
Abstract
Clonal haematopoiesis involves the expansion of certain blood cell lineages and has been associated with ageing and adverse health outcomes1,2,3,4,5. Here we use exome sequence data on 628,388 individuals to identify 40,208 carriers of clonal haematopoiesis of indeterminate potential (CHIP). Using genome-wide and exome-wide association analyses, we identify 24 loci (21 of which are novel) where germline genetic variation influences predisposition to CHIP, including missense variants in the lymphocytic antigen coding gene LY75, which are associated with reduced incidence of CHIP. We also identify novel rare variant associations with clonal haematopoiesis and telomere length. Analysis of 5,041 health traits from the UK Biobank (UKB) found relationships between CHIP and severe COVID-19 outcomes, cardiovascular disease, haematologic traits, malignancy, smoking, obesity, infection and all-cause mortality. Longitudinal and Mendelian randomization analyses revealed that CHIP is associated with solid cancers, including non-melanoma skin cancer and lung cancer, and that CHIP linked to DNMT3A is associated with the subsequent development of myeloid but not lymphoid leukaemias. Additionally, contrary to previous findings from the initial 50,000 UKB exomes6, our results in the full sample do not support a role for IL-6 inhibition in reducing the risk of cardiovascular disease among CHIP carriers. Our findings demonstrate that CHIP represents a complex set of heterogeneous phenotypes with shared and unique germline genetic causes and varied clinical implications.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
27,99 € / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
199,00 € per year
only 3,90 € per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

Data availability
Individual-level sequence data, CHIP calls and polygenic scores have been deposited with UK Biobank and are freely available to approved researchers, as done with other genetic datasets to date10. Individual-level phenotype data are already available to approved researchers for the surveys and health record datasets from which all our traits are derived. Instructions for access to UK Biobank data is available at https://www.ukbiobank.ac.uk/enable-your-research. Summary statistics from UKB trait are available in the GWAS catalogue (accession IDs are listed in Supplementary Table 33). As described10, the HapMap3 reference panel was downloaded from ftp://ftp.ncbi.nlm.nih.gov/hapmap/, GnomAD v3.1 VCFs were obtained from https://gnomad.broadinstitute.org/downloads, and VCFs for TOPMED Freeze 8 were obtained from dbGaP as described in https://topmed.nhlbi.nih.gov/topmed-whole-genome-sequencing-methods-freeze-8. Regeneron can make GHS individual-level genomic data available to qualified academic noncommercial researchers through the Regeneron pre-clinical Research portal at https://regeneron.envisionpharma.com/vt_regeneron/ under a data access agreement.
Change history
17 February 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41586-023-05803-4
08 January 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41586-024-08572-w
References
Jaiswal, S. et al. Age-related clonal hematopoiesis associated with adverse outcomes. New Engl. J. Med. 371, 2488–2498 (2014).
Jaiswal, S. et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. New Engl. J. Med. 377, 111–121 (2017).
Jaiswal, S. & Ebert, B. L. Clonal hematopoiesis in human aging and disease. Science 366, eaan4673 (2019).
Zekavat, S. M. et al. Hematopoietic mosaic chromosomal alterations increase the risk for diverse types of infection. Nat. Med. 27, 1012–1024 (2021).
Niroula, A. et al. Distinction of lymphoid and myeloid clonal hematopoiesis. Nat. Med. 27, 1921–1927 (2021).
Bick, A. G. et al. Genetic interleukin 6 signaling deficiency attenuates cardiovascular risk in clonal hematopoiesis. Circulation 141, 124–131 (2020).
Thompson, D. J. et al. Genetic predisposition to mosaic Y chromosome loss in blood. Nature 575, 652–657 (2019).
Loh, P.-R. et al. Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature 559, 350–355 (2018).
Akbari, P. et al. Sequencing of 640,000 exomes identifies GPR75 variants associated with protection from obesity. Science 373, eabf8683 (2021).
Backman, J. D. et al. Exome sequencing and analysis of 454,787 UK Biobank participants. Nature 599, 628–634 (2021).
Bick, A. G. et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 586, 763–768 (2020).
Codd, V. et al. Polygenic basis and biomedical consequences of telomere length variation. Nat. Genet. 53, 1425–1433 (2021).
Denny, J. C. et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene–disease associations. Bioinformatics 26, 1205–1210 (2010).
Nédélec, Y. et al. Genetic ancestry and natural selection drive population differences in immune responses to pathogens. Cell 167, 657–669. e21 (2016).
Joehanes, R. et al. Integrated genome-wide analysis of expression quantitative trait loci aids interpretation of genomic association studies. Genome Biol. 18, 16 (2017).
Zhernakova, D. V. et al. Identification of context-dependent expression quantitative trait loci in whole blood. Nat. Genet. 49, 139–145 (2017).
Võsa, U. et al. Large-scale cis-and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 53, 1300–1310 (2021).
Gotoh, N. et al. PARP1 V762A polymorphism affects the prognosis of myelodysplastic syndromes. Eurr. J. Haematol. 104, 526–537 (2020).
Fukaya, T. et al. Conditional ablation of CD205+ conventional dendritic cells impacts the regulation of T-cell immunity and homeostasis in vivo. Proc. Natl Acad. Sci. USA 109, 11288–11293 (2012).
Palamarchuk, A. et al. Tcl1 protein functions as an inhibitor of de novo DNA methylation in B-cell chronic lymphocytic leukemia (CLL). Proc. Natl Acad. Sci. USA 109, 2555–2560 (2012).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Finucane, H. K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015).
Sperling, A. S., Gibson, C. J. & Ebert, B. L. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat. Rev. Cancer 17, 5 (2017).
Dawoud, A. A. Z., Gilbert, R. D., Tapper, W. J. & Cross, N. C. P. Clonal myelopoiesis promotes adverse outcomes in chronic kidney disease. Leukemia 36, 507–515 (2022).
Ostrander, E. L. et al. Divergent effects of Dnmt3a and Tet2 mutations on hematopoietic progenitor cell fitness. Stem Cell Rep. 14, 551–560 (2020).
Joo, S. H., Park, J. K., Lee, E. E., Song, Y. W. & Yoon, S.-S. Changes in serum uric acid levels after allogeneic hematologic stem cell transplantation: a retrospective cohort study. Blood Res. 51, 200–203 (2016).
Reynolds, M. D. Gout and hyperuricemia associated with sickle-cell anemia. Semin. Arthritis Rheum. 12, 404–413 (1983).
McAdams-DeMarco, M. A., Maynard, J. W., Coresh, J. & Baer, A. N. Anemia and the onset of gout in a population-based cohort of adults: Atherosclerosis Risk in Communities study. Arthritis Res. Ther. 14, R193 (2012).
Bolton, K. L. et al. Clonal hematopoiesis is associated with risk of severe Covid-19. Nat. Commun. 12, 5975 (2021).
Kosmicki, J. A. et al. Pan-ancestry exome-wide association analyses of COVID-19 outcomes in 586,157 individuals. Am. J. Hum. Genet. 108, 1350–1355 (2021).
Smith, M. A., McCaffrey, R. P. & Karp, J. E. The secondary leukemias: challenges and research directions. JNCI 88, 407–418 (1996).
Fehrmann, R. S. et al. Trans-eQTLs reveal that independent genetic variants associated with a complex phenotype converge on intermediate genes, with a major role for the HLA. PLoS Genet. 7, e1002197 (2011).
Chaudhuri, A. R. & Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 18, 610–621 (2017).
Jing, C.-B. et al. Synthetic lethal targeting of TET2-mutant hematopoietic stem and progenitor cells (HSPCs) with TOP1-targeted drugs and PARP1 inhibitors. Leukemia 34, 2992–3006 (2020).
Abbotts, R. et al. DNA methyltransferase inhibitors induce a BRCAness phenotype that sensitizes NSCLC to PARP inhibitor and ionizing radiation. Proc. Natl Acad. Sci. USA 116, 22609–22618 (2019).
Moore, K. N., Mirza, M. R. & Matulonis, U. A. The poly (ADP ribose) polymerase inhibitor niraparib: management of toxicities. Gynecol. Oncol. 149, 214–220 (2018).
Jiang, W. et al. The receptor DEC-205 expressed by dendritic cells and thymic epithelial cells is involved in antigen processing. Nature 375, 151–155 (1995).
Kato, M. et al. Expression of human DEC-205 (CD205) multilectin receptor on leukocytes. Int. Immunol. 18, 857–869 (2006).
Kim, P. G. et al. Dnmt3a-mutated clonal hematopoiesis promotes osteoporosis. J. Exp. Med. 218, e20211872 (2021).
Van Hout, C. V. et al. Exome sequencing and characterization of 49,960 individuals in the UK Biobank. Nature 586, 749–756 (2020).
Ferreira, R. C. et al. Functional IL6R 358Ala allele impairs classical IL-6 receptor signaling and influences risk of diverse inflammatory diseases. PLoS Genet. 9, e1003444 (2013).
Szustakowski, J. D. et al. Advancing human genetics research and drug discovery through exome sequencing of the UK Biobank. Nat. Genet. 53, 942–948 (2021).
Benjamin, D. et al. Calling Somatic SNVs and Indels with Mutect2. Preprint at bioRxiv https://doi.org/10.1101/861054 (2019).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Pich, O., Reyes-Salazar, I., Gonzalez-Perez, A. & Lopez-Bigas, N. Discovering the drivers of clonal hematopoiesis. Nat. Commun. 13, 4267 (2022).
Costello, M. et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 41, e67 (2013).
Mbatchou, J. et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet. 53, 1097–1103 (2021).
Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021).
Watanabe, K., Taskesen, E., Van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Zhong, H. & Prentice, R. L. Correcting “winner’s curse” in odds ratios from genomewide association findings for major complex human diseases. Genet. Epidemiol. 34, 78–91 (2010).
Ghosh, A., Zou, F. & Wright, F. A. Estimating odds ratios in genome scans: an approximate conditional likelihood approach. Am. J. Hum. Genet. 82, 1064–1074 (2008).
Privé, F., Vilhjálmsson, B. J., Aschard, H. & Blum, M. G. Making the most of clumping and thresholding for polygenic scores. Am. J. Hum. Genet. 105, 1213–1221 (2019).
Yang, J. et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 44, 369–375 (2012).
Benner, C. et al. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics 32, 1493–1501 (2016).
Kikushige, Y. et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell 20, 246–259 (2011).
Quivoron, C. et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 20, 25–38 (2011).
Couronné, L., Bastard, C. & Bernard, O. A. TET2 and DNMT3A mutations in human T-cell lymphoma. New Engl. J. Med. 366, 95–96 (2012).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Author information
Authors and Affiliations
Consortia
Contributions
M.D.K., A.D., S.O., S.S., N.B., D.L., J.T. and H.M.K. performed bioinformatic and statistical genetic analysis. S.O., K.W., A.M., Y.Z., A.G., C.G., J. Mbatchou, W.S., J.G.R. and H.M.K. contributed to data engineering and bioinformatic pipeline development. J.D.O. developed and oversaw exome sequencing efforts. M.D.K., A.D., S.O., K.W., M.V.M., J.H., J.A.K., V.M.R., Y.H., C.G., R.R.W., N.V., J.B., N.N.P., M.G.L., M.J., D.J.G., L.A.L., M.N.C., G.S.A., A.E.L., M.A.R.F., R.D., G.T., C.P., A.R.S., A.A.F., W.S., J.G.R., J.D.O., J. Marchini, H.M.K., A.B., G.R.A. and E.J. contributed to experimental design, interpretation of results, and genetic programme development. M.D.K. and E.J. drafted the manuscript, with significant input from A.D., S.O., M.V.M., G.T., A.A.F., H.M.K., A.B. and G.R.A. All authors reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
M.D.K., A.D., S.O., N.B., D.L., K.W., A.M., M.V.M., S.S., J.H., J.T., J.A.K., V.M.R., Y.Z., Y.H., A.G., C.G., J. Mbatchou, R.R.W., N.V., J.B., N.N.P., M.G.L., M.J., D.J.G., L.A.L., M.N.C., G.S.A., A.E.L., M.A.R.F., R.D., C.P., A.R.S., G.T., A.A.F., W.S., J.G.R., J.D.O., J. Marchini, H.M.K., A.B., G.R.A. and E.J. are current employees and/or stockholders of Regeneron Genetics Center or Regeneron Pharmaceuticals.
Peer review
Peer review information
Nature thanks Po-Ru Loh and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Workflow to Identify CHIP and Prevalence Estimates For Carriers of CHIP Mutations.
A. Graphic depicting at a high-level the workflow used to collect and sequence the exomes of multiple large cohorts and to then identify CHIP mutations from this data. B-C. CHIP prevalence increases with age of donor at time of DNA collection in both the UKB (B, n = 484,629 individuals; one-sided F-test, P < 10−16) and GHS (C, n = 157,724 individuals; one-sided F-test, P < 10−16) cohorts, with the centre line representing the general additive model spline and the shaded region representing the 95% confidence interval. D-E. Similar to B-C, the prevalence of CHIP mutations per CHIP gene for each of the top 8 most common CHIP genes increase with age in the UKB (D, n = 484,629 individuals; one-sided F-test, P < 10−16) and in GHS (E, n = 157,724 individuals; one-sided F-test, P < 10−16).
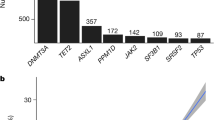
Extended Data Fig. 2 Count Distribution and Pairwise Enrichments of Clonal Hematopoiesis of Indeterminate Potential (CHIP) Gene Mutations.
A. Total number of individuals with mutations (y axis, log10 scale) in each of the 23 genes that were used to determine CHIP status across the UKB (blue) and GHS (red) CHIP callsets. B-C. Pairwise mutation counts across the UKB (B) and DiscoverEHR (C) callsets across individuals with at least two identified CHIP mutations. The color scale reflects the significance of the p-value for association between mutated CHIP gene pairs as determined by logistic regression. Per CHIP gene pair, these models included CHIP gene 1 mutation carrier status as the outcome, CHIP gene 2 mutation carrier status as the predictor, and age, sex, and smoking status (ever vs never) as covariates. P values are log10 transformed (see Table S1 for complete enrichment results).
Extended Data Fig. 3 Finemapping results at the LY75 locus on chromosome 2.
A. Fine-mapping the summary statistics from our association analysis of CHIP prioritizes the P1247L missense variant (rs78446341-A, AAF = 0.02) as highly likely to be the causal variant driving one of three causal signals at this locus (CPIP = 0.913). At the top of the panel, a locus zoom plot shows marginal association results after inverse variance weighted meta analysis across UKB and GHS (p-values are uncorrected and derive from two-sided tests performed using approximate Firth logistic regression and subsequent meta analysis). Top common variants, including those prioritized by clumping and thresholding and COJO from UKB associations are highlighted with black circles. The rs78446341-A missense variant is highlighted as well and is in low linkage disequilibrium (LD) with the other SNPs. FINEMAP estimated 3 signals were most parsimonious here (PP = 0.55). B. Fine-mapping the summary statistics from our association analysis of DNMT3A-CHIP prioritizes the P1247L missense variant (rs78446341-A, MAF = 0.02, CPIP = 0.20, CS = 4) and the rarer G525E missense variant (rs147820690-T, AAF = 0.002 CPIP = 0.60, CS = 2) as likely to be the causal variants driving the signal at two out of four causal signals at this locus. Here, FINEMAP estimated 3 signals (PP = 0.57) or 4 signals (PP = 0.41) were likely; we report results for K = 3 in Table S6 and show results from K = 4 here. The other prioritized signals are those identified by clumping and thresholding and COJO: rs12472767-C (2-159925824-T-C, CPIP = 0.99, CS = 1) and rs12472767-C (2-159821048-C-T, CPIP = 0.28, CS = 3). CS: Credible Set, PP: Posterior Probability, PIP: Posterior Inclusions Probability, CPIP: Conditional Posterior Inclusion Probability.
Extended Data Fig. 4 Results from a phenome-wide association analysis.
Results from a phenome-wide association analysis are shown for the thirty SNPs from our GWAS that had the largest number of significant associations (P < 5 x 10−8). Associations are most common among hematological, body mass, and auto-immune traits (seen across the ‘dermatology’, ‘gastroenterology’, and ‘other’ phenotypic categories). For visualization, associations with –log10(P) < 50 were set to 50. Association models were run with age, age2, sex, and age-by-sex, and 10 ancestry-informative principal components (PCs) as covariates. P-values are uncorrected and derive from two-sided tests performed using approximate Firth logistic regression. See Table S10 for full associations results.
Extended Data Fig. 5 GWAS of CHIP Subtypes.
Manhattan plot showing results from a genome-wide association analysis of CHIP subtypes. While we ran CHIP subtype analysis for each of the 8 most recurrently mutated CHIP genes (Tables S11–S19), we show Manhattan plots for the 5 CHIP subtypes that had at least 1 genome-wide significant common variant association. These included DNMT3A-CHIP (23 significant loci), TET2-CHIP (6 significant loci), ASXL1-CHIP (2 significant loci), TP53-CHIP (1 significant locus), and JAK2-CHIP (1 significant locus). Novel biologically relevant genes are labeled at each locus, with red denoting novel loci, black identifying previously identified loci and grey identifying loci with suggestive signal (P < 5 x 10−7). Association models were run with age, age2, sex, and age-by-sex, and 10 ancestry-informative principal components (PCs) as covariates. P-values are uncorrected and are from two-sided tests performed using approximate Firth logistic regression.
Extended Data Fig. 6 Results from Mendelian Randomization models and incident risk of death among CHIP carriers.
A. Forest plot of results from Two Sample Mendelian Randomization (MR) modeling of the effect of CHIP on 20 traits of interest (including the two quantitative traits BMI and ALT). Reported p-values are uncorrected, and reflect two-sided Z-tests derived from an inverse variance weighted (IVW) MR procedure. Significant causal association between CHIP and breast cancer, prostate cancer, non-melanoma skin cancer, melanoma, myeloid leukemia, and lung cancer are supported by these models. As expected, estimates of germline effect on CHIP from UKB and GHS are strongly correlated (odds ratio = 1.94 [1.76–2.13], P = 3.2 x 10−42). B. CHIP and its most common subtypes are significantly associated with death from any cause across UKB. Hazard ratio (HR) estimates from cox-proportional hazard models are shown, with error bars that represent a 95% confidence interval. P-values are uncorrected and derive from two-sided Wald tests. Models are adjusted for sex, LDL, HDL, pack years, smoking status, BMI, essential primary hypertension, type 2 diabetes mellitus, and 10 European specific genetic PCs.
Extended Data Fig. 7 CVD Incidence in IL6R Mutation Carriers with and without CHIP.
A-B. Survival curves are drawn showing that IL6R p.Asp358Ala mutation carriers (green) are not an elevated risk of CVD incidence (y-axis) compared with non-carriers (blue) in either the first 50K individuals from UKB (A) or the full 450K cohort (B). C-D. In contrast, IL6R p.Asp358Ala mutation carriers are estimated to be at a reduced risk of CVD events (C) (HR = 0.60), but only in the first 50K samples from UKB (D). Models are adjusted for sex, LDL, HDL, pack years, smoking status, BMI, essential primary hypertension, type 2 diabetes mellitus, and 10 European specific genetic PCs. Hazard ratios (HR) were estimated using cox-proportional hazard modeling, with p-values uncorrected and derived from two-sided Wald tests.
Extended Data Fig. 8 Incident risk of myeloid cancer subtypes among CHIP carriers from the UKB.
A-C. Forest plots and tables featuring hazard ratio (HR) estimates from cox-proportional hazard models are shown, with error bars that represent a 95% confidence interval. CHIP and its most common subtypes are significantly associated with acute myeloid leukemia (AML) (A), Myelodysplastic Syndromes (MDS) (B), and myeloproliferative neoplasm (MPN) (C). Here, results are depicted from analyses in which we removed samples that had a diagnosis of malignant cancer prior to sequencing collection. Models are adjusted for sex, LDL, HDL, pack years, smoking status, BMI, essential primary hypertension, type 2 diabetes mellitus, and 10 European specific genetic PCs. Hazard ratios (HR) were estimated using cox-proportional hazard modeling, with p-values uncorrected and derived from two-sided Wald tests.
Extended Data Fig. 9 Incident risk of lung cancer among CHIP carriers from the UKB and GHS cohorts.
A-D. Forest plots and tables featuring hazard ratio (HR) estimates from cox-proportional hazard models are shown, with error bars that represent a 95% confidence interval. CHIP and its most common subtypes are significantly associated with lung cancer in both smokers and non-smokers across UKB (A-B) and GHS (C-D). Here, results are depicted from analyses in which we removed samples that had a diagnosis of malignant cancer prior to DNA collection. Models are adjusted for sex, LDL, HDL, pack years, smoking status, BMI, essential primary hypertension, type 2 diabetes mellitus, and 10 European specific genetic PCs. Hazard ratios (HR) were estimated using cox-proportional hazard modeling, with p-values uncorrected and derived from two-sided Wald tests.
Supplementary information
Supplementary Information
This file contains supplementary information for numerous analyses, and includes 12 supplementary notes, which are referenced throughout the Article.
Supplementary Figures
This file contains Supplementary Figs. 1–18.
Supplementary Tables
Supplementary Tables 1–33 - see `Table Descriptions’ worksheet within the file for details.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kessler, M.D., Damask, A., O’Keeffe, S. et al. Common and rare variant associations with clonal haematopoiesis phenotypes. Nature 612, 301–309 (2022). https://doi.org/10.1038/s41586-022-05448-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-05448-9
This article is cited by
-
Genetic evidence for the causal effect of clonal hematopoiesis on pulmonary arterial hypertension
BMC Cardiovascular Disorders (2025)
-
Association of clonal hematopoiesis of indeterminate potential with cardiometabolic multimorbidity progression and mortality: a prospective study of UK Biobank
European Journal of Medical Research (2025)
-
Measurable residual disease testing and allogeneic hematopoietic cell transplantation for AML: adapting Pre-MEASURE to clinical practice
Bone Marrow Transplantation (2025)
-
The effects of loss of Y chromosome on male health
Nature Reviews Genetics (2025)
-
Clonal hematopoiesis as the intersection between genetics and resilience in Alzheimer’s disease
npj Dementia (2025)
