
Abstract
The parathyroid hormone type 1 receptor (PTH1R) is a prototypical class B1 G-protein-coupled receptor that couples to both Gq and Gs, having a crucial role in calcium homeostasis and serving as a therapeutic target for osteoporosis. Therapies targeting PTH1R face challenges because of Gq-associated prolonged signaling, which leads to bone resorption. To address this, selective activation of Gs signaling is desirable. However, the structural basis of Gq-mediated signaling remains unclear, limiting the development of signal-selective drugs. Here, we present cryo-electron microscopy structures of the PTH1R–Gq complex in two distinct extracellular conformations, demonstrating the role of N-linked glycans at N1761.28 in stabilizing the ligand-tilted conformation. Comparison with a Gs-bound PTH1R structure highlights the role of key interactions involving both the C terminus of Gα and the receptor’s intracellular loop 2 in Gq signaling. These structural insights provide a foundation for understanding the molecular mechanisms of PTH1R signaling.

Similar content being viewed by others

Main
The parathyroid hormone type 1 receptor (PTH1R) is a class B1 G-protein-coupled receptor (GPCR) that mediates the physiological effects of the endogenous peptide ligands PTH and PTH-related protein (PTHrP). PTH1R has a central role in regulating skeletal development, bone turnover and calcium homeostasis1,2. Because of its biological effects, the PTH–PTH1R signaling pathway has been investigated for the development of therapies for related diseases, including osteoporosis, hypoparathyroidism and hypophosphataemia3,4,5.
While several PTH analogs have been clinically applied to treat osteoporosis6,7, their long-term administration was found to induce osteoclastic activity, limiting their clinical utility8. The seemingly contradictory effects of bone formation and bone resorption stem from two distinct signaling events that differ in terms of duration and cellular localization9,10. One is transient signaling from the cell surface, which is consistent with the classical model of GPCR signaling. The other is prolonged signaling following internalization into early endosomes. Accumulating evidence suggests that differences in these signaling mechanisms may result in distinct biological outcomes. For example, the PTH-R25C mutant displayed decreased ligand residence time and endosomal signaling, leading to a hypocalcemic response (that is, bone formation)11. In contrast, long-acting PTH (LA-PTH) analogs that promote prolonged endosomal signaling caused a hypercalcemic response (that is, bone resorption)12.
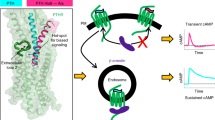
Similar to other Class B1 GPCRs, PTH1R primarily couples with Gq and Gs (Fig. 1a). In Gq-mediated signaling, phospholipase Cβ activation leads to protein kinase C signaling and an increase in intracellular Ca²⁺ concentration. On the other hand, in Gs-mediated signaling, the activated Gs stimulates cyclic adenosine monophosphate (cAMP) production through adenylyl cyclase13. In PTH1R, Gq-mediated signaling not only activates classical pathways but also facilitates prolonged cAMP production through β-arrestin-mediated endocytosis, ultimately promoting bone resorption (Fig. 1b)10,14. This process is distinct from the classical transient cAMP production at the cell surface, which induces bone formation. Therefore, a better understanding of the coupling preference of PTH1R for Gq and Gs is expected to contribute to the development of new therapies for osteoporosis that avoid the unwanted side effect of bone resorption. Several structures of Gs-bound PTH1R were recently reported, providing insights into the receptor’s ligand recognition and Gs signaling mechanisms15,16. However, the lack of structural knowledge about Gq-bound PTH1R has limited our understanding of the coupling selectivity of Gs versus Gq. Here, we present cryo-electron microscopy (cryo-EM) structures of the PTH–PTH1R–Gq complex, elucidating two distinct states in its extracellular region. Combined with functional assays, molecular dynamics (MD) simulations and kinetics measurements, these structures highlight the importance of a flexible extracellular region and yield insights into the coupling preference for Gq and Gs.

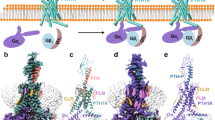
a, A schematic diagram representing the general signaling pathways of class B1 GPCRs. b, A schematic diagram representing transient signaling from the cell surface and prolonged signaling from the endosome. c,d. The combined cryo-EM density maps and ribbon models of PTH–PTH1R–Gq complexes in the upright (c) and tilted (d) states. The contour levels are set to 8.3 and 6.7, respectively. The density derived from the detergent micelle was removed by the solvent mask.
Results
Structure determination of the PTH–PTH1R–Gq complex
We used an engineered, chimeric miniGαq protein to facilitate structural determination. The miniGαq protein was previously demonstrated to reproduce Gq-coupled GPCR activation17,18. In the chimeric miniGαq protein, its N terminus is replaced by that of the Gαi1 protein, providing an additional binding site for the G-protein-stabilizing antibody scFv16 (ref. 19). The resulting chimeric miniGαq has been used for the structural studies of various Gq-coupled GPCRs20,21. Hereafter, Gq refers to the heterotrimeric G protein consisting of chimeric miniGαq, Gβ1 and Gγ2, unless otherwise stated.
To obtain a PTH–PTH1R–Gq signaling complex, we first purified PTH1R, Gq and scFv16 proteins separately. We then mixed these proteins in the presence of PTH and isolated a fraction corresponding to the PTH–PTH1R–Gq complex by Ni-affinity chromatography and gel-filtration chromatography (Extended Data Fig. 1). In cryo-EM data processing, the extracellular ___domain (ECD) of PTH1R exhibited continuous flexibility. Therefore, we performed three-dimensional (3D) classification without alignment, focusing on the ECD of PTH1R, which led to the structural determination of two distinct states: the upright state and the tilted state. These two states were solved at a resolution of 2.8 Å (Fig. 1c,d, Extended Data Fig. 2a–f and Supplementary Table 1). To facilitate model building of the ECD, we performed focused refinement on PTH1R and combined the results with the overall refined map (Extended Data Figs. 2d–f and 3a). The resulting combined density maps of the upright and tilted states had a resolution of ~3 Å in the transmembrane (TM) region and 3.5–4.5 Å in the ECD regions, allowing construction of an atomic model of most parts of the complex except for several loop regions (Extended Data Figs. 2d–f and 3b,c). The cryo-EM structures of Gq-bound PTH1R exhibit a canonical composition, consistent with previous reports. Briefly, PTH binds to the extracellular side of PTH1R and Gq binds to the intracellular side of PTH1R (Fig. 1c,d). ScFv16 stabilizes the miniGαq–Gβ interface in a manner similar to the previously described GPCR–Gi/Gq complex19,20,22,23. Comparing the upright and tilted states, they exhibit almost the same conformations, except for the extracellular region.
Conformational dynamics in the extracellular region
Relative to S17PTH as the starting point, the ECD of PTH1R and the C-terminal half of PTH are bent by approximately 40° in the tilted state compared to their positions in the upright state (Fig. 2a). Initially, we focused on the ECD of PTH1R and the C-terminal half of PTH, comparing the upright and tilted states. In both states, the hydrophobic residues L24PTH, L28PTH and V31PTH are packed closely against the hydrophobic groove of the ECD, as previously reported (Extended Data Fig. 4a,b)24. In addition, W23PTH, which is known to be involved in ligand selectivity and kinetics, is packed into the hydrophobic pocket formed by the residues of the N-terminal helix of the ECD (Extended Data Fig. 4c,d). Overall, the upright and tilted states exhibited nearly identical ECD–PTH binding modes.

a, Structural dynamics of the C-terminal half of PTH and the ECD of PTH1R in the upright and tilted states. b,c, Interaction mode related to R25PTH and the extended TM1 of PTH1R in the upright (b) and tilted (c) states. Residues involved in interactions and N-acetylglucosamine (NAG) are represented by stick models. Dashed lines indicate polar interactions. d, Schematic representation of the N-linked glycans at N1761.28 stabilizing the extension of TM1. This stabilization helps to maintain the equilibrium between the upright and tilted states, potentially influencing ligand residence time. e, Fluorescence time-lapse images of TMR-labeled PTH or PTHrP dissociation from WT and triple-mutant PTH1R-expressing cells. The white bar indicates 5 μm. f, Time-course changes in fluorescence intensity normalized at the start of perfusion. g–i, Radar charts showing the percentage of the three components (solid line, PTH; dashed line, PTHrP): fast (g), medium (h) and slow (i). j, Radar chart of time constants for the three kinetics components, displayed on a logarithmic scale (red, fast; green, medium; blue, slow; solid line, PTH; dashed line, PTHrP). Lines and error bands represent the mean and s.e.m., respectively, from 8–20 cells in two independent experiments. The exact sample sizes for f–j are provided in Supplementary Table 2. Statistical analyses are shown in Extended Data Fig. 6. k,l, Gq-coupling and Gs-coupling activities and expression levels of the PTH1R mutants. PTH-induced Gq and Gs responses were analyzed by a NanoBiT G-protein dissociation assay (k). Data are shown as the logRAi (Methods). Statistical analysis was performed with two-way ANOVA, followed by Šidák’s post hoc test (not significant (NS), P > 0.05; ***P < 0.001). Cell surface expression levels were measured by flow cytometry analysis (l). Bars and error bars represent the mean and s.e.m., respectively, from three independent experiments (k,l). vol, volume.
Conversely, we identified several remarkable differences in the conformations of the PTH1R-TM region and the N-terminal half of PTH. While both structures adopt almost identical conformations in most TM regions (Extended Data Fig. 4e,f), the tilted state reveals an extension of TM1 (Fig. 2b,c). Notably, the tilted state exhibits a clear density corresponding to the N-linked glycans from N1761.28 (superscripts indicate the Wootten class B1 GPCR numbering system25)(Extended Data Fig. 4g,h). The glycans form hydrogen bonds with T1781.30 and potentially with R1791.31, stabilizing the extension of TM1. Consequently, T1751.27 and E1771.29, which are located near the extended TM1, form polar interactions with R25PTH. Conversely, in the upright state, glycans are not observed, leading to a shortened TM1, with a potential salt bridge between E1771.29 and R1791.31 stabilizing this conformation (Fig. 2b and Extended Data Fig. 4g). As indicated by the broadened band observed in the SDS–PAGE analysis, the glycosylation of PTH1R is heterogeneous, which may result in distinct conformations (Extended Data Fig. 1).
Next, we performed MD simulations to further explore the glycan-involved interactions observed in the tilted state. The simulations were performed under three conditions: using the glycan-containing tilted state as the initial model, a glycan-free tilted state and the tilted state where T1751.27, N1761.28 and E1771.29 were substituted to alanine. Three independent production runs were performed for each condition and the overall structures remained stable across all simulations (Extended Data Fig. 5a–c). In the simulation using the glycan-containing model, E1771.29 at the terminus of the extended TM1 formed a stable α-helix conformation (Extended Data Fig. 5f–h). However, in the glycan-free model, the secondary structure of E1771.29 tended to destabilize, a trend that was even more pronounced in the triple-mutant model. Consequently, the glycan-containing model exhibited an equilibrium between the upright and tilted states, while the glycan-free and triple-mutant models predominantly favored a structure resembling the upright state (Extended Data Fig. 5i–k). Overall, the MD simulation results are consistent with the cryo-EM observations, indicating that glycans support the extension of TM1, which may stabilize the tilted state (Fig. 2d).
To investigate the effects of glycosylation on ligand-binding stability, we compared the dissociation rates of ligands from PTH1R (Fig. 2e,f and Extended Data Fig. 6a–d). HEK293A cells expressing wild-type (WT) or mutant PTH1R were equilibrated with 5 μM tetramethylrhodamine (TMR)-labeled PTH or PTHrP; then, we measured the intensity decrease after washing out unbound ligands in external solution under a fluorescence microscope. No significant TMR–PTH or TMR–PTHrP intensity was observed on mock-transfected cells, indicating limited nonspecific adsorption to the HEK293A cell surface (Extended Data Fig. 6a). Across all constructs tested, PTH exhibited a more sustained ligand residence time compared to PTHrP, consistent with previous reports (Extended Data Fig. 6b–d)9. We fitted the data using nonlinear regression with double-exponential and triple-exponential models, with the latter generally providing a better fit, as reflected by lower Akaike information criterion (AIC) values (Extended Data Fig. 6e,f and Methods). This suggests that ligand–PTH1R dissociation is not a simple two-step mechanism, consistent with previous reports9,26. Hereafter, we refer to the three kinetic components as fast, medium and slow (in order of increasing time constant, τ) and compare WT and mutant PTH1R for both TMR–PTH and TMR–PTHrP.
We first describe the differences between the two ligands in the context of WT PTH1R. For PTH, the slow component accounted for the largest fraction of the population (~60%), whereas the fast component contributed only a minor fraction (Fig. 2g–j and Extended Data Fig. 6g,h). In contrast, for PTHrP, the slow component exhibited a relatively shorter τ and a smaller fraction, accompanied by a larger fraction from the fast component. These differences are responsible for the longer ligand residence time of PTH compared to PTHrP.
Substitutions of the glycan-associated residues of PTH1R reduced the ligand residence time for both PTH and PTHrP but the effects on the dissociation kinetics were quantitatively different. For PTH, each mutant except for N1761.28A reduced the fraction of the slow and/or medium components and increased that of the fast component (Fig. 2g–i, Extended Data Fig. 6h and Supplementary Table 2). The triple mutant (T1751.27A;N1761.28A;E1771.29A) induced a more pronounced population shift from the slow to the fast component (Fig. 2f–i). For PTHrP, all the mutants exhibited a decreased fraction of the slow component and tended to increase that of the medium component. These results suggest that glycan-associated residues of PTH1R regulate the kinetics of both PTH and PTHrP, albeit through different mechanisms.
We further examined the role of these residues in determining the preference for Gq and Gs. In our previous structural study of the Gs-bound PTH1R, we also observed continuous flexibility in the extracellular region (Extended Data Fig. 4i,j)16. However, the resolution of the tilted state in the Gq-bound structure (3.5–4.5 Å) is notably better than that of class 4 in the Gs-bound structure (~7 Å; corresponding to tilted state) (Extended Data Fig. 4k,l). To functionally validate these findings, we conducted a mutagenesis study using the NanoBiT G-protein dissociation assay27. As controls, we tested two plasmid volume conditions for the WT PTH1R. We confirmed that the G-protein coupling activities for Gq and Gs were nearly equivalent in the half-volume condition as compared to the original volume condition (Fig. 2k,l and Supplementary Table 3). While the single mutants T1751.27A, N1761.28A, N1761.28Q and E1771.29A did not show a significant difference in the Gq versus Gs coupling preference, we found that the triple-alanine mutant of these residues reduced the G-protein coupling activity (logarithm of relative intrinsic activity (logRAi); Methods) more severely for Gq than for Gs (Fig. 2k,l). Although the contribution of these residues to each signaling pathway appears modest in our assay system, these structural and functional insights imply that Gq coupling is relatively more dependent on the tilted state compared to Gs coupling. Although this study offers some insights into the physiological importance of the tilted state, it remains only partially understood and will require further investigation in future research.
Gq binding expands the intracellular cavity of PTH1R
Our structural examination revealed that the upright and tilted states exhibit very similar conformations in their TM and intracellular regions (Extended Data Fig. 4e,f). Hereafter, Gq-bound PTH1R refers to the upright state, unless otherwise noted.
The structures of Gq-bound and Gs-bound PTH1R showed that TMs 1–4 and 7 adopt nearly identical conformations (Fig. 3a), whereas TMs 5 and 6 of Gq-bound PTH1R exhibit outward shifts of 3.3 Å and 4.4 Å at the intracellular ends, respectively, compared to those of Gs-bound PTH1R (Fig. 3b). This additional expansion results in increased size of the intracellular cavity of PTH1R. Additionally, the α5 helix (α5h) in Gq-bound PTH1R is positioned more vertically by 3.5 Å at the N terminus of α5h compared to its position in Gs-bound PTH1R. These structural features of Gq-bound PTH1R result in fewer interactions involving TM5, TM6 and α5h (Fig. 3c,d). More specifically, in Gs-bound PTH1R, the hydrophobic residues L378G.H5.20 (superscript indicates the common Gα numbering system28), L383G.H5.25 and L384G.H5.26 of Gs pack closely against the cytoplasmic hydrophobic surface of PTH1R, consisting of I3815.57, L3855.61, L3895.65, S4096.41, V4126.44 and L4136.45. By contrast, Gq-bound PTH1R does not closely pack these residues, instead exhibiting a gap between them.

a, Overall comparison of the TM regions of Gq-bound and Gs-bound PTH1R. b, Comparison of the degree of opening in TMs 5 and 6 of the receptor and the angle of α5h of G protein in Gq-bound and Gs-bound PTH1R. c,d, Interactions between the PTH1R intracellular cavity and the G-protein α5h for Gq (c) and Gs (d). Notable residues are shown as stick and Corey–Pauling–Koltun (CPK) models. e,f, Contact frequencies between PTH1R and α5h measured in MD simulations. Residues with a contact frequency below 30% are omitted. Both heat maps represent the combined data from three independent replicate simulations. g,h. The SASA calculated in the MD simulations for Gq (g) and Gs (h). The total SASA was computed as the sum of I3815.57, L3855.61, L3895.65, S4096.41, V4126.44 and L4136.45 throughout three replicate simulations. Average values, calculated every 80 frames, are shown in bold.
To investigate the hydrophobic interactions involving TM5, TM6 and α5h, we performed 200-ns MD simulations of PTH1R bound to Gq or Gs. In each condition, the conformation was stable throughout three replicate simulations (Extended Data Fig. 5d,e). By analyzing the interaction dynamics involving TM5, TM6 and α5h, we observed lower contact frequency in Gq-bound PTH1R than in Gs-bound PTH1R (Fig. 3e,f). Moreover, our analysis of the solvent-accessible surface area (SASA) across TM5, TM6 and α5h revealed that the total SASA for this region was generally higher in Gq-bound PTH1R, suggesting a tendency for a more open conformation at the cytosolic site of PTH1R (Fig. 3g). By contrast, Gs-bound PTH1R showed relatively low SASA during the simulations, indicating that the region packs more closely (Fig. 3h). Taken together, these results suggest that, in Gq-bound PTH1R, the intracellular cavity is largely expanded and α5h is embedded relatively vertically, resulting in fewer interactions between them, whereas Gs-bound PTH1R forms a stable hydrophobic interaction involving TM5, TM6 and α5h.
To functionally validate the structural findings, we performed a mutagenesis study using the NanoBiT G-protein dissociation assay27. As controls, we titrated the transfection volumes of the WT PTH1R plasmid by one half and one fourth and confirmed that the G-protein coupling activities were equal overall for Gq and Gs across these expression levels (Extended Data Fig. 7a–c). Similar approaches were adopted in subsequent sections. We observed no significant differences with a single-residue substitution. This is likely because the hydrophobic interactions involving TM5 and TM6 consist of weak interactions from multiple residues. We, therefore, designed multiple-point mutants for TM5 and TM6. As expected, we observed a more pronounced reduction in Gs coupling with the triple mutant in TM5. Although the double mutant in TM6 did not match the structural findings, this discrepancy is likely because of the ___location of these residues near the base of the TM6 kink, where they are less influenced by the extent of TM6 expansion.
Interaction of α5h C terminus with intracellular cavity
We next focused on interactions between the C terminus of α5h and the intracellular cavity of PTH1R. In both Gq-bound and Gs-bound PTH1R, YG.H5.23 is tightly accommodated within the intracellular pocket of PTH1R (Fig. 4a,b). Additionally, E242G.H5.22 in Gq and Q380G.H5.22 in Gs could form hydrophilic interactions (Fig. 4c,d). The NanoBiT G-protein dissociation assay also demonstrated that many of these residues are crucial for both Gq and Gs signaling (Fig. 4e,f). The differences in the reduction of Gq and Gs activity observed in several mutants, such as H2232.50A, are likely because of subtle conformational differences, including variations in rotamer conformations. Overall, no consistent trend in Gq versus Gs preference was observed in this region.

a,b, Interactions between YG.H5.23 and PTH1R in the Gq-bound (a) and Gs-bound (b) states. Residues involved in these interactions are shown as stick and CPK models. c,d, Interactions between EG.H5.22 (c) and QG.H5.22 (d) and PTH1R. Interacting residues in PTH1R–Gq and the corresponding residues in PTH1R–Gs are shown as stick models. Ionic interactions are represented by dashed lines. e,f, Gq and Gs coupling activities and expression levels of the PTH1R mutants. PTH-induced Gq and Gs responses were analyzed by the NanoBiT G-protein dissociation assay (e). Data are shown as the logRAi (Methods). Statistical analysis was performed using a two-way ANOVA, followed by Šidák’s post hoc test (NS, P > 0.05; ***P < 0.001). Cell surface expression levels were measured by flow cytometry analysis (f). Bars and error bars represent the mean and s.e.m., respectively, from 3–7 independent experiments. The exact sample sizes for e,f are provided in Supplementary Table 3.
Intracellular loop 2 (ICL2) contributes to Gq signaling
We next examined the interactions between ICL2 of PTH1R and G protein. Note that we used the miniGαs/q71 construct as a Gαq surrogate for the structural study17. This construct was designed on the basis of miniGαs but the receptor-interacting residues were substituted by those from Gαq. In fact, the superimposition of miniGαq with native Gαq showed a high level of similarity in their conformations and revealed that the receptor-interacting residues were derived from Gαq rather than from miniGαs or the Gαi N terminus. This indicates that our structure closely reproduces the binding mode with Gαq (Extended Data Fig. 8a).
Compared to that in Gs-bound PTH1R, the β2–β3 loop of the G protein in Gq-bound PTH1R is positioned closer to the intracellular cavity because of the more vertical binding of α5h (Fig. 5a). Near the TM4 side of ICL2, both complexes exhibit similar conformations; however, Gq possesses polar residues such as R31G.hns1.02 and R32 G.hns1.03 in proximity to ICL2, potentially allowing for the formation of polar interactions with E3174.38 (Fig. 5b,c). A previous study reported that a series of substitutions (E3174.38D, K3184.39S, K3194.40E and Y3204.41L) selectively eliminated Gq signaling while preserving Gs signaling29. Thus, polar interactions along this region are considered to contribute to Gq signaling. Near the TM3 side of ICL2, F314ICL2 is deeply embedded in a pocket formed by hydrophobic residues of Gα in both Gq and Gs (Fig. 5d,e). In addition, F315ICL2 is buried in the gap between TM3 and TM4, suggesting a contribution to the structural stability of ICL2. These interactions are largely identical in Gq and Gs.

a, Conformational differences between Gq-bound and Gs-bound PTH1R around ICL2. b,c. Interactions between the TM4 side of ICL2 and the G protein in Gq (b) and Gs (c). Residues potentially engaged in ionic interactions are shown as stick models. d,e. Interactions between the TM3 side of ICL2 and the G protein in Gq (d) and Gs (e). Notable residues are shown as stick and CPK models. f, Gq and Gs coupling activities of the PTH1R mutants. PTH-induced Gq and Gs responses were analyzed by the NanoBiT G-protein dissociation assay. Bars and error bars represent the mean and s.e.m., respectively, from 3–6 independent experiments. The exact sample sizes are provided in Supplementary Table 3. Statistical analysis was performed using a two-way ANOVA, followed by Šidák’s post hoc test (NS, P > 0.05; *P < 0.05, **P < 0.01 and ***P < 0.001). g,h, Schematic representation of the G-protein coupling preference for Gq (g) and Gs (h).
We performed the NanoBiT G-protein dissociation assay to examine the contributions of these residues to Gq and Gs activations. Substitutions of residues on the TM4 side of ICL2, especially E3174.38A, preferentially reduced Gq response, as expected (Fig. 5f and Extended Data Fig. 7d). Surprisingly, F314ICL2A and F315ICL2A substitutions also caused a substantial reduction in Gq coupling while having little effect on Gs coupling. Thus, mutations affecting ICL2 preferentially reduce Gq signaling overall. These results indicate that Gq signaling is more dependent than Gs signaling on interactions involving ICL2.
Gq-bound PTH1R shows point-to-point interactions that involve the intracellular cavity and C terminus of α5h but not the lateral sides of TMs 5 and 6 (Figs. 3c and 4a,c). In sharp contrast, Gs-bound PTH1R shows face-to-face interactions that involve TM5, TM6 and the lateral side of α5h, along with point-to-point interactions as observed in the Gq complex (Figs. 3d and 4b,d). Thus, for Gq coupling, anchoring of the G protein by ICL2 is necessary to stabilize the relative positions of the receptor and G protein, whereas it is dispensable for Gs coupling (Fig. 5g,h).
Structural comparison with other GPCRs
To examine the generality of the Gq versus Gs preference observed in our PTH1R structures, we compared PTH1R to other receptors. Among the class B1 GPCRs, only a relatively low-resolution structure of corticotropin-releasing factor type-2 receptor (CRF2R) bound to G11, a member of the Gq subfamily30, has been reported. G11-bound CRF2R exhibits additional expansion of TM5 and TM6, along with a more vertically inserted α5h, compared to Gs-bound CRF2R (Extended Data Fig. 9a). Hence, the Gs-bound state shows more extensive hydrophobic interactions involving TM5, TM6 and α5h compared to the G11-bound state, similar to PTH1R (Extended Data Fig. 9b–e). A previous study indicated that G11 signaling depends on ICL2 to some extent, whereas Gs signaling does not30. More specifically, polar residues within ICL2, such as E220ICL2 and R2234.41 in CRF2R, contribute to the G11 signal, whereas their contribution to the Gs signal is relatively limited. Thus, CRF2R shows coupling selectivity mechanisms similar to those of PTH1R. Then, we compared structural features observed in PTH1R to other classes of GPCRs. The structures of Gq-bound and Gs-bound adhesion GPCR L3 (ADGRL3), a class B2 GPCR, were reported previously31. Although TM5 marginally moves toward TM6, Gq-bound and Gs-bound ADGRL3 show almost the same conformations (Extended Data Fig. 9f,g). Among the class A GPCRs, we compared the Gq versus Gs coupling preference for neurokinin 1 receptor (NK1R) and cholecystokinin 1 receptor (CCK1R)23,32. The structures of Gq-bound and Gs-bound NK1R are almost identical (Extended Data Fig. 9h). On the other hand, α5h in Gs-bound CCK1R is more protruded from the receptor core compared to that in Gq-bound CCK1R, although the conformation of the TM region is almost identical in both complexes (Extended Data Fig. 9i). These classes of GPCRs exhibit structural features different from those of PTH1R. Taken together, the evidence suggests that the mechanisms demonstrated in this study are conserved only in class B1 GPCRs.
Discussion
We determined the high-resolution structure of the PTH–PTH1R–Gq complex. By comparing the structures of Gq-bound PTH1R and Gs-bound PTH1R, we identified regions that have specific and crucial roles in each coupling, suggesting mechanisms for coupling preference between Gq and Gs. Specifically, Gq-bound PTH1R recognizes the C-terminal YG.H5.23 of Gq and anchors it through ICL2. In Gs binding, the recognition of YG.H5.23 remains crucial but binding is supported by surface hydrophobic interactions involving TM5, TM6 and α5h (Fig. 5g,h).
This study provides comprehensive data to understand the allosteric mechanism that links the ligand binding to G-protein preference. Specifically, we present an interpretable structure of the ligand-tilted state and, through MD simulations, demonstrate the role of glycans in stabilizing this conformation (Fig. 2b,c and Extended Data Fig. 5i–k). Furthermore, our kinetic measurements revealed that mutations at glycan-associated residues of PTH1R tend to reduce the ligand residence time of PTH. Notably, the triple-mutant PTH1R (T1751.27A;N1761.28A;E1771.29A), which exhibited the most pronounced reduction in ligand residence time, also showed significant preference for Gs over Gq in the NanoBiT G-protein dissociation assay (Fig. 2f,k,l). These findings raise the possibility that the glycan stabilizes the tilted state and extends ligand residence time, which could in turn influence G-protein preference. A series of studies have shown that other factors also influence allosteric signal transmission. For instance, several studies highlighted the importance of Ca2+-mediated interactions with extracellular loop 1 (ECL1) for prolonged signaling33,34,35. Other studies also indicated that position 5 of the ligands is a critical determinant in distinguishing the properties of PTH and PTHrP36,37,38. Furthermore, although PTH1R is currently the only class B1 GPCR for which the structure of the tilted state has been reported, it remains unclear whether a corresponding conformation exists in other receptors. These aspects need to be investigated in future structural studies.
Furthermore, during the revision of this manuscript, a cryo-EM structure of the LA-PTH-bound PTH1R–Gq complex was reported by another group39. Consistent with our observations, Gq and Gs exhibit distinct binding modes, contributing to a growing understanding of G-protein preference among class B1 GPCRs.
This study provides a structural basis for understanding the Gq versus Gs coupling preference of PTH1R, which may facilitate the development of drugs for osteoporosis and hypocalcemia. Furthermore, as many class B1 GPCRs are reported to couple with both Gq and Gs40,41, these findings enhance the understanding not only of PTH1R but also of other class B1 GPCRs. However, further studies are needed for a comprehensive understanding of the coupling preference of class B1 GPCRs.
Methods
Constructs
The full-length human PTH1R gene with its N-terminal signal sequence replaced with an HA signal sequence was subcloned into the pEG BacMam vector42, with a C-terminal eGFP–His8 tag and a 3C protease cleavage site. The FLAG epitope tag (DYKDDDDK) was introduced between the HA signal sequence and PTH1R. Rat Gβ1 and bovine Gγ2 were subcloned into the pFastBac Dual vector and engineered miniGαqiN17 was subcloned into the C terminus of the bovine Gγ2 with a 9-aa GSAGSAGSA linker. A His6 tag was inserted into N terminus of Gβ1. The resulting pFastBac dual vector can express the miniGq trimer. RIC8A, a guanine nucleotide exchange factor for Gα subunits, was subcloned into pFastBac vector without any tags. The gene encoding scFv16 was synthesized (GeneArt) and subcloned into a pFastBac vector, with the resulting construct encoding the GP67 secretion signal sequence at the N terminus and a His8 tag followed by a TEV protease cleavage site at the C terminus.
Expression and purification of human PTH1R
Bacmid preparation and virus production were performed according to the Bac-to-Bac baculovirus system manual (Gibco, Invitrogen). HEK293S N-acetylglucosaminyl-transferase I-negative cells (American Type Culture Collection, CRL-3022) were cultured in Freestyle 293 medium (Invitrogen) supplemented with 2% FBS (Sigma-Aldrich) in the presence of 8% CO2 and infected by P2 baculovirus at a density of approximately 4 × 106 cells per ml. After a 16-h incubation at 37 °C, the culture was supplemented with 10 mM sodium butyrate and incubated at 30 °C for 72 h. The cultured cells were collected by centrifugation (5,000g, 12 min, 4 °C) and disrupted by sonication in lysis buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl and 10% glycerol). The membrane fraction was collected by ultracentrifugation (186,000g, 1 h, 4 °C) and solubilized for 1 h at 4 °C in solubilization buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 10% glycerol, 1% LMNG (Anatrace), 0.1% CHS (Sigma-Aldrich) and 1 μM PTH). Insoluble materials were removed by ultracentrifugation (186,000g, 1 h, 4 °C) and incubated with Ni-NTA resin (Qiagen) for 45 min. Ni-NTA resin was washed with ten column volumes of wash buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.03% LMNG, 0.003% CHS, 100 nM PTH and 30 mM imidazole) and then eluted with the same buffer supplemented with 270 mM imidazole. The eluate was treated with 3C protease and dialyzed with dialysis buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 10% glycerol, 0.03% LMNG and 0.003% CHS). The cleaved GFP–His8 tag and the 3C protease were removed with Ni-NTA resin. The receptor was purified by size-exclusion chromatography (SEC) on a Superdex 200 10/300 Increase column equilibrated in SEC buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.03% LMNG, 0.003% CHS and 100 nM PTH). The peak fractions were collected and concentrated to about 2.3 mg ml−1. The receptor was frozen in liquid nitrogen after supplementing with 10% glycerol.
Expression and purification of miniGq trimer
The miniGq heterotrimer was expressed and purified using the Bac-to-Bac baculovirus expression system as previously described43. Spodoptera frugiperda (Sf9; Gibco) cells at a density of 3 × 106 cells per ml were coinfected with baculoviruses encoding miniGq trimer and RIC8A. The infected Sf9 cells were incubated in Sf900II medium at 27 °C for 48 h. The Sf9 cells were collected by centrifugation (5,000g, 12 min, 4 °C) and disrupted by sonication in lysis buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 10% glycerol, 1 mM MgCl2, 1 μM GDP (Roche) and 100 μM TCEP). Insoluble materials were removed by ultracentrifugation (186,000g, 1 h, 4 °C) and incubated with Ni-NTA resin (Qiagen) for 30 min. Ni-NTA resin was washed with 15 column volumes of wash buffer (20 mM Tris-HCl pH 8.0, 500 mM NaCl, 10% glycerol, 1 mM MgCl2, 1 μM GDP (Roche), 100 μM TCEP and 30 mM imidazole) and then eluted with the elution buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 10% glycerol, 1 mM MgCl2, 1 μM GDP (Roche), 100 μM TCEP and 300 mM imidazole). The eluate was treated with 3C protease and dialyzed with lysis buffer. The cleaved His6 tag and the 3C protease were removed with Ni-NTA resin. The Gq trimer was purified by SEC on a Superdex 200 10/300 Increase column equilibrated in SEC buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM MgCl2, 1 μM GDP (Roche) and 100 μM TCEP). The peak fractions were collected and concentrated to approximately 3.5 mg ml−1. The protein was frozen in liquid nitrogen after supplementing with 10% glycerol.
Expression and purification of scFv16
The His8-tagged scFv16 was expressed and secreted by Sf9 insect cells, as previously reported44,45. The infected Sf9 cells were incubated in Sf900II medium at 27 °C for 48 h. The Sf9 cells were removed by centrifugation (5,000g, 12 min, 4 °C). The secreta-containing supernatant was pH-balanced and chelating agents were quenched with a buffer containing 20 mM HEPES–NaOH pH 7.5, 150 mM NaCl, 5 mM CaCl2 and 1 mM NiCl2). The supernatant was mixed with Ni Superflow resin (GE Healthcare Life Sciences) and stirred for 1 h at 4 °C. The collected resin was washed with ten column volumes of wash buffer (20 mM HEPES–NaOH pH 7.5, 500 mM NaCl and 20 mM imidazole) and then scFv16 was eluted with elution buffer (20 mM HEPES–NaOH pH 7.5, 150 mM NaCl and 400 mM imidazole). The eluate was collected and purified by SEC on a Superdex 75 Increase 10/300 column (GE) using SEC buffer (20 mM HEPES–NaOH pH 7.5 and 150 mM NaCl). The peak fractions were collected and concentrated to approximately 2.6 mg ml−1. The protein was frozen in liquid nitrogen after supplementing with 10% glycerol.
Complex formation and purification of PTH1R–miniGq–scFv16
First, 500 μg of purified PTH1R was mixed with 1 mg of miniGq trimer in the presence of 10 μM of PTH and apyrase. After 1 h of incubation, 400 μg of scFv16 was supplemented before incubating on ice overnight. The mixture was incubated with Ni-NTA resin (Qiagen) for 30 min, which was then washed with three column volumes of wash buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 10% glycerol, 0.03% LMNG, 0.003% CHS, 1 μM PTH and 20 mM imidazole) and eluted with elution buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 10% glycerol, 0.03% LMNG, 0.003% CHS, 1 μM PTH and 300 mM imidazole). The eluate was purified by SEC on a Superdex 200 10/300 Increase column equilibrated in SEC buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.03% LMNG, 0.003% CHS and 1 μM PTH). The peak fractions were collected and concentrated to about 6.2 mg ml−1.
Grid preparation and cryo-EM data collection
The purified PTH1R–miniGq–scFv16 complex was applied onto a freshly glow-discharged Quantifoil holey carbon grid (R1.2/1.3, UltraAu, 300-mesh) and blotted at 4 °C in 100% humidity for 4.0 s. The grid was then plunge-frozen in liquid ethane using a Vitrobot Mark IV (FEI). Data collections were performed on a Titan Krios G4i microscope (Thermo Fisher Scientific) running at 300 kV and equipped with a BioQuantum K3 imaging filter and a K3 direct electron detector (Gatan) in electron counting mode. In total, 3,222 videos were acquired at a nominal magnification of 105,000 with a calibrated pixel size of 0.83 Å per pixel. All videos were acquired using the SerialEM (version 4) software46 with a defocus range of −0.8 to −1.6 μm and an objective aperture of 10 μm. Each video was acquired for 2.3 s and split into 48 frames, resulting in an accumulated exposure of about 50.939 e− per Å2 at the micrograph.
Cryo-EM image processing
All acquired movies were dose-fractionated and subjected to beam-induced motion correction implemented in RELION 3.1 (ref. 47). Then, dataset was exported to cryoSPARC version 3.1 (ref. 48) and the contrast transfer function (CTF) parameters were calculated using patch CTF estimation. A total of 1,991,608 particles were detected using topaz49 and extracted. The particles were subjected to two rounds of two-dimensional classifications to remove ice contamination and deviated particles. Next, the particles were re-extracted and exported to RELION 4.0 (ref. 47) and then subjected to two rounds of 3D classification to classify on the basis of extracellular heterogeneity. The resulting two sets of particles (upright state, 265,775; tilted state, 259,479) were subjected to Bayesian polishing. The particle sets were exported to cryoSPARC and subjected to CTF and nonuniform refinement, which yielded maps with a global nominal resolution of 2.77 Å (the upright state) and 2.76 Å (the tilted state), according to the Fourier shell correlation = 0.143 criteria. To resolve the extracellular region, receptor-focused local refinement was performed, resulting in maps with a nominal resolution of 2.80 Å (the upright state) and 2.75 Å (the tilted state). Finally, the overall refined map and local refined map were combined by phenix.combine_focused_maps program and sharpened by phenix.auto_sharpen program (version 1.20)50. The processing strategy is described in Extended Data Figs. 2 and 3.
Model building and refinement
The quality of the density map was sufficient to build an atomic model. The previously reported high-resolution cryo-EM structure of Gs-bound PTH1R (Protein Data Bank (PDB) 7VVL), crystal structure of PTH1R-ECD (PDB 3C4M), cryo-EM structure of MRGPRX2–Gq complex (PDB 7S8L) and cryo-EM structure of MT1–Gi complex (PDB 7DB6) were used as the initial models for the model building of PTH1R-TM, PTH1R-ECD, Gαq and Gβγ–scFv16, respectively16,21,22,24. Initially, the models were fitted into the density map by jiggle fit using Coot (version 0.9.8)51. Then, atomic models were manually readjusted into the density map using Coot. Finally, atomic models were refined using phenix.real_space_refine (version 1.20) with the secondary-structure restraints using phenix.secondary_structure_restraints. The statistics of the 3D reconstruction and model refinement are summarized in Supplementary Table 1. All molecular graphics figures were prepared with CueMol (version 2.3; http://www.cuemol.org) and UCSF ChimeraX (version 1.3)52.
MD simulation
MD simulations were performed with GROMACS 2020.3 and 2024 (ref. 53). The simulation system was set with a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) membrane bilayer, 150 mM NaCl and TIP3P water molecules using Charmm GUI54. The net charge was neutralized by adding Cl− ions. The molecular topology and force field parameters from CHARMM36m were used for all simulations55. The simulation results were analyzed with PyMOL (version 2.50; https://pymol.org/2/), getcontacts scripts (https://getcontacts.github.io/) and mdtraj (version 1.9.7)56.
For PTH–PTH1R–Gq/Gs complex simulations, the initial structure of PTH1R was prepared using the program Coot (version 0.9.8). For PTH1R, GSAGSAGSA and GSAGSA linkers were replaced with the missing loop region of V55ECD to R104ECD and ECL1, respectively. N and C termini were truncated as observed in the cryo-EM structures. For Gαq, GSAGSAGSA and GSAGS linkers were added to mimic the missing loop regions of K51 to G68 and D85 to R93. ScFv16 was removed to facilitate efficient calculation. For the PTH–PTH1R–Gq complex, initial system dimensions were 146 × 146 × 180 Å and comprised 570 POPC lipids, 92,203 water molecules, 351 chloride ions and 345 sodium ions. For PTH–PTH1R–Gs simulations, a similar strategy was used. The long missing regions were replaced with GSA linkers and antibody Nanobody 35 was removed for efficient calculation. The composition of the system was generally similar. The simulation systems were energy-minimized until the maximum force was reduced below 1,000 kJ mol−1 nm−2 with fixed positions of the nonhydrogen atoms. After minimization, equilibration were performed under NVT and NPT ensembles for 10 and 50 ns, respectively, with 10 kcal mol−1 Å−2 restraints for heavy atoms of the protein. Before production simulation, another 50-ns equilibration was performed under the NPT ensemble without any restraints. The production runs of the equilibrium simulations were performed three times for each 200 ns without any restraints. The detailed conditions for the production runs are as follows: a 2-fs timestep in the NPT ensemble with semi-isotropic coupling at 310 K and 1 bar, maintained by the Nose–Hoover thermostat and Parrinello–Rahman barostat. The long-range electrostatic interactions were calculated by the particle mesh Ewald method.
For the glycan-containing PTH–PTH1R complex simulations, the initial structure was prepared by completing the missing loop using the program Coot in the same manner as the PTH–PTH1R–Gq/Gs complex simulations. In the glycan-free model, the glycans were removed and, in the triple-mutant model, T1751.27, N1761.28 and E1771.29 were modified to alanine. The initial system was created using Charmm GUI54 under the following conditions: a simulation box of 100 × 100 × 148 Å, comprising 256 POPC lipids, approximately 33,000 water molecules, 98 chloride ions (99 in the triple-mutant model) and 88 sodium ions. Before the production simulations, energy minimization was carried out until the maximum force was reduced below 1,000 kJ mol−1 nm−2, followed by equilibration in the NVT and NPT ensembles for a total of 51.8 ns, while gradually reducing the position restraints on each atom. The production runs of the equilibrium simulations were performed three times for 500 ns each, without any restraints. The detailed conditions for the production runs are as follows: a 2-fs timestep in the NPT ensemble with semi-isotropic coupling at 300 K and 1 bar, maintained by the v-rescale thermostat and C-rescale barostat. Long-range electrostatic interactions were calculated using the particle mesh Ewald method.
Ligand dissociation rate measurement
Dissociation rates of TMR-labeled PTH or PTHrP from the PTH1R-expressing cell surface were measured using fluorescence microscopy with a perfusion system. PTH and PTHrP were synthesized and labeled with TMR at K13 by GenScript, according to a previous report9. To prevent cellular responses derived from mechanical stimuli associated with perfusion, a TRPV4-knockout HEK293A cell line (Thermo Fisher Scientific)57 was used in the dissociation rate measurements.
Transfection was performed using Lipofectamine 3000 (Thermo Fisher Scientific) as previously reported, with some modifications58. Transfection solution was prepared by mixing solution A (9 µl of Opti-MEM (Thermo Fisher Scientific) with 100 ng of plasmid DNA (N-terminally FLAG-tagged PTH1R/pcDNA3.1, LgBiT–AT1R/pFC15A for photobleaching control or an empty pCAGGS vector for nonspecific binding control) and 0.5 µl of P3000 reagent) and solution B (9 µl of Opti-MEM with 0.5 µl of Lipofectamine 3000 reagent) and incubated for 15 min at 25 °C before mixing with cell suspension. The transfection solution was mixed with 400 µl of cell suspension (1 × 106 cells per ml) in DMEM (Nissui) supplemented with 10% FBS (Gibco), glutamine, penicillin and streptomycin. Immediately after mixing, the cell suspension was seeded into µ-slide 6(VI) (80606, ibidi; 150 µl per chamber) and incubated for 16 h at 37 °C and 5% CO2.
Fluorescence imaging was performed using a custom-built fluorescence microscope system (Ti2E, Nikon; ×20 objective, MRH07241, Nikon; image-splitting optics, W-view Gemini-2C and W-view Gemini, Hamamatsu; dichroic filters, ZT405/488/561/640/705rpc, Chroma in the microscope, FF640-FDi01, Semrock in W-view-2C and T560lpxr-UF21 in W-view; emission filter, FF01-600/52, Semrock; camera, ORCA-Fusion BT, Hamamatsu; laser, OBIS LS 561 nm, 100 mW, Coherent)58. Image acquisition and microscope control were managed using AIS 2.5 software (Zido). Culture medium was replaced with solution C (HBSS containing 0.01% BSA (fatty acid-free grade; SERVA), 5 mM HEPES (pH 7.4) and 30 µM Dyngo-4a (S7163, Selleck)) to inhibit endocytosis after ligand stimulation. Then, 50 µl of 5 µM PTH–TMR or PTHrP–TMR diluted in solution C was loaded in each chamber of µ-slide 6(VI) and connected with a 20-ml syringe (SS-20ESZ, Terumo) by silicone tubing (7318212, Bio-Rad) with an adaptor (10831, ibidi) and a joint (WR0525, Terumo). The syringe was set on a syringe pump (STC-523, Terumo) and the flow path was filled with solution C. The perfusion was initiated at a flow rate of 10 µl s−1 and synchronized with the start of image acquisition under the following conditions: exposure time, 500 ms per frame; frame rate, 2 fps; total frames, 600; laser power, 50 mW at 561 nm with 5% ND filter; temperature, room temperature (25 °C). For photobleaching control, N-terminally LgBiT-fused AT1R was expressed and stained by TMR–FiBiT instead of the TMR-labeled PTH or PTHrP58.
The acquired images were processed and quantified using ImageJ (version 1.52a). First, the acquired multi-tif files were background subtracted using the ImageJ rolling-ball subtraction algorithm with a ball radius of 50 pixels. Then, the cell areas were manually enclosed and registered in the region of interest manager and the average intensity of each cell in each frame was quantified. The fold change in the fluorescence intensity was normalized to the average fluorescence intensity of the initial five frames and the background drift because of changes in TMR concentration in the perfusion solution was corrected by referring to the control measurement of 5 µM TMR–FiBiT on LgBiT–AT1R-expressing cells. Minimal photobleaching of TMR–FiBiT specifically bound on LgBiT–AT1R-expressing cells was observed (Fig. 2e).
The time-dependent changes in normalized intensity of cells were fitted by the following double-exponential and triple-exponential functions using smDynamicsAnalyzer59 (https://github.com/masataka-yanagawa/IgorPro8-smDynamicsAnalyzer), an Igor Pro 9 (WaveMetrix)-based homemade program.
where m is the number of states, t is time, Ai and τi are coefficients that are related to the fraction of the ith state with time constant τi. Then, AIC values were compared between two-state and three-state models. AIC is defined as follows:
where n is the number of data points for the curve fitting, σ2 is the residual sum of squares and p is the number of free parameters. Depending on the cell, either the two-state or the three-state model was selected; however, to compare parameters under the same fitting model across all conditions and cells, the three-state model, which was selected for more cells, was used in the present study (Extended Data Fig. 6e,f). The three-state model is consistent with previous fluorescence resonance energy transfer measurements of TMR–PTH and TMR–PTHrP dissociation rates from GFP-fused PTH1R-expressing cells9. In the present study, we used a three-state exponential fitting model where the slow component \({A}_{3}\exp \left(\frac{-t}{{\tau }_{3}}\right)\) corresponds to the constant term \({A}_{0}\) used in the previous two-state model9, which assumed that the time constant of the slowest state was infinite.
NanoBiT G-protein dissociation assay
PTH-induced G-protein dissociation was measured by the NanoBiT G-protein dissociation assay27 with some modifications. Gq/11-deficient HEK293 cells60, which provided clearer NanoBiT G-protein responses in PTH1R than parental HEK293 cells, were seeded in a six-well culture plate at a concentration of 2 × 105 cells per ml (2 ml per well in DMEM (Nissui) supplemented with 5% FBS (Gibco), glutamine, penicillin and streptomycin) 1 day before transfection. A NanoBiT G-protein sensor of the Gα subunit fused with a large fragment (LgBiT) at the α-helical ___domain (Gαs–LgBiT or Gαq–LgBiT) and the N-terminally small fragment (SmBiT)-fused Gγ2 subunit with a C68S substitution (SmBiT–Gγ2-CS) was expressed along with untagged Gβ1 subunit and the G-protein chaperone RIC8A (for NanoBiT–Gq) or RIC8B (for NanoBiT–Gs). The transfection solution was prepared by combining 6 µl (per well hereafter) of polyethylenimine Max solution (1 mg ml−1; Polysciences), 200 µl of Opti-MEM (Thermo Fisher Scientific) and a plasmid mixture consisting of 200 ng of the N-terminally FLAG-tagged PTH1R (or an empty plasmid for mock transfection), 100 ng of Gα–LgBiT, 500 ng of Gβ1 subunit, 500 ng of SmBiT–Gγ2-CS subunit and 100 ng of RIC8A or RIC8B. After incubation for 1 day, the transfected cells were harvested with 0.5 mM EDTA-containing Dulbecco’s PBS (D-PBS), centrifuged and suspended in 2 ml of HBSS containing 0.01% BSA (fatty acid-free grade; SERVA) and 5 mM HEPES (pH 7.4) (assay buffer). The cell suspension was dispensed in a white 96-well plate at a volume of 80 µl per well and loaded with 20 µl of 50 µM coelenterazine (Angene) diluted in the assay buffer. After a 2-h incubation at room temperature, the plate was measured for baseline luminescence (SpectraMax L, Molecular Devices) and titrated concentrations of PTH (20 µl; 6× final concentration) were manually added. The plate was immediately read for the second measurement in kinetic mode and luminescence counts recorded from 5 min to 10 min after PTH addition were averaged and normalized to the initial counts. The fold change was further normalized to vehicle-treated samples and used to plot the G-protein dissociation response. Using the Prism software (versions 8 and 9; GraphPad Prism), the G-protein dissociation signals were fitted to a four-parameter sigmoidal concentration–response curve with a constraint of the Hill slope to absolute values less than 2. For each replicate experiment, from the parameters span (top–bottom) and half-maximal effective concentration (EC50) values, the parameter span/EC50 was calculated and values of the individual PTH1R mutants were normalized to that of WT PTH1R performed in parallel. The resulting parameter (span_mutant/EC50_mutant)/(span_WT/EC50_WT), known as RAi, was log-transformed and a resulting logRAi value was used to denote G-protein coupling activity of the mutants. For each replicate experiment, span and pEC50 values of PTH1R mutants were normalized to those of WT PTH1R performed in parallel and the resulting Emax and ∆pEC50 values are summarized in Supplementary Table 3.
Flow cytometry
Transfection was performed according to the same procedure described above. Then, 1 day after transfection, the cells were collected by adding 200 μl of 0.53 mM EDTA-containing D-PBS, followed by 200 μl of 5 mM HEPES (pH 7.4)-containing HBSS. The cell suspension was transferred to a 96-well V-bottom plate in duplicate and fluorescently labeled with an anti-FLAG epitope (DYKDDDDK) tag monoclonal antibody (Clone 1E6, FujiFilm Wako Pure Chemicals; 10 μg per ml diluted in D-PBS containing 2% goat serum and 2 mM EDTA (blocking buffer)) and a goat anti-mouse IgG secondary antibody conjugated with Alexa Fluor 488 (Thermo Fisher Scientific; 10 μg per ml diluted in the blocking buffer). After washing with D-PBS, the cells were resuspended in 200 μl of 2 mM EDTA-containing D-PBS and filtered through a 40-μm filter. The fluorescence intensity of single cells was quantified by an EC800 flow cytometer equipped with a 488-nm laser (Sony). The fluorescence signal derived from Alexa Fluor 488 was recorded in the FL1 channel and the flow cytometry data were analyzed with FlowJo software (version 10). Live cells were gated with a forward scatter (FS-Peak-Lin) cutoff at the 390 setting, with a gain value of 1.7. Values of mean fluorescence intensity (MFI) from approximately 20,000 cells per sample were used for analysis. For each experiment, we normalized the MFI value of the mutants by that of the WT performed in parallel and denoted relative levels.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Cryo-EM maps generated in this study were deposited to the EM Data Bank under accession codes EMD-61746 (upright state; composite map), EMD-61795 (upright state; consensus refinement map), EMD-61796 (upright state; focused refinement map), EMD-61747 (tilted state; composite map), EMD-61797 (tilted state; consensus refinement map) and EMD-61798 (tilted state; focused refinement map). The atomic coordinates generated in this study were deposited to the PDB under accession codes 9JR2 (upright state) and 9JR3 (tilted state). The structural models used in this study are available from the PDB under the following accession codes: PTH1R–Gs complex (class 2), 7VVL; PTH1R–Gs complex (class 4), 7VVN; H1R–Gq complex, 7DFL; CRF2R–Gs complex, 6PB1; CRF2R–G11 complex, 7TRY; NK1R–Gs complex, 7RMH; NK1R–Gq complex, 7RMG; CCK1R–Gs complex, 7MBX; CCK1R–Gq complex, 7MBY; ADGRL3–Gs complex, 7WY8; ADGRL3–Gq complex, 7WY5; PTH1R-ECD, 3C4M; MRGPRX2–Gq complex, 7S8L; MT1–Gi complex, 7DB6. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. Source data are provided with this paper.
References
Jüppner, H. et al. A G-protein-linked receptor for parathyroid hormone and parathyroid hormone-related peptide. Science 254, 1024–1026 (1991).
Potts, J. T., Kronenberg, H. M. & Rosenblatt, M. Parathyroid hormone: chemistry, biosynthesis, and mode of action. Adv. Protein Chem. 35, 323–396 (1982).
Leder, B. Z. Parathyroid hormone and parathyroid hormone-related protein analogs in osteoporosis therapy. Curr. Osteoporos. Rep. 15, 110–119 (2017).
Bastepe, M. & Gensure, R. C. Hypoparathyroidism and pseudohypoparathyroidism. In Endotext (eds Feingold, K. R. et al.) (MDText.com, 2000).
Ito, N., Hidaka, N. & Kato, H. The pathophysiology of hypophosphatemia. Best Pract. Res. Clin. Endocrinol. Metab. 38, 101851 (2024).
Lindsay, R., Krege, J. H., Marin, F., Jin, L. & Stepan, J. J. Teriparatide for osteoporosis: importance of the full course. Osteoporos. Int. 27, 2395–2410 (2016).
Brent, M. B. Abaloparatide: a review of preclinical and clinical studies. Eur. J. Pharmacol. 909, 174409 (2021).
Silva, B. C., Costa, A. G., Cusano, N. E., Kousteni, S. & Bilezikian, J. P. Catabolic and anabolic actions of parathyroid hormone on the skeleton. J. Endocrinol. Invest. 34, 801–810 (2011).
Ferrandon, S. et al. Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat. Chem. Biol. 5, 734–742 (2009).
Vilardaga, J.-P., Jean-Alphonse, F. G. & Gardella, T. J. Endosomal generation of cAMP in GPCR signaling. Nat. Chem. Biol. 10, 700–706 (2014).
Lee, S. et al. A homozygous [Cys25]PTH(1–84) mutation that impairs PTH/PTHrP receptor activation defines a novel form of hypoparathyroidism. J. Bone Miner. Res. 30, 1803–1813 (2015).
Shimizu, M. et al. Pharmacodynamic actions of a long-acting PTH analog (LA-PTH) in thyroparathyroidectomized (TPTX) rats and normal monkeys. J. Bone Miner. Res. 31, 1405–1412 (2016).
Sutkeviciute, I. & Vilardaga, J.-P. Structural insights into emergent signaling modes of G-protein-coupled receptors. J. Biol. Chem. 295, 11626–11642 (2020).
White, A. D. et al. Gq/11-dependent regulation of endosomal cAMP generation by parathyroid hormone class B GPCR. Proc. Natl Acad. Sci. USA 117, 7455–7460 (2020).
Lh, Z. et al. Structure and dynamics of the active human parathyroid hormone receptor-1. Science 364, 148–153 (2019).
Kobayashi, K. et al. Endogenous ligand recognition and structural transition of a human PTH receptor. Mol. Cell 82, 3468–3483 (2022).
Nehmé, R. et al. Mini-G proteins: novel tools for studying GPCRs in their active conformation. PLoS One 12, e0175642 (2017).
Wan, Q. et al. Mini-G-protein probes for active G-protein-coupled receptors (GPCRs) in live cells. J. Biol. Chem. 293, 7466–7473 (2018).
Koehl, A. et al. Structure of the µ-opioid receptor–Gi protein complex. Nature 558, 547–552 (2018).
Kim, K. et al. Structure of a hallucinogen-activated Gq-coupled 5-HT2A serotonin receptor. Cell 182, 1574–1588 (2020).
Cao, C. et al. Structure, function and pharmacology of human itch GPCRs. Nature 600, 170–175 (2021).
Okamoto, H. H. et al. Cryo-EM structure of the human MT1–Gi signaling complex. Nat. Struct. Mol. Biol. 28, 694–701 (2021).
Mobbs, J. I. et al. Structures of the human cholecystokinin 1 (CCK1) receptor bound to G and Gmimetic proteins provide insight into mechanisms of G-protein selectivity. PLoS Biol. 19, e3001295 (2021).
Pioszak, A. A. & Xu, H. E. Molecular recognition of parathyroid hormone by its G-protein-coupled receptor. Proc. Natl Acad. Sci. USA 105, 5034–5039 (2008).
Wootten, D., Simms, J., Miller, L. J., Christopoulos, A. & Sexton, P. M. Polar transmembrane interactions drive formation of ligand-specific and signal pathway-biased family B G protein-coupled receptor conformations. Proc. Natl Acad. Sci. USA 110, 5211–5216 (2013).
Clark, L. J. et al. Allosteric interactions in the parathyroid hormone GPCR–arrestin complex formation. Nat. Chem. Biol. 16, 1096–1104 (2020).
Inoue, A. et al. Illuminating G-protein-coupling selectivity of GPCRs. Cell 177, 1933–1947 (2019).
Flock, T. et al. Universal allosteric mechanism for Gα activation by GPCRs. Nature 524, 173–179 (2015).
Guo, J., Chung, U.-I., Kondo, H., Bringhurst, F. R. & Kronenberg, H. M. The PTH/PTHrP receptor can delay chondrocyte hypertrophy in vivo without activating phospholipase C. Dev. Cell 3, 183–194 (2002).
Zhao, L.-H. et al. Structure insights into selective coupling of G protein subtypes by a class B G-protein-coupled receptor. Nat. Commun. 13, 6670 (2022).
Qian, Y. et al. Structural insights into adhesion GPCR ADGRL3 activation and Gq, Gs, Gi, and G12 coupling. Mol. Cell 82, 4340–4352 (2022).
Harris, J. A. Selective G-protein signaling driven by substance P-neurokinin receptor dynamics. Nat. Chem. Biol. 18, 109–115 (2022).
White, A. D. et al. Ca2+ allostery in PTH-receptor signaling. Proc. Natl Acad. Sci. USA 116, 3294–3299 (2019).
Li, M., Li, M. & Guo, J. Molecular mechanism of Ca2+ in the allosteric regulation of human parathyroid hormone receptor-1. J. Chem. Inf. Model. 62, 5110–5119 (2022).
Li, M., Bao, Y., Li, M. & Guo, J. GPCR allostery: a view from computational biology. Curr. Med. Chem. 30, 4533–4553 (2023).
Gardella, T. J. et al. Parathyroid hormone (PTH)–PTH-related peptide hybrid peptides reveal functional interactions between the 1–14 and 15–34 domains of the ligand. J. Biol. Chem. 270, 6584–6588 (1995).
Behar, V. et al. Histidine at position 5 is the specificity ‘switch’ between two parathyroid hormone receptor subtypes. Endocrinology 137, 4217–4224 (1996).
Dean, T., Vilardaga, J.-P., Potts, J. T. & Gardella, T. J. Altered selectivity of parathyroid hormone (PTH) and PTH-related protein (PTHrP) for distinct conformations of the PTH/PTHrP receptor. Mol. Endocrinol. Baltim. Md 22, 156–166 (2008).
Zhang, X. et al. Allosteric mechanism in the distinctive coupling of Gq and Gs to the parathyroid hormone type 1 receptor. Proc. Natl Acad. Sci. USA 122, e2426178122 (2025).
Thompson, A. & Kanamarlapudi, V. Agonist-induced internalisation of the glucagon-like peptide-1 receptor is mediated by the Gαq pathway. Biochem. Pharmacol. 93, 72–84 (2015).
Shioda, S. et al. Pleiotropic functions of PACAP in the CNS: neuroprotection and neurodevelopment. Ann. N. Y. Acad. Sci. 1070, 550–560 (2006).
Goehring, A. et al. Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat. Protoc. 9, 2574–2585 (2014).
Iwama, A. et al. Structure and dynamics of the pyroglutamylated RF-amide peptide QRFP receptor GPR103. Nat. Commun. 15, 4769 (2024).
Sano, F. K., Akasaka, H., Shihoya, W. & Nureki, O. Cryo-EM structure of the endothelin-1–ETB–Gi complex. eLife 12, e85821 (2023).
Akasaka, H. et al. Structure of the active Gi-coupled human lysophosphatidic acid receptor 1 complexed with a potent agonist. Nat. Commun. 13, 5417 (2022).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Kimanius, D., Dong, L., Sharov, G., Nakane, T. & Scheres, S. H. W. New tools for automated cryo-EM single-particle analysis in RELION-4.0. Biochem. J. 478, 4169–4185 (2021).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. et al. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in PHENIX. Acta Crystallogr. D 75, 861–877 (2019).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. et al. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Pronk, S. et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 (2013).
Jo, S. et al. CHARMM-GUI 10 years for biomolecular modeling and simulation. J. Comput. Chem. 38, 1114–1124 (2017).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017).
McGibbon, R. T. et al. MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys. J. 109, 1528–1532 (2015).
Kuwashima, Y. et al. Comparative analysis of single-molecule dynamics of TRPV1 and TRPV4 channels in living cells. Int. J. Mol. Sci. 22, 8473 (2021).
Yoda, T., Sako, Y., Inoue, A. & Yanagawa, M. Four-color single-molecule imaging system for tracking GPCR dynamics with fluorescent HiBiT peptide. Biophys. Physicobiol. 21, e210020 (2024).
Yanagawa, M. & Sako, Y. Workflows of the single-molecule imaging analysis in living cells: tutorial guidance to the measurement of the drug effects on a GPCR. In Live Cell Imaging: Methods and Protocols (ed. Kim, S.-B.) (Springer, 2021).
Schrage, R. et al. The experimental power of FR900359 to study Gq-regulated biological processes. Nat. Commun. 6, 10156 (2015).
Acknowledgements
We thank K. Ogomori, C. Harada and Y. Matsuzaki at the University of Tokyo for their technical and editorial assistance in structural analysis, K. Sato, S. Nakano and A. Inoue at Tohoku University for their help with plasmid preparation, the cell-based GPCR assays and the flow cytometry analysis and T. Kambara, Y. Inutsuka, S. Xu, Y. Onishi, M. Hayashi, S. Yamamoto, J. Asada, M. Kakiuchi and T. Furuya at RIKEN and the University of Tokyo for their assistance in kinetic experiments. This work was supported by grants from the Platform for Drug Discovery, Informatics and Structural Life Science by the Ministry of Education, Culture, Sports, Science and Technology and Japan Society for the Promotion of Science (JSPS) KAKENHI grants JP21H05037 (O.N.), JP23KJ0491 (F.K.S.), 24KJ0906 (H.A.), JP22K19371 (W.S.) and JP22H02751 (W.S.), the ONO Medical Research Foundation (W.S.), the Kao Foundation for Arts and Sciences (W.S.), the Takeda Science Foundation (W.S.), the Uehara Memorial Foundation (W.S.) and the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research) from AMED under grant number JP22ama121002 (support no. 3272, O.N.). T.K. was funded by JSPS KAKENHI JP20K15754, JP22K15072 and JP24K01961 and JST PRESTO JPMJPR22E4. K. Kobayashi was funded by JSPS KAKENHI JP24K18060, JP25H02243, the Astellas Foundation for Research on Metabolic Disorders and the Kazato Research Foundation. M.Y. was funded by JSPS KAKENHI JP24K01982 and JP24H01266 and JST PRESTO JPMJPR20EF. A.I. was funded by JSPS KAKENHI JP21H04791, JP21H05113, JP21H05037 and JP24K2128 and AMED under grant numbers JP22ama121038 and JP22zf012700 and the Japan Science and Technology Agency (JST) under grant numbers JPMJFR215T and JPMJMS2023 and The Uehara Memorial Foundation. Y.O. was funded by JSPS KAKENHI JP19H03394, JP19H05794, JP19H05795, JP22H02798 and JP22H04926, JST-CREST JPMJCR1852 and JPMJCR20E2 and JST-Moonshot R&D grant JPMJMS2025-14. Y.K. was funded by JSPS KAKENHI 22K19263 and 22H02555 and AMED under grant number JP22gm6410026 and JP223fa627001.
Author information
Authors and Affiliations
Contributions
F.K.S. performed the cryo-EM structural study and MD simulation. F.K.S., K. Kobayashi and H.H.O. constructed and purified the plasmids. F.K.S. purified the miniGq heterotrimer, scFv16 and PTH-bound PTH1R with the assistance of K. Kobayashi. F.K.S. prepared the cryo-EM grids. F.K.S. collected the cryo-EM data with the assistance of T.T., T.K. and Y.K. F.K.S. processed the cryo-EM data. F.K.S. built the atomic models with the assistance of H.H. and Y.I. F.K.S. performed the MD simulation with the assistance of T.A.K. and R.I. T.A. performed the kinetic experiments with the assistance of Y.O. A.I. and K. Kawakami performed the mutation assays. K.S., Y.S. and M.Y. performed the dissociation rate measurements of PTH and PTHrP from PTH1R. F.K.S prepared the initial paper and F.K.S., H.A. and W.S. wrote the paper with input from all authors. A.I. and O.N. supervised the research.
Corresponding authors
Ethics declarations
Competing interests
O.N. is a cofounder and scientific advisor for Curreio. All other authors declare no competing interests.
Peer review
Peer review information
Nature Chemical Biology thanks Sanduo Zheng and the other, anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Purification of PTH–PTH1R–Gq complex.
Size-exclusion chromatography profiles of the PTH–PTH1R–Gq complex. The peak fraction was analysed by SDS-PAGE. Protein purification was performed once for structural analysis and was not independently repeated.
Extended Data Fig. 2 Cryo-EM analysis and map quality of the PTH–PTH1R–Gq complex.
a. Cryo-EM workflow of the PTH–PTH1R–Gq complex. b, c. Representative cryo-EM micrograph (b) and 2D averages (c) of the PTH–PTH1R–Gq complex. A total of 3,222 similar micrographs were acquired and subsequently used for further analysis (b). d–f. Local-resolution cryo-EM density maps (d), Fourier shell correlation (FSC) curve (e), and Directional 3DFSC plots (f) calculated by the 3DFSC processing server (https://3dfsc.salk.edu/upload/info/) of the overall refined upright state, the focused refined upright state, the overall refined tilted state, and the focused refined tilted state.
Extended Data Fig. 3 Model quality of the PTH–PTH1R–Gq complex.
a. A workflow of map postprocessing and model building. b. FSC between model and map for the upright and tilted states. c. Cryo-EM densities fitted to the model.
Extended Data Fig. 4 Structural comparison of the ligand binding site and extracellular domains in PTH-bound PTH1R.
a–d. Hydrophobic interactions between PTH and the ECD of PTH1R in the upright (a, c) and tilted (b, d) states. e, f. Comparison of the ligand binding modes in the transmembrane ___domain of PTH1R between the upright and tilted states. Representative interacting residues are shown as stick models. g, h. Atomic model and cryo-EM density maps around N-acetylglucosamine (NAG) modified on N1761.28 in the upright (g) and tilted (h) states. The contour levels are set to 6.2 and 7.3, respectively. i, j. Comparison of the upright (i) and tilted (j) states with the corresponding classes of Gs-bound PTH1R. k, l. Local-resolution cryo-EM density maps of the tilted state (k) and class 4 (Gs-bound) (l). The contour levels are set to 5.6 and 0.013, respectively.
Extended Data Fig. 5 Analysis of molecular dynamics (MD) simulations.
a–e. Root mean square deviation of Cα atoms in MD simulations for the glycan-containing tilted model (a), glycan-free tilted model (b), triple-mutated tilted model (c), PTH1R-Gq complex (d), and PTH1R-Gs complex (e). Average values, calculated every 10 frames, are shown in bold. f–h. Ramachandran plot of E1771.29 in MD simulations for the glycan-containing (f), glycan-free (g), and triple-mutated (h) tilted models. i–k. Angles among the Cα atoms of S3PTH, S17PTH, and H32PTH for the glycan-containing (i), glycan-free (j), and triple-mutated (k) tilted models. Dashed lines indicate the angles in the cryo-EM structures. Average values, calculated every 10 frames, are shown in bold.
Extended Data Fig. 6 PTH1R kinetic experiments.
a. Control fluorescence time-lapse images of TMR-FiBiT on LgBiT-AT1R-expressing cells (upper panels) with and without perfusion flow and of TMR-PTH/PTHrP on mock transfected cells (lower panels) with flow. Images show representative data from 10-14 cells across two independent experiments. b, c. Fluorescence time-lapse images of TMR-PTH (b) and PTHrP (c) dissociation from wild-type and mutant PTH1R-expressing cells. The images of wild-type and the triple mutant are the same as Fig. 2e for comparison. The white bar indicates 5 μm (a–c). d. Time-course changes in fluorescence intensity normalized at the start of perfusion (red: PTH, blue: PTHrP). Lines and error bands represent mean and SEM, respectively, from 8-20 cells done in two independent experiments (d). e, f. AIC comparison between double- and triple-exponential fitting for PTH (e) and PTHrP (f). Dots with a line show the AICs calculated from the same cell data. Red lines show mean AIC of all cells plotted. Statistical analysis was performed with paired t-test. g, h. Logarithmic time constants (g) and percentage (h) of three kinetic components (upper: fast, middle: medium, lower: slow). Mean and SEM are plotted as bar graphs (n = 8-20 cells). The exact sample sizes are provided in Supplementary Tables 2. Statistical analysis was performed with two-way ANOVA, followed by Sidak’s post-hoc test for PTH and PTHrP comparison, and Dunnett’s post-hoc test for wild-type and mutants. In all panels, p-values are shown as follows: ns, p > 0.05; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. The exact p-values are provided in source data file.
Extended Data Fig. 7 NanoBiT-G-protein-dissociation assay.
a. The Gq and Gs coupling activity levels titrated by transfection plasmid volumes of the wild-type PTH1R. b, c. Gq- and Gs-coupling activities and expression levels of the PTH1R mutants. PTH-induced Gq and Gs responses were analysed by a NanoBiT-G-protein-dissociation assay (b). Data are shown as logarithms of relative intrinsic activities (Log RAi; see the Methods). Statistical analysis was performed with two-way ANOVA, followed by Sidak’s post-hoc test (ns, p > 0.05; *, p < 0.01; ***, p < 0.001). The exact p-values are provided in source data file. Cell-surface expression levels were measured by flow cytometry analysis (c). Bars and error bars represent mean and SEM, respectively, from 3 independent experiments (b,c). d. The expression levels of the PTH1R mutants. Cell-surface expression levels were measured by flow cytometry analysis. Bars and error bars represent mean and SEM, respectively, from 3–6 independent experiments. The exact sample sizes are provided in Supplementary Tables 3 (a,d).
Extended Data Fig. 8 Structural Comparison of miniGq and Native Gq, and Sequence Alignment of PTH and PTHrP.
a. Structural superimposition of miniGq with native Gq in complex with H1R. ICL2-interacting residues are shown as stick models. b. Sequence alignment of PTH and PTHrP, calculated using ESPript 3.0 (https://espript.ibcp.fr/ESPript/ESPript/).
Extended Data Fig. 9 Structural comparison of Gq (G11) and Gs among class A, B1, and B2 GPCRs.
a. Structural comparison of the opening degrees in TMs 5 and 6 of the receptor, and the angles of α5h in the G-protein in G11-bound and Gs-bound CRF2R. b–e. Interfaces between TM5, TM6, and α5h in G11-bound CRF2R (b), Gs-bound CRF2R (c), Gq-bound PTH1R (d), and Gs-bound PTH1R (e). f–i. Structural comparison of Gq and Gs regarding the opening degrees of TMs 5 and 6 in the receptor, and the angles of α5h in the G-protein for PTH1R (f), ADGRL3 (g), NK1R (h), and CCK1R (i).
Supplementary information
Supplementary Tables 1–3
Supplementary Table 1: Cryo-EM data collection, refinement and validation statistics. Supplementary Table 2: Fitting parameters for kinetic experiments of PTH and PTHrP. Supplementary Table 3: Detailed parameters of NanoBiT G-protein dissociation assay.
Source data
Source Data Figs. 2, 4 and 5 and Extended Data Figs. 6 and 7
Source data related to the NanoBiT G-protein dissociation assay and kinetic measurements.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sano, F.K., Shimizume, K., Kobayashi, K. et al. Insights into G-protein coupling preference from cryo-EM structures of Gq-bound PTH1R. Nat Chem Biol (2025). https://doi.org/10.1038/s41589-025-01957-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41589-025-01957-6
