iwasaki, H., Ohjimi, Y. & Kaneko, Y. Establishment and characterization of a renal cell carcinoma cell line (FU-UR-1) with the reciprocal ASPL-TFE3 fusion transcript. Oncol. Rep. 11, 1169–1175 (2004).
Calandrini, C. et al. An organoid biobank for childhood kidney cancers that captures disease and tissue heterogeneity. Nat. Commun. 11, 1310 (2020).
Baba, M. et al. TFE3 Xp11.2 translocation renal cell carcinoma mouse model reveals novel therapeutic targets and identifies GPNMB as a diagnostic marker for human disease. Mol. Cancer Res. 17, 1613–1626 (2019).
Argani, P. et al. Translocation carcinomas of the kidney after chemotherapy in childhood. J. Clin. Oncol. 24, 1529–1534 (2006).
Chen, Y.-B. Update on selected high-grade renal cell carcinomas of the kidney: FH-deficient, ALK-rearranged, and medullary carcinomas. Adv. Anat. Pathol. 31, 118–125 (2024).
Bezwada, D. & Brugarolas, J. Reporting on FH-deficient renal cell carcinoma using circulating succinylated metabolites. J. Clin. Invest. 133, e170195 (2023).
Kiuru, M. et al. Familial cutaneous leiomyomatosis is a two-hit condition associated with renal cell cancer of characteristic histopathology. Am. J. Pathol. 159, 825–829 (2001).
Geller, J. I. et al. Characterization of adolescent and pediatric renal cell carcinoma: a report from the Children’s Oncology Group study AREN03B2. Cancer 121, 2457–2464 (2015).
Beckermann, K. E. et al. Renal medullary carcinoma: establishing standards in practice. J. Oncol. Pract. 13, 414–421 (2017).
Baniak, N., Tsai, H. & Hirsch, M. S. The differential diagnosis of medullary-based renal masses. Arch. Pathol. Lab. Med. 145, 1148–1170 (2021).
Bahadoram, S. et al. Renal cell carcinoma: an overview of the epidemiology, diagnosis, and treatment. G. Ital. Nefrol. 39, 2022–vol3 (2022).
Davis, C. J. Jr, Mostofi, F. K. & Sesterhenn, I. A. Renal medullary carcinoma. The seventh sickle cell nephropathy. Am. J. Surg. Pathol. 19, 1–11 (1995).
Avery, R. A. et al. Renal medullary carcinoma: clinical and therapeutic aspects of a newly described tumor. Cancer 78, 128–132 (1996).
Shah, A. Y. et al. Management and outcomes of patients with renal medullary carcinoma: a multicentre collaborative study. BJU Int. 120, 782–792 (2016).
Msaouel, P., Tannir, N. M. & Walker, C. L. A model linking sickle cell hemoglobinopathies and SMARCB1 loss in renal medullary carcinoma. Clin. Cancer Res. 24, 2044–2049 (2018).
Alvarez, O., Rodriguez, M. M., Jordan, L. & Sarnaik, S. Renal medullary carcinoma and sickle cell trait: a systematic review. Pediatr. Blood Cancer 62, 1694–1699 (2015).
Cheng, J. X. et al. Renal medullary carcinoma: rhabdoid features and the absence of INI1 expression as markers of aggressive behavior. Mod. Pathol. 21, 647–652 (2008).
Hong, A. L. et al. Renal medullary carcinomas depend upon SMARCB1 loss and are sensitive to proteasome inhibition. eLife 8, e44161 (2019).
Su, Y. & Hong, A. L. Recent advances in renal medullary carcinoma. Int. J. Mol. Sci. 23, 7097 (2022).
Colombo, P. et al. Unclassified renal cell carcinoma with medullary phenotype versus renal medullary carcinoma: lessons from diagnosis in an Italian man found to harbor sickle cell trait. Urol. Case Rep. 3, 215–218 (2015).
Amin, M. B. et al. Collecting duct carcinoma versus renal medullary carcinoma: an appeal for nosologic and biological clarity. Am. J. Surg. Pathol. 38, 871–874 (2014).
van der Beek, J. N. et al. A pediatric and young adult case of unclassified renal cell carcinoma with medullary phenotype (RCCU-MP): clinical course and treatment. J. Onco-Nephrol. 8, 49–57 (2024).
van der Beek, J. N. et al. MRI characteristics of pediatric renal tumors: a SIOP-RTSG radiology panel Delphi study. J. Magn. Reson. Imaging 55, 543–552 (2022).
Jackson, T. J. et al. How we approach paediatric renal tumour core needle biopsy in the setting of preoperative chemotherapy: a review from the SIOP Renal Tumour Study Group. Pediatr. Blood Cancer 69, e29702 (2022).
Beek, J. Nvander et al. MRI characteristics of pediatric and young-adult renal cell carcinoma: a single-center retrospective study and literature review. Cancers 15, 1401 (2023).
Walsh, A., Kelly, D. R., Vaid, Y. N., Hilliard, L. M. & Friedman, G. K. Complete response to carboplatin, gemcitabine, and paclitaxel in a patient with advanced metastatic renal medullary carcinoma. Pediatr. Blood Cancer 55, 1217–1220 (2010).
Strouse, J. J. et al. Significant responses to platinum-based chemotherapy in renal medullary carcinoma. Pediatr. Blood Cancer 44, 407–411 (2005).
Tan, K.-T. et al. Haplotype-resolved germline and somatic alterations in renal medullary carcinomas. Genome Med. 13, 114 (2021).
Calderaro, J. et al. Balanced translocations disrupting SMARCB1 are hallmark recurrent genetic alterations in renal medullary carcinomas. Eur. Urol. 69, 1055–1061 (2016).
Bratslavsky, G. et al. Comprehensive genomic profiling of metastatic collecting duct carcinoma, renal medullary carcinoma, and clear cell renal cell carcinoma. Urol. Oncol. 39, 367.e1–367.e5 (2021).
Tsuzuki, S. et al. A case of renal cell carcinoma unclassified with medullary phenotype without detectable gene deletion. Pathol. Int. 69, 710–714 (2019).
Liu, Q. et al. Renal medullary carcinoma: molecular, immunohistochemistry, and morphologic correlation. Am. J. Surg. Pathol. 37, 368–374 (2013).
Stahlschmidt, J., Cullinane, C., Roberts, P. & Picton, S. V. Renal medullary carcinoma: prolonged remission with chemotherapy, immunohistochemical characterisation and evidence of bcr/abl rearrangement. Med. Pediatr. Oncol. 33, 551–557 (1999).
Msaouel, P. et al. Comprehensive molecular characterization identifies distinct genomic and immune hallmarks of renal medullary carcinoma. Cancer Cell 37, 720–734.e13 (2020).
Wei, D. et al. Novel renal medullary carcinoma cell lines, UOK353 and UOK360, provide preclinical tools to identify new therapeutic treatments. Genes. Chromosomes Cancer 59, 472–483 (2020).
Carugo, A. et al. p53 is a master regulator of proteostasis in SMARCB1-deficient malignant rhabdoid tumors. Cancer Cell 35, 204–220.e9 (2019).
Msaouel, P. et al. Updated recommendations on the diagnosis, management, and clinical trial eligibility criteria for patients with renal medullary carcinoma. Clin. Genitourin. Cancer 17, 1–6 (2019).
Gangireddy, V., Liles, G. B., Sostre, G. D. & Coleman, T. Response of metastatic renal medullary carcinoma to carboplatinum and paclitaxel chemotherapy. Clin. Genitourin. Cancer 10, 134–139 (2012).
Wilson, N. R. et al. Efficacy and safety of gemcitabine plus doxorubicin in patients with renal medullary carcinoma. Clin. Genitourin. Cancer 19, e401–e408 (2021).
Amjad, A. I. et al. Renal medullary carcinoma: case report of an aggressive malignancy with near-complete response to dose-dense methotrexate, vinblastine, doxorubicin, and cisplatin chemotherapy. Case Rep. Oncol. Med. 2014, 615895 (2014).
Carden, M. A. et al. Platinum plus bortezomib for the treatment of pediatric renal medullary carcinoma: two cases. Pediatr. Blood Cancer 64, e26402 (2017).
Ryan, A., Tawagi, K., VanderVeen, N., Matrana, M. & Vasquez, R. Combination therapy with bortezomib in renal medullary carcinoma: a case series. Clin. Genitourin. Cancer 19, e395–e400 (2021).
Beckermann, K. E. et al. Clinical and immunologic correlates of response to PD-1 blockade in a patient with metastatic renal medullary carcinoma. J. Immunother. Cancer 5, 1 (2017).
Chi, S. N. et al. Tazemetostat for tumors harboring SMARCB1/SMARCA4 or EZH2 alterations: results from NCI-COG pediatric MATCH APEC1621C. J. Natl Cancer Inst. 115, 1355–1363 (2023).
Ruiz-Cordero, R. et al. Hybrid oncocytic/chromophobe renal tumors are molecularly distinct from oncocytoma and chromophobe renal cell carcinoma. Mod. Pathol. 32, 1698–1707 (2019).
Mayr, J. A. et al. Loss of complex I due to mitochondrial DNA mutations in renal oncocytoma. Clin. Cancer Res. 14, 2270–2275 (2008).
Joshi, S. et al. The genomic landscape of renal oncocytoma identifies a metabolic barrier to tumorigenesis. Cell Rep. 13, 1895–1908 (2015).
Durinck, S. et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat. Genet. 47, 13–21 (2015).
Abualjadayel, M. H. et al. A rare benign tumor in a 14-year-old girl. Case Rep. Nephrol. 2018, 1548283 (2018).
Speer, S., Wiseman, D., Moussa, M. & Bütter, A. Renal oncocytosis in a pediatric patient: case report and review of the literature. J. Pediatr. Surg. Case Rep. 3, 481–484 (2015).
Wei, X., Wang, Y., Fang, Y. & Chen, L. Renal oncocytoma in a 13-year-old girl: a case report and literature review. Indian J. Pathol. Microbiol. 66, 868–870 (2023).
Waisman, J. & Löwhagen, T. Oncocytic renal tubular adenoma (so-called oncocytoma) in seventeen-year-old girl. Urology 36, 449–451 (1990).
Suherman, S. et al. Multiple renal oncocytoma and bilateral cystic kidney. Eur. J. Pediatr. 144, 406–409 (1985).
Seyedzadeh, A., Parashar, K., Raafat, F., Alton, H. M. & Milford, D. V. Bilateral multifocal renal oncocytoma. Pediatr. Nephrol. 18, 1286–1288 (2003).
Ciftci, A. O. et al. Renal oncocytoma: diagnostic and therapeutic aspects. J. Pediatr. Surg. 35, 1396–1398 (2000).
Gibson, A. & Ray, A. Rare case of hybrid oncocytoma and chromophobe renal cell carcinoma in a pediatric patient. Pediatr. Blood Cancer 63, 1127 (2016).
Kesik, V. et al. A rare type of renal cell carcinoma in a girl: hybrid renal cell carcinoma. Pediatr. Hematol. Oncol. 27, 228–232 (2010).
Beckwith, J. B. & Palmer, N. F. Histopathology and prognosis of Wilms tumors: results from the First National Wilms’ Tumor Study. Cancer 41, 1937–1948 (1978).
Haas, J. E., Palmer, N. F., Weinberg, A. G. & Beckwith, J. B. Ultrastructure of malignant rhabdoid tumor of the kidney: a distinctive renal tumor of children. Hum. Pathol. 12, 646–657 (1981).
Custers, L. et al. Somatic mutations and single-cell transcriptomes reveal the root of malignant rhabdoid tumours. Nat. Commun. 12, 1407 (2021).
Weeks, D. A., Beckwith, J. B., Mierau, G. W. & Luckey, D. W. Rhabdoid tumor of kidney. A report of 111 cases from the National Wilms’ Tumor Study Pathology Center. Am. J. Surg. Pathol. 13, 439–458 (1989).
Van Den Heuvel‐Eibrink, M. M. et al. Malignant rhabdoid tumours of the kidney (MRTKs), registered on recent SIOP protocols from 1993 to 2005: a report of the SIOP renal tumour study group. Pediatr. Blood Cancer 56, 733–737 (2011).
Tomlinson, G. E. et al. Rhabdoid tumor of the kidney in the National Wilms’ Tumor Study: age at diagnosis as a prognostic factor. J. Clin. Oncol. 23, 7641–7645 (2005).
Abstracts From the 49th Congress of the International Society of Paediatric Oncology (SIOP) Washington, DC, USA October 12–15, 2017. Pediatr. Blood Cancer 64, e26772 (2017).
Kieran, M. W. et al. Absence of oncogenic canonical pathway mutations in aggressive pediatric rhabdoid tumors. Pediatr. Blood Cancer 59, 1155–1157 (2012).
Holsten, T. et al. Germline variants in SMARCB1 and other members of the BAF chromatin-remodeling complex across human disease entities: a meta-analysis. Eur. J. Hum. Genet. 26, 1083–1093 (2018).
Eaton, K. W., Tooke, L. S., Wainwright, L. M., Judkins, A. R. & Biegel, J. A. Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr. Blood Cancer 56, 7–15 (2011).
Kordes, U. et al. Clinical and molecular features in patients with atypical teratoid rhabdoid tumor or malignant rhabdoid tumor. Genes. Chromosomes Cancer 49, 176–181 (2010).
Brennan, B. et al. Outcome of extracranial malignant rhabdoid tumours in children registered in the European Paediatric Soft Tissue Sarcoma Study Group Non-Rhabdomyosarcoma Soft Tissue Sarcoma 2005 Study-EpSSG NRSTS 2005. Eur. J. Cancer 60, 69–82 (2016).
Schneppenheim, R. et al. Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am. J. Hum. Genet. 86, 279–284 (2010).
Hasselblatt, M. et al. Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am. J. Surg. Pathol. 35, 933–935 (2011).
Ammerlaan, A. C. J. et al. Long-term survival and transmission of INI1-mutation via nonpenetrant males in a family with rhabdoid tumour predisposition syndrome. Br. J. Cancer 98, 474–479 (2008).
Brennan, B., Stiller, C. & Bourdeaut, F. Extracranial rhabdoid tumours: what we have learned so far and future directions. Lancet Oncol. 14, e329–e336 (2013).
Lee, R. S. et al. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J. Clin. Invest. 122, 2983–2988 (2012).
Johann, P. D. et al. Atypical teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell 29, 379–393 (2016).
Chun, H. J. et al. Genome-wide profiles of extra-cranial malignant rhabdoid tumors reveal heterogeneity and dysregulated developmental pathways. Cancer Cell 29, 394–406 (2016).
Chun, H.-J. E. et al. Identification and analyses of extra-cranial and cranial rhabdoid tumor molecular subgroups reveal tumors with cytotoxic T cell infiltration. Cell Rep. 29, 2338–2354.e7 (2019).
Leruste, A. et al. Clonally expanded T cells reveal immunogenicity of rhabdoid tumors. Cancer Cell 36, 597–612.e8 (2019).
Roberts, C. W. M., Galusha, S. A., McMenamin, M. E., Fletcher, C. D. M. & Orkin, S. H. Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc. Natl Acad. Sci. USA 97, 13796–13800 (2000).
Klochendler-Yeivin, A. et al. The murine SNF5/INI1 chromatin remodeling factor is essential for embryonic development and tumor suppression. EMBO Rep. 1, 500–506 (2000).
Guidi, C. J. et al. Disruption of Ini1 leads to peri-implantation lethality and tumorigenesis in mice. Mol. Cell. Biol. 21, 3598–3603 (2001).
Zhang, Z.-K. et al. Cell cycle arrest and repression of cyclin D1 transcription by INI1/hSNF5. Mol. Cell. Biol. 22, 5975–5988 (2002).
Versteege, I., Medjkane, S., Rouillard, D. & Delattre, O. A key role of the hSNF5/INI1 tumour suppressor in the control of the G1-S transition of the cell cycle. Oncogene 21, 6403–6412 (2002).
McKenna, E. S. et al. Epigenetic inactivation of the tumor suppressor BIN1 drives proliferation of SNF5-deficient tumors. Cell Cycle 11, 1956–1965 (2012).
Betz, B. L., Strobeck, M. W., Reisman, D. N., Knudsen, E. S. & Weissman, B. E. Re-expression of hSNF5/INI1/BAF47 in pediatric tumor cells leads to G1 arrest associated with induction of p16ink4a and activation of RB. Oncogene 21, 5193–5203 (2002).
Kia, S. K., Gorski, M. M., Giannakopoulos, S. & Verrijzer, C. P. SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol. Cell. Biol. 28, 3457–3464 (2008).
Gadd, S., Sredni, S. T., Huang, C.-C. & Perlman, E. J. Renal Tumor Committee of the Children’s Oncology Group. Rhabdoid tumor: gene expression clues to pathogenesis and potential therapeutic targets. Lab. Investig. J. Tech. Methods Pathol. 90, 724–738 (2010).
Albanese, P., Belin, M.-F. & Delattre, O. The tumour suppressor hSNF5/INI1 controls the differentiation potential of malignant rhabdoid cells. Eur. J. Cancer 42, 2326–2334 (2006).
Jagani, Z. et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat. Med. 16, 1429–1433 (2010).
Mora-Blanco, E. L. et al. Activation of β-catenin/TCF targets following loss of the tumor suppressor SNF5. Oncogene 33, 933–938 (2014).
Wang, X. et al. TCR-dependent transformation of mature memory phenotype T cells in mice. J. Clin. Invest. 121, 3834–3845 (2011).
Liu, N. Q. et al. SMARCB1 loss activates patient-specific distal oncogenic enhancers in malignant rhabdoid tumors. Nat. Commun. 14, 7762 (2023).
Isakoff, M. S. et al. Inactivation of the Snf5 tumor suppressor stimulates cell cycle progression and cooperates with p53 loss in oncogenic transformation. Proc. Natl Acad. Sci. USA 102, 17745–17750 (2005).
Tsikitis, M., Zhang, Z., Edelman, W., Zagzag, D. & Kalpana, G. V. Genetic ablation of Cyclin D1 abrogates genesis of rhabdoid tumors resulting from Ini1 loss. Proc. Natl Acad. Sci. Usa. 102, 12129–12134 (2005).
Kurmasheva, R. T., Erickson, S. W., Earley, E., Smith, M. A. & Houghton, P. J. In vivo evaluation of the EZH2 inhibitor (EPZ011989) alone or in combination with standard of care cytotoxic agents against pediatric malignant rhabdoid tumor preclinical models — a report from the Pediatric Preclinical Testing Consortium. Pediatr. Blood Cancer 68, e28772 (2021).
Tajbakhsh, M. et al. Initial testing of cisplatin by the pediatric preclinical testing program. Pediatr. Blood Cancer 50, 992–1000 (2008).
Maris, J. M. et al. Initial testing of the aurora kinase A inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP). Pediatr. Blood Cancer 55, 26–34 (2010).
Carol, H. et al. Initial testing of the MDM2 inhibitor RG7112 by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 60, 633–641 (2013).
Kolb, E. A. et al. Initial testing (stage 1) of a monoclonal antibody (SCH 717454) against the IGF-1 receptor by the pediatric preclinical testing program. Pediatr. Blood Cancer 50, 1190–1197 (2008).
Lowery, C. D. et al. Broad spectrum activity of the checkpoint kinase 1 inhibitor prexasertib as a single agent or chemopotentiator across a range of preclinical pediatric tumor models. Clin. Cancer Res. 25, 2278–2289 (2019).
Howard, T. P. et al. MDM2 and MDM4 are therapeutic vulnerabilities in malignant rhabdoid tumors. Cancer Res. 79, 2404–2414 (2019).
Howard, T. P. et al. Rhabdoid tumors are sensitive to the protein-translation inhibitor homoharringtonine. Clin. Cancer Res. 26, 4995–5006 (2020).
Kim, K. H. & Roberts, C. W. M. Mechanisms by which SMARCB1 loss drives rhabdoid tumor growth. Cancer Genet. 207, 365–372 (2014).
Henssen, A. G. et al. Therapeutic targeting of PGBD5-induced DNA repair dependency in pediatric solid tumors. Sci. Transl. Med. 9, eaam9078 (2017).
Chauvin, C. et al. High-throughput drug screening identifies pazopanib and clofilium tosylate as promising treatments for malignant rhabdoid tumors. Cell Rep. 21, 1737–1745 (2017).
Calandrini, C. et al. Organoid-based drug screening reveals neddylation as therapeutic target for malignant rhabdoid tumors. Cell Rep. 36, 109568 (2021).
Morin, A. et al. Proteasome inhibition as a therapeutic approach in atypical teratoid/rhabdoid tumors. Neurooncol. Adv. 2, vdaa051 (2020).
Wang, X. et al. BRD9 defines a SWI/SNF sub-complex and constitutes a specific vulnerability in malignant rhabdoid tumors. Nat. Commun. 10, 1881 (2019).
Radko-Juettner, S. et al. Targeting DCAF5 suppresses SMARCB1-mutant cancer by stabilizing SWI/SNF. Nature 628, 442–449 (2024).
Knutson, S. K. et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl Acad. Sci. Usa. 110, 7922–7927 (2013).
Margueron, R. & Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349 (2011).
Cao, R. et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298, 1039–1043 (2002).
Chang, C.-J. & Hung, M.-C. The role of EZH2 in tumour progression. Br. J. Cancer 106, 243–247 (2012).
Drosos, Y. et al. NSD1 mediates antagonism between SWI/SNF and polycomb complexes and is required for transcriptional activation upon EZH2 inhibition. Mol. Cell 82, 2472–2489.e8 (2022).
Kazansky, Y. et al. Overcoming clinical resistance to EZH2 inhibition using rational epigenetic combination therapy. Cancer Discov. 14, 965–981 (2024).
Argani, P. et al. Clear cell sarcoma of the kidney: a review of 351 cases from the National Wilms Tumor Study Group Pathology Center. Am. J. Surg. Pathol. 24, 4–18 (2000).
Suzuki, H. et al. Clear-cell sarcoma of the kidney seen in a 3-day-old newborn. Z. Kinderchir. 38, 422–424 (1983).
Cao, M., Zhang, J., Ma, H. & Liang, Y. Clear cell sarcoma of the kidney in an adult: a case report and literature review. Transl. Cancer Res. 11, 288–294 (2022).
Kidd, J. M. Exclusion of certain renal neoplasms from the category of Wilms’ tumor (abstract). Am. J. Pathol. 59, 16a (1970).
Morgan, E. & Kidd, J. M. Undifferentiated sarcoma of the kidney: a tumor of childhood with histopathologic and clinical characteristics distinct from Wilms’ tumor. Cancer 42, 1916–1921 (1978).
Benedetti, D. J. et al. Treatment and outcomes of clear cell sarcoma of the kidney: a report from the Children’s Oncology Group studies AREN0321 and AREN03B2. Cancer 130, 2361–2371 (2024).
Marsden, H. B. & Lawler, W. Bone-metastasizing renal tumour of childhood. Br. J. Cancer 38, 437–441 (1978).
Manchanda, V., Mohta, A., Khurana, N., Gupta, C. R. & Neogi, S. Bilateral clear cell sarcoma of the kidney. J. Pediatr. Surg. 45, 1927–1930 (2010).
Zekri, W. et al. Bilateral clear cell sarcoma of the kidney. J. Egypt. Natl Cancer Inst. 27, 97–100 (2015).
Punnett, H. H., Halligan, G. E., Zaeri, N. & Karmazin, N. Translocation 10;17 in clear cell sarcoma of the kidney. A first report. Cancer Genet. Cytogenet. 41, 123–128 (1989).
Rakheja, D., Weinberg, A. G., Tomlinson, G. E., Partridge, K. & Schneider, N. R. Translocation (10;17)(q22;p13): a recurring translocation in clear cell sarcoma of kidney. Cancer Genet. Cytogenet. 154, 175–179 (2004).
Brownlee, N. A. et al. Recurring translocation (10;17) and deletion (14q) in clear cell sarcoma of the kidney. Arch. Pathol. Lab. Med. 131, 446–451 (2007).
O’Meara, E. et al. Characterization of the chromosomal translocation t(10;17)(q22;p13) in clear cell sarcoma of kidney. J. Pathol. 227, 72–80 (2012).
Karlsson, J. et al. Activation of human telomerase reverse transcriptase through gene fusion in clear cell sarcoma of the kidney. Cancer Lett. 357, 498–501 (2015).
Lee, C.-H. et al. 14-3-3 fusion oncogenes in high-grade endometrial stromal sarcoma. Proc. Natl Acad. Sci. USA 109, 929–934 (2012).
Kenny, C. et al. Dysregulated mitogen-activated protein kinase signalling as an oncogenic basis for clear cell sarcoma of the kidney. J. Pathol. 244, 334–345 (2018).
Ueno-Yokohata, H. et al. Consistent in-frame internal tandem duplications of BCOR characterize clear cell sarcoma of the kidney. Nat. Genet. 47, 861–863 (2015).
Roy, A. et al. Recurrent internal tandem duplications of BCOR in clear cell sarcoma of the kidney. Nat. Commun. 6, 8891 (2015).
Astolfi, A. et al. Whole transcriptome sequencing identifies BCOR internal tandem duplication as a common feature of clear cell sarcoma of the kidney. Oncotarget 6, 40934–40939 (2015).
Kenny, C. et al. Mutually exclusive BCOR internal tandem duplications and YWHAE-NUTM2 fusions in clear cell sarcoma of kidney: not the full story. J. Pathol. 238, 617–620 (2016).
Argani, P. et al. Primary renal sarcomas with BCOR-CCNB3 gene fusion: a report of 2 cases showing histologic overlap with clear cell sarcoma of kidney, suggesting further link between BCOR-related sarcomas of the kidney and soft tissues. Am. J. Surg. Pathol. 41, 1702–1712 (2017).
Han, H. et al. BCOR-CCNB3 fusion-positive clear cell sarcoma of the kidney. Pediatr. Blood Cancer 67, e28151 (2020).
Dorwal, P. et al. Clear cell sarcoma of the kidney (CCSK) with BCOR-CCNB3 fusion: a rare case report with a brief review of the literature. Pediatr. Dev. Pathol. 26, 149–152 (2023).
Wong, M. K. et al. Clear cell sarcomas of the kidney are characterised by BCOR gene abnormalities, including exon 15 internal tandem duplications and BCOR-CCNB3 gene fusion. Histopathology 72, 320–329 (2018).
Santiago, T. et al. Clear cell sarcoma of kidney involving a horseshoe kidney and harboring EGFR internal tandem duplication. Pediatr. Blood Cancer 64, e26602 (2017).
Kao, Y.-C. et al. Recurrent BCOR internal tandem duplication and YWHAE-NUTM2B fusions in soft tissue undifferentiated round cell sarcoma of infancy: overlapping genetic features with clear cell sarcoma of kidney. Am. J. Surg. Pathol. 40, 1009–1020 (2016).
Antonescu, C. R. et al. Undifferentiated round cell sarcoma with BCOR internal tandem duplications (ITD) or YWHAE fusions: a clinicopathologic and molecular study. Mod. Pathol. 33, 1669–1677 (2020).
Fehr, A., Hansson, M. C., Kindblom, L.-G. & Stenman, G. YWHAE-FAM22 gene fusion in clear cell sarcoma of the kidney. J. Pathol. 227, e5–e7 (2012).
Weeks, D. A., Malott, R. L., Zuppan, C., Mierau, G. W. & Beckwith, J. B. Primitive pelvic sarcoma resembling clear cell sarcoma of kidney. Ultrastruct. Pathol. 15, 403–408 (1991).
Karlsson, J. et al. Aberrant epigenetic regulation in clear cell sarcoma of the kidney featuring distinct DNA hypermethylation and EZH2 overexpression. Oncotarget 7, 11127–11136 (2016).
Ueno, H. et al. DNA methylation profile distinguishes clear cell sarcoma of the kidney from other pediatric renal tumors. PLoS ONE 8, e62233 (2013).
Gooskens, S. L. et al. TCF21 hypermethylation in genetically quiescent clear cell sarcoma of the kidney. Oncotarget 6, 15828–15841 (2015).
Huang, C.-C. et al. Classification of malignant pediatric renal tumors by gene expression. Pediatr. Blood Cancer 46, 728–738 (2006).
Cutcliffe, C. et al. Clear cell sarcoma of the kidney: up-regulation of neural markers with activation of the sonic hedgehog and Akt pathways. Clin. Cancer Res. 11, 7986–7994 (2005).
Karlsson, J. et al. Clear cell sarcoma of the kidney demonstrates an embryonic signature indicative of a primitive nephrogenic origin. Genes. Chromosomes Cancer 53, 381–391 (2014).
Kao, Y.-C. et al. NTRK3 overexpression in undifferentiated sarcomas with YWHAE and BCOR genetic alterations. Mod. Pathol. 33, 1341–1349 (2020).
Fiore, M. et al. Molecular signature of biological aggressiveness in clear cell sarcoma of the kidney (CCSK). Int. J. Mol. Sci. 24, 3743 (2023).
Balarezo, F. S. & Joshi, V. V. Clear cell sarcoma of the pediatric kidney: detailed description and analysis of variant histologic patterns of a tumor with many faces. Adv. Anat. Pathol. 8, 98–108 (2001).
Kao, Y.-C. et al. BCOR overexpression is a highly sensitive marker in round cell sarcomas with BCOR genetic abnormalities. Am. J. Surg. Pathol. 40, 1670–1678 (2016).
Argani, P. et al. Diffuse strong BCOR immunoreactivity is a sensitive and specific marker for clear cell sarcoma of the kidney (CCSK) in pediatric renal neoplasia. Am. J. Surg. Pathol. 42, 1128–1131 (2018).
Kenny, C. et al. Immunophenotype-genotype correlations in clear cell sarcoma of kidney-an evaluation of diagnostic ancillary studies. Pediatr. Dev. Pathol. 23, 345–351 (2020).
Furtwängler, R. et al. Clear cell sarcomas of the kidney registered on International Society of Pediatric Oncology (SIOP) 93-01 and SIOP 2001 protocols: a report of the SIOP Renal Tumour Study Group. Eur. J. Cancer 49, 3497–3506 (2013).
England, R. J. et al. Mesoblastic nephroma: a report of the United Kingdom Children’s Cancer and Leukaemia Group (CCLG). Pediatr. Blood Cancer 56, 744–748 (2011).
Gooskens, S. L. et al. Congenital mesoblastic nephroma 50 years after its recognition: a narrative review. Pediatr. Blood Cancer 64, e26437 (2017).
Ganick, D. J., Gilbert, E. F., Beckwith, J. B. & Kiviat, N. Congenital cystic mesoblastic nephroma. Hum. Pathol. 12, 1039–1043 (1981).
O’Malley, D. P., Mierau, G. W., Beckwith, J. B. & Weeks, D. A. Ultrastructure of cellular congenital mesoblastic nephroma. Ultrastruct. Pathol. 20, 417–427 (1996).
Nadasdy, T. et al. Congenital mesoblastic nephroma: an immunohistochemical and lectin study. Hum. Pathol. 24, 413–419 (1993).
Shao, L., Hill, D. A. & Perlman, E. J. Expression of WT-1, Bcl-2, and CD34 by primary renal spindle cell tumors in children. Pediatr. Dev. Pathol. 7, 577–582 (2004).
Arva, N. C., Bonadio, J., Perlman, E. J. & Cajaiba, M. M. Diagnostic utility of Pax8, Pax2, and NGFR immunohistochemical expression in pediatric renal tumors. Appl. Immunohistochem. Mol. Morphol. 26, 721–726 (2018).
Bolande, R. P., Brough, A. J. & Izant, R. J. Congenital mesoblastic nephroma of infancy. A report of eight cases and the relationship to Wilms’ tumor. Pediatrics 40, 272–278 (1967).
Beckwith, J. B. Mesenchymal renal neoplasms of infancy revisited. J. Pediatr. Surg. 9, 803–805 (1974).
Joshi, V. V., Kasznica, J. & Walters, T. R. Atypical mesoblastic nephroma. Pathologic characterization of a potentially aggressive variant of conventional congenital mesoblastic nephroma. Arch. Pathol. Lab. Med. 110, 100–106 (1986).
Argani, P. & Beckwith, J. B. Metanephric stromal tumor: report of 31 cases of a distinctive pediatric renal neoplasm. Am. J. Surg. Pathol. 24, 917–926 (2000).
Argani, P. et al. Frequent BRAF V600E mutations in metanephric stromal tumor. Am. J. Surg. Pathol. 40, 719–722 (2016).
Rudzinski, E. R. et al. Pan-Trk immunohistochemistry identifies NTRK rearrangements in pediatric mesenchymal tumors. Am. J. Surg. Pathol. 42, 927–935 (2018).
Misove, A. et al. An unusual fusion gene EML4-ALK in a patient with congenital mesoblastic nephroma. Genes. Chromosomes Cancer 60, 837–840 (2021).
Wegert, J. et al. Recurrent intragenic rearrangements of EGFR and BRAF in soft tissue tumors of infants. Nat. Commun. 9, 2378 (2018).
Church, A. J. et al. Recurrent EML4–NTRK3 fusions in infantile fibrosarcoma and congenital mesoblastic nephroma suggest a revised testing strategy. Mod. Pathol. 31, 463–473 (2018).
Hornick, J. L. Replacing molecular genetic testing with immunohistochemistry using antibodies that recognize the protein products of gene rearrangements: ‘next-generation’ immunohistochemistry. Am. J. Surg. Pathol. 45, 584–586 (2021).
Vokuhl, C. et al. ETV6–NTRK3 in congenital mesoblastic nephroma: a report of the SIOP/GPOH nephroblastoma study. Pediatr. Blood Cancer 65, e26925 (2018).
El Demellawy, D. et al. Congenital mesoblastic nephroma: a study of 19 cases using immunohistochemistry and ETV6-NTRK3 fusion gene rearrangement. Pathology 48, 47–50 (2016).
Anderson, J., Gibson, S. & Sebire, N. J. Expression of ETV6-NTRK in classical, cellular and mixed subtypes of congenital mesoblastic nephroma. Histopathology 48, 748–753 (2006).
Rubin, B. P. et al. Congenital mesoblastic nephroma t(12;15) is associated with ETV6-NTRK3 gene fusion: cytogenetic and molecular relationship to congenital (infantile) fibrosarcoma. Am. J. Pathol. 153, 1451–1458 (1998).
Knezevich, S. R. et al. ETV6-NTRK3 gene fusions and trisomy 11 establish a histogenetic link between mesoblastic nephroma and congenital fibrosarcoma. Cancer Res. 58, 5046–5048 (1998).
van Spronsen, R. et al. Infantile fibrosarcoma with an EGFR kinase ___domain duplication: underlining a close relationship with congenital mesoblastic nephroma and highlighting a similar morphological spectrum. Ann. Diagn. Pathol. 57, 151885 (2022).
Vanoli, F. et al. Generating in vitro models of NTRK-fusion mesenchymal neoplasia as tools for investigating kinase oncogenic activation and response to targeted therapy. Oncogenesis 12, 8 (2023).
Tongsong, T., Palangmonthip, W., Chankhunaphas, W. & Luewan, S. Prenatal course and sonographic features of congenital mesoblastic nephroma. Diagnostics 12, 1951 (2022).
Siemer, S. et al. Prenatal diagnosis of congenital mesoblastic nephroma associated with renal hypertension in a premature child. Int. J. Urol. 11, 50–52 (2004).
Montaruli, E. & Fouquet, V. Prenatal diagnosis of congenital mesoblastic nephroma. Fetal Diagn. Ther. 33, 79–80 (2013).
Chen, W.-Y. et al. Prenatal diagnosis of congenital mesoblastic nephroma in mid-second trimester by sonography and magnetic resonance imaging. Prenat. Diagn. 23, 927–931 (2003).
Woolfield, N. F., Abbott, G. D. & McRae, C. U. A mesoblastic nephroma with hypercalcaemia. Aust. Paediatr. J. 24, 309–310 (1988).
Fung, T. Y., Fung, Y. M., Ng, P. C., Yeung, C. K. & Chang, M. Z. Polyhydramnios and hypercalcemia associated with congenital mesoblastic nephroma: case report and a new appraisal. Obstet. Gynecol. 85, 815–817 (1995).
Jayabose, S. et al. Hypercalcemia in childhood renal tumors. Cancer 61, 788–791 (1988).
Malone, P. S. et al. Congenital mesoblastic nephroma, renin production, and hypertension. J. Pediatr. Surg. 24, 599–600 (1989).
Cook, H. T., Taylor, G. M., Malone, P. & Risdon, R. A. Renin in mesoblastic nephroma: an immunohistochemical study. Hum. Pathol. 19, 1347–1351 (1988).
Loeb, D. M., Hill, D. A. & Dome, J. S. Complete response of recurrent cellular congenital mesoblastic nephroma to chemotherapy. J. Pediatr. Hematol. Oncol. 24, 478–481 (2002).
Jehangir, S., Kurian, J. J., Selvarajah, D., Thomas, R. J. & Holland, A. J. A. Recurrent and metastatic congenital mesoblastic nephroma: where does the evidence stand? Pediatr. Surg. Int. 33, 1183–1188 (2017).
Orbach, D. et al. Spotlight on the treatment of infantile fibrosarcoma in the era of neurotrophic tropomyosin receptor kinase inhibitors: international consensus and remaining controversies. Eur. J. Cancer 137, 183–192 (2020).
Nagasubramanian, R. et al. Infantile fibrosarcoma with NTRK3–ETV6 fusion successfully treated with the tropomyosin-related kinase inhibitor LOXO-101. Pediatr. Blood Cancer 63, 1468–1470 (2016).
Laetsch, T. W. et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 19, 705–714 (2018).
DuBois, S. G. et al. The use of neoadjuvant larotrectinib in the management of children with locally advanced TRK fusion sarcomas. Cancer 124, 4241–4247 (2018).
Treece, A. L. Pediatric renal tumors: updates in the molecular era. Surg. Pathol. Clin. 13, 695–718 (2020).
Li, Y., Pawel, B. R., Hill, D. A., Epstein, J. I. & Argani, P. Pediatric cystic nephroma is morphologically, immunohistochemically, and genetically distinct from adult cystic nephroma. Am. J. Surg. Pathol. 41, 472–481 (2017).
Schultz, K. A. P. et al. DICER1 and associated conditions: identification of at-risk individuals and recommended surveillance strategies. Clin. Cancer Res. 24, 2251–2261 (2018).
Guillerman, R. P., Foulkes, W. D. & Priest, J. R. Imaging of DICER1 syndrome. Pediatr. Radiol. 49, 1488–1505 (2019).
van Peer, S. E. et al. Clinical and molecular characteristics and outcome of cystic partially differentiated nephroblastoma and cystic nephroma: a narrative review of the literature. Cancers 13, 997 (2021).
Wu, M. K. et al. Tumor progression in DICER1-mutated cystic nephroma-witnessing the genesis of anaplastic sarcoma of the kidney. Hum. Pathol. 53, 114–120 (2016).
Kao, J.-L., Tsung, S.-H. & Shiao, C.-C. Rare anaplastic sarcoma of the kidney: a case report. World J. Clin. Cases 8, 1495–1501 (2020).
Chen, C.-C. & Liao, K.-S. Anaplastic sarcoma of the kidney: case report and literature review. Tzu Chi Med. J. 31, 129–132 (2019).
Arabi, H., Al-Maghraby, H., Yamani, A., Yousef, Y. & Huwait, H. Anaplastic sarcoma of the kidney: a rare unique renal neoplasm. Int. J. Surg. Pathol. 24, 556–561 (2016).
Vujanić, G. M., Kelsey, A., Perlman, E. J., Sandstedt, B. & Beckwith, J. B. Anaplastic sarcoma of the kidney: a clinicopathologic study of 20 cases of a new entity with polyphenotypic features. Am. J. Surg. Pathol. 31, 1459–1468 (2007).
Argani, P. et al. Primary renal synovial sarcoma: molecular and morphologic delineation of an entity previously included among embryonal sarcomas of the kidney. Am. J. Surg. Pathol. 24, 1087–1096 (2000).
Thorner, P. S. et al. PRAME protein expression in DICER1-related tumours. J. Pathol. Clin. Res. 8, 294–304 (2022).
Wu, M. K. et al. Evolution of renal cysts to anaplastic sarcoma of kidney in a child with DICER1 syndrome. Pediatr. Blood Cancer 63, 1272–1275 (2016).
Wu, M. K. et al. Anaplastic sarcomas of the kidney are characterized by DICER1 mutations. Mod. Pathol. 31, 169–178 (2018).
Doros, L. A. et al. DICER1 mutations in childhood cystic nephroma and its relationship to DICER1-renal sarcoma. Mod. Pathol. 27, 1267–1280 (2014).
Kroll-Wheeler, L. & Heider, A. Anaplastic sarcoma of the kidney with heterologous ganglioneuroblastic differentiation: another DICER1-associated tumor. Pediatr. Dev. Pathol. 25, 186–191 (2022).
Galliani, C. A., Bisceglia, M., Del Giudice, A. & Cretì, G. Desmoplastic small round cell tumor of the kidney: report of a case, literature review, and comprehensive discussion of the distinctive morphologic, immunohistochemical, and molecular features in the differential diagnosis of small round cell tumors affecting the kidney. Adv. Anat. Pathol. 27, 408–421 (2020).
Ertoy Baydar, D., Armutlu, A., Aydin, O., Dagdemir, A. & Yakupoglu, Y. K. Desmoplastic small round cell tumor of the kidney: a case report. Diagn. Pathol. 15, 95 (2020).
Eklund, M. J., Cundiff, C., Shehata, B. M. & Alazraki, A. L. Desmoplastic small round cell tumor of the kidney with unusual imaging features. Clin. Imaging 39, 904–907 (2015).
Rao, P., Tamboli, P., Fillman, E. P. & Meis, J. M. Primary intra-renal desmoplastic small round cell tumor: expanding the histologic spectrum, with special emphasis on the differential diagnostic considerations. Pathol. Res. Pract. 210, 1130–1133 (2014).
da Silva, R. C. et al. Desmoplastic small round cell tumor of the kidney mimicking Wilms tumor: a case report and review of the literature. Appl. Immunohistochem. Mol. Morphol. 17, 557–562 (2009).
Eaton, S. H. & Cendron, M. A. Primary desmoplastic small round cell tumor of the kidney in a 7-year-old girl. J. Pediatr. Urol. 2, 52–54 (2006).
Collardeau-Frachon, S. et al. Primary desmoplastic small round cell tumor of the kidney: a case report in a 14-year-old girl with molecular confirmation. Pediatr. Dev. Pathol. 10, 320–324 (2007).
Wang, L. L. et al. Desmoplastic small round cell tumor of the kidney in childhood. Am. J. Surg. Pathol. 31, 576–584 (2007).
Barnoud, R. et al. Immunohistochemical expression of WT1 by desmoplastic small round cell tumor: a comparative study with other small round cell tumors. Am. J. Surg. Pathol. 24, 830–836 (2000).
Gerald, W. L. et al. Clinical, pathologic, and molecular spectrum of tumors associated with t(11;22)(p13;q12): desmoplastic small round-cell tumor and its variants. J. Clin. Oncol. 16, 3028–3036 (1998).
Magrath, J. W. et al. Comprehensive transcriptomic analysis of EWSR1::WT1 targets identifies CDK4/6 inhibitors as an effective therapy for desmoplastic small round cell tumors. Cancer Res. 84, 1426–1442 (2024).
Slotkin, E. K. et al. Comprehensive molecular profiling of desmoplastic small round cell tumor. Mol. Cancer Res. 19, 1146–1155 (2021).
Chow, W. A. et al. Recurrent secondary genomic alterations in desmoplastic small round cell tumors. BMC Med. Genet. 21, 101 (2020).
Honoré, C. et al. Can we cure patients with abdominal desmoplastic small round cell tumor? Results of a retrospective multicentric study on 100 patients. Surg. Oncol. 29, 107–112 (2019).
Quezado, M., Benjamin, D. R. & Tsokos, M. EWS/FLI-1 fusion transcripts in three peripheral primitive neuroectodermal tumors of the kidney. Hum. Pathol. 28, 767–771 (1997).
Rodriguez-Galindo, C. et al. Is primitive neuroectodermal tumor of the kidney a distinct entity? Cancer 79, 2243–2250 (1997).
Liang, L. et al. Renal Ewing’s sarcoma/primitive neuroectodermal tumor (PNET): a case series of 7 patients and literature review. Transl. Androl. Urol. 10, 548–554 (2021).
Qureshi, S. S. et al. Incidence, treatment, and outcomes of primary and recurrent Non-Wilms renal tumors in children: report of 109 patients treated at a single institution. J. Pediatr. Urol. 16, 475.e1–475.e9 (2020).
Murugan, P., Rao, P., Tamboli, P., Czerniak, B. & Guo, C. C. Primary Ewing sarcoma/primitive neuroectodermal tumor of the kidney: a clinicopathologic study of 23 cases. Pathol. Oncol. Res. 24, 153–159 (2018).
Zöllner, S., Dirksen, U., Jürgens, H. & Ranft, A. Renal Ewing tumors. Ann. Oncol. 24, 2455–2461 (2013).
Rowe, R. G. et al. Ewing sarcoma of the kidney: case series and literature review of an often overlooked entity in the diagnosis of primary renal tumors. Urology 81, 347–353 (2013).
Karpate, A. et al. Ewing sarcoma/primitive neuroectodermal tumor of the kidney: clinicopathologic analysis of 34 cases. Ann. Diagn. Pathol. 16, 267–274 (2012).
Popov, S. D., Sebire, N. J., Pritchard-Jones, K. & Vujanić, G. M. Renal tumors in children aged 10–16 years: a report from the United Kingdom Children’s Cancer and Leukaemia Group. Pediatr. Dev. Pathol. 14, 189–193 (2011).
Thyavihally, Y. B. et al. Primitive neuroectodermal tumor of the kidney: a single institute series of 16 patients. Urology 71, 292–296 (2008).
Ellison, D. A., Parham, D. M., Bridge, J. & Beckwith, J. B. Immunohistochemistry of primary malignant neuroepithelial tumors of the kidney: a potential source of confusion? A study of 30 cases from the National Wilms Tumor Study Pathology Center. Hum. Pathol. 38, 205–211 (2007).
Jimenez, R. E. et al. Primary Ewing’s sarcoma/primitive neuroectodermal tumor of the kidney: a clinicopathologic and immunohistochemical analysis of 11 cases. Am. J. Surg. Pathol. 26, 320–327 (2002).
Parham, D. M. et al. Primary malignant neuroepithelial tumors of the kidney: a clinicopathologic analysis of 146 adult and pediatric cases from the National Wilms’ Tumor Study Group Pathology Center. Am. J. Surg. Pathol. 25, 133–146 (2001).
Tarek, N. et al. Primary Ewing sarcoma/primitive neuroectodermal tumor of the kidney: the MD Anderson Cancer Center Experience. Cancers 12, 2927 (2020).
Lalwani, N. et al. Pediatric and adult primary sarcomas of the kidney: a cross-sectional imaging review. Acta Radiol. 52, 448–457 (2011).
Radhakrishnan, V. et al. Synovial sarcoma of kidney in a child: a rare presentation. J. Indian. Assoc. Pediatr. Surg. 21, 75–77 (2016).
Mastoraki, A. et al. Primary synovial sarcoma of the kidney: diagnostic approach and therapeutic modalities for a rare nosological entity. J. Pers. Med. 12, 1450 (2022).
Feng, X. et al. The role of SYT-SSX fusion gene in tumorigenesis of synovial sarcoma. Pathol. Res. Pract. 222, 153416 (2021).
Kadoch, C. & Crabtree, G. R. Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell 153, 71–85 (2013).
Li, J. et al. A role for SMARCB1 in synovial sarcomagenesis reveals that SS18–SSX induces canonical BAF destruction. Cancer Discov. 11, 2620–2637 (2021).
Brien, G. L. et al. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. eLife 7, e41305 (2018).
Michel, B. C. et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat. Cell Biol. 20, 1410–1420 (2018).
Livingston, J. A. et al. A phase 1 study of FHD-609, a heterobifunctional degrader of bromodomain-containing protein 9, in patients with advanced synovial sarcoma or SMARCB1-deficient tumors. Clin. Cancer Res. https://doi.org/10.1158/1078-0432.CCR-24-2583 (2024).
de Jel, D. V. C. et al. Paediatric metanephric tumours: a clinicopathological and molecular characterisation. Crit. Rev. Oncol. Hematol. 150, 102970 (2020).
Arroyo, M. R., Green, D. M., Perlman, E. J., Beckwith, J. B. & Argani, P. The spectrum of metanephric adenofibroma and related lesions: clinicopathologic study of 25 cases from the National Wilms Tumor Study Group Pathology Center. Am. J. Surg. Pathol. 25, 433–444 (2001).
Chami, R. et al. BRAF mutations in pediatric metanephric tumors. Hum. Pathol. 46, 1153–1161 (2015).
Choueiri, T. K. et al. BRAF mutations in metanephric adenoma of the kidney. Eur. Urol. 62, 917–922 (2012).
Fejes, Z. et al. Angiomyolipoma of the kidney-clinicopathological analysis of 52 cases. Pathol. Oncol. Res. 28, 1610831 (2022).
Warncke, J. C. et al. Pediatric renal angiomyolipomas in tuberous sclerosis complex. J. Urol. 197, 500–506 (2017).
Bissler, J. J. & Kingswood, J. C. Renal angiomyolipomata. Kidney Int. 66, 924–934 (2004).
Dabora, S. L. et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am. J. Hum. Genet. 68, 64–80 (2001).
Muto, Y. et al. Genotype-phenotype correlation of renal lesions in the tuberous sclerosis complex. Hum. Genome Var. 9, 1–5 (2022).
Martin, K. R. et al. The genomic landscape of tuberous sclerosis complex. Nat. Commun. 8, 15816 (2017).
Henske, E. P. et al. Loss of heterozygosity in the tuberous sclerosis (TSC2) region of chromosome band 16p13 occurs in sporadic as well as TSC-associated renal angiomyolipomas. Genes. Chromosomes Cancer 13, 295–298 (1995).
Qin, W. et al. Angiomyolipoma have common mutations in TSC2 but no other common genetic events. PLoS ONE 6, e24919 (2011).
Agaram, N. P. et al. Dichotomy of genetic abnormalities in PEComas with therapeutic implications. Am. J. Surg. Pathol. 39, 813–825 (2015).
Malinowska, I. et al. Perivascular epithelioid cell tumors (PEComas) harboring TFE3 gene rearrangements lack the TSC2 alterations characteristic of conventional PEComas: further evidence for a biological distinction. Am. J. Surg. Pathol. 36, 783–784 (2012).
Garami, A. et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 11, 1457–1466 (2003).
Dibble, C. C. & Cantley, L. C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 25, 545–555 (2015).
Pleniceanu, O., Omer, D., Azaria, E., Harari-Steinberg, O. & Dekel, B. mTORC1 inhibition is an effective treatment for sporadic renal angiomyolipoma. Kidney Int. Rep. 3, 155–159 (2018).
Bissler, J. J. et al. Everolimus long-term use in patients with tuberous sclerosis complex: four-year update of the EXIST-2 study. PLoS ONE 12, e0180939 (2017).
Banerjee, A., Reddy K, A., Munghate, G., Bodhanwala, M. & Bendre, P. S. Ossifying renal tumor of infancy — a case report. J. Pediatr. Surg. Case Rep. 93, 102650 (2023).
Schelling, J., Schröder, A., Stein, R. & Rösch, W. H. Ossifying renal tumor of infancy. J. Pediatr. Urol. 3, 258–261 (2007).
Lee, S. H., Choi, Y. H., Kim, W. S., Cheon, J.-E. & Moon, K. C. Ossifying renal tumor of infancy: findings at ultrasound, CT and MRI. Pediatr. Radiol. 44, 625–628 (2014).
Sotelo-Avila, C., Beckwith, J. B. & Johnson, J. E. Ossifying renal tumor of infancy: a clinicopathologic study of nine cases. Pediatr. Pathol. Lab. Med. 15, 745–762 (1995).
Liu, J. et al. Clonal trisomy 4 cells detected in the ossifying renal tumor of infancy: study of 3 cases. Mod. Pathol. 26, 275–281 (2013).
Hu, J., Wu, Y., Qi, J., Zhang, C. & Lv, F. Ossifying renal tumor of infancy (ORTI): a case report and review of the literature. J. Pediatr. Surg. 48, e37–e40 (2013).
Inam, R., Gandhi, J., Joshi, G., Smith, N. L. & Khan, S. A. Juxtaglomerular cell tumor: reviewing a cryptic cause of surgically correctable hypertension. Curr. Urol. 13, 7–12 (2019).
Trnka, P., Orellana, L., Walsh, M., Pool, L. & Borzi, P. Reninoma: an uncommon cause of renin-mediated hypertension. Front. Pediatr. 2, 89 (2014).
Kuroda, N. et al. Juxtaglomerular cell tumor: a morphological, immunohistochemical and genetic study of six cases. Hum. Pathol. 44, 47–54 (2013).
Treger, T. D. et al. Targetable NOTCH1 rearrangements in reninoma. Nat. Commun. 14, 5826 (2023).
Author information
Authors and Affiliations
Contributions
All authors researched data for the article. A.L.H., D.P., M.J.O.S., A.L.W., J.D., J.I.G. and M.M.V.D.H.E. contributed substantially to discussion of the content. All authors wrote the article. A.L.H., D.P., M.J.O.S., A.L.W., J.D., D.J.B., N.G.C., J.S.D., E.A.M., A.J.M., M.V.O., J.N.V.D.B., J.W., F.S., J.I.G. and M.M.V.D.H.E. reviewed and/or edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
N.G.C.’s spouse is a Senior Medical Officer for Jannsen. The other authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Urology thanks Harold Lovvorn and the other, anonymous, reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Perotti, D., O’Sullivan, M.J., Walz, A.L. et al. Hallmark discoveries in the biology of non-Wilms tumour childhood kidney cancers. Nat Rev Urol (2025). https://doi.org/10.1038/s41585-024-00993-6
Accepted:
Published:
DOI: https://doi.org/10.1038/s41585-024-00993-6
