
Abstract
Bacillus Calmette–Guérin (BCG) induction and maintenance (I+M) after transurethral resection of bladder tumor is standard of care (SOC) in high-risk non-muscle invasive bladder cancer (NMIBC). However, disease recurrence/progression occurs in approximately 40% of patients at 2 years, with unfavorable prognosis. Limited bladder-sparing therapeutic options exist, and no improvements to response durability have been observed in decades. CREST is a global, phase 3, randomized trial evaluating subcutaneous sasanlimab in combination with BCG-I+M (Arm A), sasanlimab in combination with BCG-I (Arm B) or BCG-I+M (Arm C) in BCG-naive high-risk NMIBC. The primary endpoint was investigator-assessed event-free survival (EFS) for Arm A versus Arm C; key secondary endpoints were EFS (Arm B versus Arm C) and overall survival. Patients were randomized 1:1:1 to Arm A (N = 352), Arm B (N = 352) and Arm C (N = 351). The trial met its primary endpoint with a statistically significant and clinically meaningful prolongation of EFS (Arm A versus Arm C); hazard ratio, 0.68 (95% confidence interval: 0.49–0.94); one-sided P = 0.0095. The 36-month estimated EFS rates were 82.1% (Arm A) and 74.8% (Arm C). EFS benefit for Arm A versus Arm C was observed across prespecified subgroups, including carcinoma in situ (CIS) and T1. The safety profile of the combination was consistent with the known profiles. To our knowledge, sasanlimab is the first anti-PD-1 antibody to show a clinically meaningful prolongation of EFS when combined with BCG-I+M versus SOC in patients with BCG-naive high-risk NMIBC. Sasanlimab combined with BCG-I+M has the potential to redefine the treatment paradigm and clinical decision-making for patients with BCG-naive high-risk NMIBC. ClinicalTrials.gov identifier: NCT04165317.
Similar content being viewed by others

Main
Bladder cancer is the ninth most prevalent cancer worldwide1, and approximately 75% of cases are non-muscle invasive bladder cancer (NMIBC)2,3. High-risk NMIBC is treated with transurethral resection of bladder tumor (TURBT) followed by intravesical induction and maintenance with Bacillus Calmette–Guérin (BCG-I+M), the current standard of care (SOC)2,3. The SOC schedule for BCG is once-weekly induction for 6 weeks, followed by maintenance for up to 3 years2,3. However, disease recurrence and/or progression are common and may result in patients requiring subsequent treatment, including radical cystectomy and systemic therapies for muscle-invasive bladder cancer and advanced disease2,3. Treatments that allow bladder preservation remain limited; therefore, there is a high unmet need for enhanced treatment options that provide durable disease control by delaying disease recurrence and progression while maintaining quality of life (QOL)3.
Clinical trials have examined the efficacy and safety of PD-(L)1 inhibitors in BCG-naive and post-BCG NMIBC settings4, resulting in US Food and Drug Administration approval of pembrolizumab in BCG-unresponsive carcinoma in situ (CIS) NMIBC5,6. Sasanlimab is a humanized, monoclonal antibody specific for human PD-1 that blocks the interaction between PD-1 and PD-L1/PD-L2 (ref. 7). In a phase 1 trial (NCT02573259), sasanlimab showed durable anti-tumor activity and a manageable safety profile in patients with advanced or metastatic solid tumors8,9. Exposure to BCG is associated with increased PD-L1 expression in preclinical models and tumors from patients with high-risk NMIBC10,11,12. Enhanced PD-L1 expression might contribute to the immune escape mechanism in bladder cancer cells13, thus justifying the combination of PD-(L)1 inhibition with BCG to improve therapeutic efficacy.
CREST (NCT04165317) is a global, phase 3, randomized, open-label, three-arm trial examining the efficacy and safety of sasanlimab administered subcutaneously in combination with BCG compared to BCG for BCG-naive high-risk NMIBC. The primary objective was to demonstrate that sasanlimab in combination with BCG-I+M (Arm A) is superior to BCG-I+M (Arm C) in prolonging event-free survival (EFS) in patients with high-risk NMIBC. The primary endpoint was investigator-assessed EFS for Arm A versus Arm C, defined as time from randomization to recurrence of high-grade disease, progression of disease, persistence of CIS (for patients with CIS at randomization) or death due to any cause, whichever occurred first. Key secondary endpoints included investigator-assessed EFS (Arm B versus Arm C) and overall survival (OS; Arms A and B versus Arm C). An interactive infographic is available at https://www.crestphase3-infographic.com.
Results
Patients
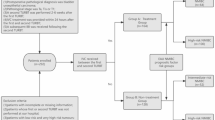
A total of 1,394 patients were screened for enrollment; 1,055 patients with BCG-naive high-risk NMIBC (high-grade Ta, T1 and/or CIS) at 140 centers in 14 countries were randomized to Arm A (N = 352), Arm B (N = 352) or Arm C (N = 351) (Fig. 1). The first patient was randomized on 20 January 2020, and the last patient was randomized on 16 November 2021. Randomization was stratified by the presence of CIS (yes or no) and geographic region (United States, Western Europe and Canada or rest of the world). Patient demographics and disease characteristics at baseline were balanced among the three arms and were representative of the overall patient population with high-risk NMIBC (Table 1). The median age was 67 years (range, 31–91); 81.8% of patients were men; and 61.2% of patients were White, 35.5% were Asian and 0.9% were Black or African American. Urothelial carcinoma was reported in 96.4% of patients; 54.2% had T1 tumor as the highest grade; and 25.5% had CIS with or without papillary tumors. The proportion of analyzed patients whose tumor tissue had PD-L1 high or low expression at baseline was 21.5% and 75.0%, respectively, and was similar across treatment arms.

aThe first patient was randomized on 20 January 2020, and the last patient was randomized on 16 November 2021. bOne patient was a screen failure but was randomized in error. cTwo patients withdrew from study treatment in the sasanlimab + BCG-I arm due to recurrence of low-grade disease. dLack of efficacy refers to patients who experience an EFS event of recurrence of high-grade disease, persistence of CIS or progression of disease. CRF, case report form.
Treatment
Sasanlimab was administered as a subcutaneous injection once every 4 weeks for up to 25 cycles. The median duration of sasanlimab treatment was 80.3 weeks (range, 4.0–103.9) in Arm A and 84.8 weeks (range, 4.0–104.4) in Arm B (Extended Data Table 1). Sasanlimab treatment completion of 25 cycles was 46.3% and 45.7% in Arms A and B, respectively; the most frequent reason for sasanlimab discontinuation was adverse events (AEs; Arm A (31.8%) and Arm B (25.0%)) (Supplementary Table 1).
The median duration of treatment and number of doses of BCG-I+M were 98.1 weeks (range, 2.0–125.1) and 18.0 doses (range, 2.0–23.0), respectively, in Arm A and 98.9 weeks (range, 2.0–110.0) and 21.0 doses (range, 2.0–25.0), respectively, in Arm C. In Arm B, the median duration of treatment and number of doses of BCG-I were 6.0 weeks (range, 1.0–34.9) and 6.0 doses (range, 1.0–12.0), respectively (Extended Data Table 1). BCG-I+M treatment completion at 2 years was 48.6% and 57.8% in Arms A and C, respectively; the most frequent reasons for BCG maintenance discontinuation were AEs in Arm A (21.9%) and lack of efficacy in Arm C (15.4%) (Supplementary Table 1).
Primary endpoint
Investigator-assessed EFS for Arm A versus Arm C
The primary endpoint of the trial was investigator-assessed EFS for Arm A versus Arm C, defined as time from randomization to recurrence of high-grade disease, progression of disease, persistence of CIS (for patients with CIS at randomization) or death due to any cause, whichever occurred first. As of the data cutoff date of 2 December 2024, based on 150 EFS events (61 in Arm A and 89 in Arm C (Extended Data Table 2)), the trial met its primary objective and demonstrated a clinically meaningful and statistically significant improvement in EFS for Arm A versus Arm C. The risk of experiencing an EFS event was 32% lower in Arm A versus Arm C (stratified hazard ratio (HR), 0.68 (95% confidence interval (CI): 0.49–0.94); one-sided P = 0.0095) (Fig. 2a). The median follow-up for the overall population was 36.3 months for EFS. Median EFS was not reached for any arm. The probability of being event free at 36 months was 82.1% for Arm A and 74.8% for Arm C (Fig. 2a and Extended Data Table 2). The results of the EFS analyses were consistent between the intent-to-treat population and all prespecified subgroups, including those defined according to geographic regions and tumor stage at randomization (Fig. 3). For patients with CIS at randomization (with or without papillary tumors), the unstratified HR was 0.53 (95% CI: 0.29–0.98), and the probability of being event free at 36 months was 83.0% for Arm A (N = 88) and 71.8% for Arm C (N = 88) (Fig. 3 and Extended Data Fig. 1). In the subgroup of patients with T1 tumor (with or without CIS) at randomization, unstratified HR was 0.63 (95% CI: 0.41–0.96), and the probability of being event free at 36 months was 81.3% for Arm A (N = 204) and 72.2% for Arm C (N = 193) (Fig. 3 and Extended Data Fig. 2).

For EFS, an event was defined as the first of recurrence of high-grade disease, progression of disease, persistence of CIS (for patients with CIS at randomization) or death due to any cause. a, Kaplan–Meier estimates of EFS for sasanlimab + BCG-I+M versus BCG-I+M according to treatment arm in the intent-to-treat population. The dashed lines indicate EFS at 24 months and 36 months. b, Kaplan–Meier estimates of EFS for sasanlimab + BCG-I versus BCG-I+M according to treatment arm in the intent-to-treat population. The dashed lines indicate EFS at 24 months and 36 months.

The HR for EFS in all patients was calculated on the basis of an analysis stratified by the presence of CIS at randomization (yes or no) and geography (United States, Western Europe and Canada or rest of world). In each subgroup, the HR for EFS was estimated with the use of unstratified Cox proportional hazards models. Data are presented as HR (center) with 95% CIs (error bars).
Key secondary endpoints
Investigator-assessed EFS for Arm B versus Arm C
EFS was not significantly different for Arm B versus Arm C (stratified HR, 1.16; 95% CI: 0.87–1.55; one-sided P = 0.8439; Fig. 2b and Extended Data Table 2), and this outcome was consistent across all prespecified subgroups (Extended Data Fig. 3).
OS
At the time of final EFS analysis, an interim OS analysis was performed: 91 deaths (8.6%) had occurred across arms, with 32 in Arm A, 30 in Arm B and 29 in Arm C (Supplementary Table 2), out of which five, four and 10 were attributed to bladder cancer, respectively. The median follow-up for the overall population was 40.9 months for OS. The median OS had not been reached for any arm, with no difference between arms (stratified HR for Arm A versus Arm C, 1.13 (95% CI: 0.68–1.87); one-sided P = 0.6791; Fig. 4a and stratified HR for Arm B versus Arm C, 1.07 (95% CI: 0.64–1.79); one-sided P = 0.6791; Fig. 4b).

a, Kaplan–Meier estimates of OS for sasanlimab + BCG-I+M versus BCG-I+M. b, Kaplan–Meier estimates of OS for sasanlimab + BCG-I versus BCG-I+M.
Additional secondary endpoints
Complete response and duration of complete response for Arm A versus Arm C
For patients with CIS at randomization, the complete response (CR) rate achieved at any time was 89.8% in Arm A and 85.2% in Arm C (Extended Data Table 3). A confirmatory biopsy was required by week 24 or collected as soon as possible within 53 weeks from randomization for the assessment of CR; 84.8% in Arm A and 84.0% in Arm C had a confirmatory biopsy as assessed by the investigator. The proportion of investigator-assessed CR with a CR biopsy confirmation also by blinded independent central review (BICR) was 86.6% in Arm A and 85.7% in Arm C (Extended Data Table 3). For patients who achieved a CR, median duration of CR was not reached for any arm, and the probability of remaining in CR at 36 months from the first time of documentation of CR was 91.7% and 67.7% for Arms A and C, respectively (Extended Data Fig. 4 and Extended Data Table 3).
Patient-reported outcomes for Arm A versus Arm C
Completion rates for the European Organisation for Research and Treatment of Cancer Core Quality of Life Questionnaire (EORTC QLQ-C30) were greater than 84% for Arms A and C through end of treatment. Differences of 10 or more points were considered to represent a clinically meaningful difference for the EORTC QLQ-C30 (refs. 14,15). The global health status QOL score mean change from baseline at the end of treatment visit was −5.7 (s.d.: 21.27; 95% CI: −8.34 to −3.03) for Arm A and −1.0 (s.d.: 19.68; 95% CI: −3.35 to 1.37) for Arm C, thereby not reaching the threshold for a clinically meaningful difference between arms (Extended Data Fig. 5 and Supplementary Table 3).
Additional planned secondary endpoints not reported in this paper are time to recurrence of low-grade disease as assessed by the investigator, time to cystectomy, disease-specific survival as assessed by the investigator, health-related QOL as measured by EORTC QLQ-NMIBC24 and the Patient Treatment Administration Burden Questionnaire, trough concentration of sasanlimab (Arms A and B), level of anti-drug antibodies (Arms A and B) and baseline PD-L1 expression level.
Safety
Of 1,055 randomized patients, 1,047 received at least one dose of trial treatment (Arm A (N = 350), Arm B (N = 349), Arm C (N = 349)). Treatment-related adverse events (TRAEs) of any grade occurred in 87.1% of patients in Arm A, 79.0% in Arm B and 70.2% in Arm C (Table 2 and Extended Data Table 4). The most common were dysuria (29.4%), pollakiuria (22.9%) and hematuria (20.9%) in Arm A; dysuria (13.2%), hypothyroidism (12.6%) and lipase increased (12.1%) in Arm B; and dysuria (32.1%), hematuria (20.1%) and urinary tract infection (20.1%) in Arm C (Table 2). TRAEs of grade 3 or higher occurred in 29.1% of patients in Arm A, 21.8% in Arm B and 6.3% in Arm C (Table 2 and Extended Data Table 4). The most common were lipase increased (6.0%), hematuria (4.0%), amylase increased (2.3%) and alanine transaminase increased (2.3%) in Arm A; hematuria (1.4%), lipase increased (1.4%) and aspartate aminotransferase increased (1.1%) in Arm B; and hematuria (3.2%), urinary tract infection (0.6%) and alanine aminotransferase increased (0.3%) in Arm C (Table 2). No treatment-related deaths occurred in Arms A or C; treatment-related deaths occurred in two patients in Arm B (pneumonia bacterial: n = 1 and myocarditis: n = 1; Extended Data Table 4). In Arm A, 26.3% and 16.9% of patients discontinued sasanlimab and BCG, respectively, due to TRAEs. In Arm B, 16.7% and 2.3% of patients discontinued sasanlimab and BCG, respectively, due to TRAEs. In Arm C, 8.6% of patients discontinued BCG due to TRAEs (Extended Data Table 4). The rates of any-grade and grade 3 or higher immune-related adverse events (irAEs) were 42.6% and 15.7%, respectively, in Arm A and 46.8% and 14.1%, respectively, in Arm B. The most common irAEs of any grade were thyroid disorders (Arm A: 17.7%; Arm B: 20.4%) and rash (Arm A: 13.1%, Arm B: 13.8%) (Extended Data Table 5). The most common grade 3/4 irAE was immune-related hepatitis (Arm A: 3.4%; Arm B: 3.2%) (Extended Data Table 5). Grade 5 irAEs occurred in no patients in Arm A and in one patient in Arm B (myocarditis). Systemic corticosteroids were administered in 69 patients in Arm A and in 70 patients in Arm B (Extended Data Table 4). Injection site reactions related to sasanlimab occurred in 2.3% of patients in Arm A and in 3.7% of patients in Arm B (all grade 1) (Extended Data Table 4).
Sensitivity analysis
EFS by BICR for Arm A versus Arm C
Although the sensitivity analysis of EFS by BICR assessment was not statistically significant at the one-sided 0.025 significance level (stratified HR = 0.75; 95% CI: 0.52–1.06; one-sided P = 0.0517), the observed HR remained below 1 in favor of Arm A, supporting the findings of the intent-to-treat population (Supplementary Table 4).
Discussion
The CREST trial showed a statistically significant and clinically meaningful benefit for sasanlimab in combination with BCG-I+M over the SOC (BCG-I+M) in prolonging EFS in patients with BCG-naive high-risk NMIBC, particularly for patients with CIS or T1 tumors. The risk of an EFS event, defined as recurrence of high-grade disease, persistence of CIS (for patients with CIS at randomization), disease progression or death due to any cause, was 32% lower in Arm A than in Arm C. The efficacy benefit was seen across all prespecified subgroups, including the geographic regions and the diagnosis of CIS or T1 at randomization. The EFS evaluation by BICR was consistent with the primary analysis.
New treatment options that delay high-grade disease recurrence and progression are required to improve the treatment outcomes in patients with high-risk NMIBC. Indeed, patients with high-risk or very-high-risk NMIBC who have recurrence before radical cystectomy have shorter cancer-specific survival and OS compared to those with primary high-risk or very-high-risk NMIBC16. When the cancer reaches the MIBC stage, there is substantial negative impact on metastasis-free and cancer-specific survival: approximately 50% of patients with MIBC will progress to metastatic urothelial cancer within 2–3 years after cystectomy17.
At data cutoff, OS follow-up was ongoing, with deaths occurring in 8.6% of patients, most of which were non-bladder cancer and not treatment related as assessed by the investigator. Overall, the interim survival data suggest no difference between treatment arms.
For patients with CIS, achievement of CR is associated with reduced risk of progression and death and may be a useful indicator of long-term outcome18. Patients in Arm A had a CR rate that was approximately 5% higher than Arm C. More importantly, improved durability of the observed CRs with the combination was reflected by the higher proportion of patients sustaining meaningful CR over time for Arm A than in Arm C. Currently, limited bladder-sparing therapeutic options exist after recurrence of CIS, emphasizing the importance of durable disease control in this population3.
Arm B was investigated as BCG-I with the intent to reduce the burden of BCG treatment for patients. Consensus on best practice management for the use of intravesical immunotherapy with BCG concluded that continuing treatment with BCG maintenance improved outcomes and is superior to induction-only treatment in NMIBC19. Sasanlimab combined with BCG-I did not result in prolongation of EFS versus BCG-I+M, underscoring the need for BCG maintenance not only as a component of SOC treatment but also in combination with sasanlimab.
The clinical benefit observed with sasanlimab combined with BCG-I+M is substantiated by evidence from preclinical models and tumor samples from patients after BCG treatment10,11,12. In preclinical models, it has been observed that BCG exposure is associated with increased PD-L1 expression, and changes from PD-L1–negative status to PD-L1–positive status have been observed in patients after BCG exposure12,19,20. The role of PD-L1 status in NMIBC as a predictive factor of response to BCG therapy is mixed, but several studies have shown an association with higher tumor cell PD-L1 expression and poorer outcomes21. Elevated PD-L1 levels may contribute to BCG resistance via the immune escape mechanism13, providing a potential mechanism by which sasanlimab combined with BCG-I+M enhances efficacy compared to BCG-I+M.
The observed safety profile was consistent with the known safety profile for each individual agent. As expected, higher frequencies of patients with any-grade TRAEs and grade 3 or higher TRAEs, including irAEs, were observed in Arm A versus Arm C and were similar in both combination arms, suggesting no relevant safety differences between a shorter versus a longer exposure to BCG. Treatment durations and total completion rates for BCG maintenance suggest that the addition of sasanlimab to BCG-I+M did not have a clinically meaningful impact on the administration of the individual treatments. The benefit–risk of adding sasanlimab to BCG in clinical practice should be considered by urologists based on the patients’ ability to tolerate these AEs22. Patient and caregiver education and healthcare provider monitoring of AEs remain fundamental to optimal patient care22.
QOL was generally maintained in Arm A versus Arm C. Despite the numerical reduction in QOL compared to baseline in both treatment arms, this reduction was below the threshold of a clinically meaningful change14. Of note, QOL questionnaires are not specifically designed to assess the impact of immunotherapy on patient-reported outcomes (PROs)23. Studies from other molecules in oncology have suggested that patients generally prefer the subcutaneous route of administration to other routes24; therefore, the use of subcutaneous sasanlimab may offer convenience to patients and greater efficiency for clinic implementation.
One limitation of the trial was the open-label design, which was mitigated by the retrospective BICR of tumor biopsy and imaging and by the blinding of the sponsor to the aggregate/cumulative data summaries by treatment arm until primary analysis. Another limitation was related to the low number of death events observed at the time of OS interim analysis. Given the early disease setting, it was unlikely to observe an impact on OS outcomes, supporting the EFS primary endpoint as a reliable measurement of efficacy to be used in randomized phase 3 trials for the treatment of patients with high-risk NMIBC25.
In summary, the statistically significant reduction in the risk of recurrence of high-grade disease, persistence of CIS, disease progression or death led to durable disease control in patients receiving BCG-I+M in combination with sasanlimab. The safety profile of this combination was manageable, and QOL was maintained. Sasanlimab in combination with BCG-I+M has the potential to redefine the treatment paradigm and clinical decision-making for patients with BCG-naive high-risk NMIBC, particularly for patients with aggressive CIS or T1 tumors.
Methods
Inclusion and ethics
The trial was sponsored by Pfizer and was designed by the sponsor and steering committee; it was approved by the institutional review board or ethics committee at each site and was conducted in accordance with the principles of the Declaration of Helsinki, Good Clinical Practice guidelines and applicable regulatory requirements. The protocol was approved by the Human Genetic Resources Administration of China (HGRAC). Patients provided written informed consent before trial entry. Data were collected by participating investigators, analyzed at Pfizer and interpreted by all authors. All authors vouch for the accuracy and completeness of the data reported and adherence to the trial protocol. Medical writers funded by the sponsor provided medical writing in accordance with Good Publication Practice guidelines.
Detailed methods are described in the protocol and the statistical analysis plan provided with the protocol (Supplementary Information).
Patients
Eligible adult patients had BCG-naive high-risk (submucosal invasive (T1) tumor, high-grade non-invasive papillary carcinomas (Ta), and/or CIS) NMIBC. Patients must have had complete resection of Ta/T1 papillary disease, with the most recent TURBT ≤12 weeks before randomization. Exclusion criteria included evidence of muscle-invasive, locally advanced or metastatic urothelial cancer or concurrent extravesical urothelial carcinoma (urethral or upper tract).
Trial design and treatments
Patients were randomized 1:1:1 to receive sasanlimab in combination with BCG induction and maintenance (BCG-I+M; Arm A), sasanlimab with BCG induction only (BCG-I; Arm B) or BCG-I+M (Arm C). Randomization was stratified by the presence of CIS (yes or no) and geographic region (United States, Western Europe and Canada or rest of the world).
Subcutaneous sasanlimab (300 mg) was administered in a 2-ml prefilled syringe on day 1 of each 4-week cycle, for up to 25 cycles. Intravesical BCG induction occurred as one dose weekly via instillation for six consecutive weeks; re-induction was permitted (persistence of CIS and high-grade Ta after induction). For patients who did not undergo re-induction, BCG maintenance (Arms A and C) occurred on days 1, 8 and 15 of cycles 4, 7, 13, 19 and 25. For patients who had a re-induction period, maintenance cycles occurred at cycles 7, 13, 19 and 25. Patients could continue receiving one treatment independently if the other had been discontinued. BCG and sasanlimab dose reductions were not permitted.
Endpoints
The primary endpoint was investigator-assessed EFS for Arm A versus Arm C, defined as time from randomization to recurrence of high-grade disease, progression of disease, persistence of CIS (for patients with CIS at randomization) or death due to any cause, whichever occurred first. Key secondary endpoints included investigator-assessed EFS (Arm B versus Arm C) and OS (Arms A and B versus Arm C). Additional secondary endpoints were investigator-assessed CR and duration of CR for patients with CIS at randomization and who achieved CR. Safety and health-related QOL PROs were also assessed.
Assessments
Cytology and cystoscopy, followed by biopsy and imaging when needed, were performed at screening, every 12 weeks for 2 years after randomization and every 24 weeks thereafter until recurrence of high-grade disease, persistence of CIS or progression of disease, regardless of initiation of subsequent anti-cancer therapy. In the event of a positive cystoscopy and/or cytology result, biopsy and imaging (computerized tomography or magnetic resonance imaging) were performed.
QOL was assessed using the EORTC QLQ-C30 throughout treatment and during the safety follow-up periods.
Safety assessments involved monitoring and recording AEs and laboratory abnormalities throughout treatment and up to 90 days after the last dose of trial drug. Potential immune-related adverse events (irAEs) were identified based on a prespecified list of preferred terms and additional medical review by the sponsor. AEs were characterized by type and severity according to National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 5.0.
BICR assessment
BICR provided retrospective central assessment of imaging and biopsies and supported the sensitivity analysis of EFS and the confirmation of CR assessed by the investigator.
PD-L1 expression
Tumor tissue from the most recent TURBT was used for the assessment of PD-L1 expression at baseline. PD-L1 expression (high or low) was reported as detected by a pathologist and assisted by image analysis. PD-L1 expression was defined as the number of PD-L1–positive cells and/or qualitative assessment of PD-L1 staining on tumor and immune cells in regions of interest that are defined by tumor cell morphology. PD-L1 status was assessed by the VENTANA PD-L1 immunohistochemistry SP263 assay and used the VENTANA OptiView PD-L1 detection kit. High was defined as ≥25% tumor cell or (immune cells present in the tumor area >1% and PD-L1–positive immune cells ≥25%) or (immune cells present in the tumor area equal to 1% and PD-L1–positive immune cells equal to 100%). Low was defined as <25% tumor cell and (immune cells present in the tumor area >1% and PD-L1–positive immune cells <25%) or (immune cells present in the tumor area equal to 1% and PD-L1–positive immune cells <100%) or immune cells present in the tumor area equal to 0.
Statistical analysis
Efficacy analyses were performed in the intent-to-treat population, defined as all patients who had been randomized to a treatment arm. EFS, OS and duration of CR were estimated for each treatment arm using the Kaplan–Meier method. The treatment effect on EFS and OS was estimated using a Cox proportional hazards model stratified by the randomization strata to calculate the HR along with 95% CIs. Comparisons between Arms A and B versus Arm C were conducted using the stratified log-rank test. EFS subgroup analyses were prespecified.
CR rates and two-sided 95% CI using the Clopper–Pearson method were calculated for each arm. The difference in CR rates between Arms A and B versus Arm C were tested using a Mantel–Haenszel test stratified by the randomization stratum of geographic region. The one-sided Mantel–Haenszel test P value and the Mantel–Haenszel confidence limits of the difference were calculated.
The trial was initially designed to test two parallel independent primary endpoints—investigator-assessed EFS for Arm A versus Arm C and Arm B versus Arm C—with approximately 999 patients to be randomized. The study-wise type I error was maintained at a one-sided alpha level ≤ 0.025, allocating an alpha level of 0.0125 to each comparison. A three-look group sequential design with Lan–DeMets (O’Brien–Fleming) α-spending function was used to preserve the overall type I error rate and determine the efficacy boundary. The first interim analysis for EFS was for futility only. The trial was to be considered to be positive if the stratified log-rank test for EFS was significant for either comparison. Under the assumptions of an HR of 0.69 and a median EFS of 24 months in Arm C, 389 EFS events would be required for each comparison to provide 90% power to detect a difference between treatment arms (for Arm A versus Arm C and Arm B versus Arm C) using a one-side log-rank test at a significance level of 0.0125.
During the conduct of the trial, the design was modified to (1) demote EFS for Arm B versus Arm C to a key secondary endpoint and (2) remove the interim efficacy analysis of EFS and set a calendar-based data cutoff date for EFS final analysis to allow for approximately 3 years of follow-up after the last patient was randomized. The study-wise type I error was maintained at or below the one-sided alpha level of 0.025. The initial one-sided alpha allocated to the primary endpoint was 0.025. If statistical significance of the primary endpoint was met, EFS between Arm B versus Arm C and OS between Arm A versus Arm C could be formally tested with one-sided alpha of 0.023 and 0.002, respectively. If EFS for Arm B versus Arm C was significant, alpha recycling would be applied to evaluate OS between Arm A versus Arm C at the one-sided 0.025 level. If OS for Arm A versus Arm C was significant, all the alpha would be recycled to formally test OS for Arm B versus Arm C. One interim analysis on the OS endpoint was planned at the time of the EFS primary analysis, with the efficacy boundary determined by a Haybittle–Peto α-spending function (P < 0.0001).
For QOL, mean, standard deviation, median, range and 95% CI of absolute scores and change from baseline were calculated. Safety analyses included all patients who received at least one dose of the trial treatment and were performed with the use of descriptive statistics.
The InForm Database (Oracle, version 7.0) was used for electronic data collection, and ePRO electronic devices were used for collection of questionnaire data. Analyses were performed using SAS version 9.4 software.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The analyses in this paper were based on a data cutoff of 2 December 2024.
Upon reasonable request and subject to review, Pfizer will provide the data that support the findings of this trial. Subject to certain criteria, conditions and exceptions, Pfizer may also provide access to the related individual deidentified participant data from Pfizer-sponsored global interventional clinical trials conducted for medicines, vaccines and medical devices (1) for indications that have been approved in the United States and/or European Union or (2) in programs that have been terminated (that is, development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary and statistical analysis plan. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.
Data may be requested from Pfizer trials 24 months after trial completion. The deidentified participant data will be made available to researchers whose proposals meet the research criteria and other conditions and, for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
References
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 74, 229–263 (2024).
Holzbeierlein, J. M. et al. Diagnosis and treatment of non-muscle invasive bladder cancer: AUA/SUO Guideline: 2024 Amendment. J. Urol. 211, 533–538 (2024).
Gontero, P. et al. European Association of Urology Guidelines on non-muscle-invasive bladder cancer (TaT1 and carcinoma in situ)—a summary of the 2024 Guidelines Update. Eur. Urol. 86, 531–549 (2024).
Audisio, A. et al. New perspectives in the medical treatment of non-muscle-invasive bladder cancer: immune checkpoint inhibitors and beyond. Cells 11, 357 (2022).
Balar, A. V. et al. Pembrolizumab monotherapy for the treatment of high-risk non-muscle-invasive bladder cancer unresponsive to BCG (KEYNOTE-057): an open-label, single-arm, multicentre, phase 2 study. Lancet Oncol. 22, 919–930 (2021).
US Food and Drug Administration. Keytruda. Highlights of prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/125514s110lbl.pdf
Youssef, S. et al. Abstract 2667: In vitro properties and pre-clinical activity of PF-06801591, a high-affinity engineered anti-human PD-1. Cancer Res. 77, 2667 (2017).
Johnson, M. L. et al. Assessment of subcutaneous vs intravenous administration of anti–PD-1 antibody PF-06801591 in patients with advanced solid tumors. JAMA Oncol. 5, 999–1007 (2019).
Cho, B. C. C. et al. 1055P Updated results of subcutaneous (SC) anti-programmed cell death 1 (PD-1) receptor antibody PF-06801591 for locally advanced or metastatic non-small cell lung cancer (NSCLC) or urothelial carcinoma (UC). Ann. Oncol. 31, S721 (2020).
Hashizume, A. et al. Enhanced expression of PD-L1 in non-muscle-invasive bladder cancer after treatment with Bacillus Calmette-Guerin. Oncotarget 9, 34066–34078 (2018).
Maas, M. et al. PD-L1 expression in high-risk non-muscle-invasive bladder cancer is influenced by intravesical Bacillus Calmette–Guérin (BCG) therapy. Cancers (Basel) 16, 1356 (2024).
Wang, Y. et al. Bacillus Calmette–Guérin and anti-PD-L1 combination therapy boosts immune response against bladder cancer. Onco Targets Ther. 11, 2891–2899 (2018).
Cui, J. W. et al. Tumor immunotherapy resistance: revealing the mechanism of PD-1 / PD-L1-mediated tumor immune escape. Biomed. Pharmacother. 171, 116203 (2024).
Osoba, D., Rodrigues, G., Myles, J., Zee, B. & Pater, J. Interpreting the significance of changes in health-related quality-of-life scores. J. Clin. Oncol. 16, 139–144 (1998).
King, M. T. The interpretation of scores from the EORTC quality of life questionnaire QLQ-C30. Qual. Life Res. 5, 555–567 (1996).
Grossmann, N. C. et al. Comparative outcomes of primary versus recurrent high-risk non–muscle-invasive and primary versus secondary muscle-invasive bladder cancer after radical cystectomy: results from a retrospective multicenter study. Eur. Urol. Open Sci. 39, 14–21 (2022).
Tian, J. et al. Population-based outcome of muscle-invasive bladder cancer following radical cystectomy: who can benefit from adjuvant chemotherapy? Transl. Androl. Urol. 10, 356–373 (2021).
Lerner, S. P. et al. Failure to achieve a complete response to induction BCG therapy is associated with increased risk of disease worsening and death in patients with high risk non-muscle invasive bladder cancer. Urol. Oncol. 27, 155–159 (2009).
Kamat, A. M. et al. Expert consensus document: consensus statement on best practice management regarding the use of intravesical immunotherapy with BCG for bladder cancer. Nat. Rev. Urol. 12, 225–235 (2015).
Gerald, T. et al. Impact of baseline PD-L1 status in BCG naive nonmuscle invasive bladder cancer on outcomes and changes after BCG exposure. Urol. Oncol. https://doi.org/10.1016/j.urolonc.2025.01.015 (2025).
Sanguedolce, F. et al. Integrating the PD-L1 prognostic biomarker in non-muscle invasive bladder cancer in clinical practice—a comprehensive review on state-of-the-art advances and critical issues. J. Clin. Med. 13, 2182 (2024).
Schneider, B. J. et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: ASCO Guideline Update. J. Clin. Oncol. 39, 4073–4126 (2021).
Voon, P. J., Cella, D. & Hansen, A. R. Health-related quality-of-life assessment of patients with solid tumors on immuno-oncology therapies. Cancer 127, 1360–1368 (2021).
Cappuzzo, F. et al. 244MO Primary results from IMscin002: a study to evaluate patient (pt)- and healthcare professional (HCP)-reported preferences for atezolizumab (atezo) subcutaneous (SC) vs intravenous (IV) for the treatment of NSCLC. ESMO Open 9, 102706 (2024).
Kamat, A. M. et al. Definitions, end points, and clinical trial designs for bladder cancer: recommendations from the Society for Immunotherapy of Cancer and the International Bladder Cancer Group. J. Clin. Oncol. 41, 5437–5447 (2023).
Acknowledgements
The authors thank the participants and their families and all staff at the participating sites. The CREST trial was funded by Pfizer. Medical writing support, including writing of the first draft, was provided by M. Gamboa and E. Porteous of Nucleus Global, an Inizio Company, and was funded by Pfizer.
Author information
Authors and Affiliations
Contributions
Conceptualization and methodology: N.D.S., T.B.P., J. Bedke, M.D.G., J.P.R. and G.D.S. Investigation: N.D.S., T.B.P., J. Bedke, M.D.G., J.P.R., J.H.K., M.K., E.X., B.A., D.Y., F.G.-R., A.B., G.K. and G.D.S. Formal analysis: C.W. Interpretation: all authors. Supervision: J. Brinkmann, A.-M.C., R.C. and J.V. Writing (original draft, reviewing and editing): all authors.
Corresponding author
Ethics declarations
Competing interests
N.D.S. reports research/consulting: Allessa, Amgen, Artera, Astellas, AstraZeneca, Aura Biosciences, Bayer, Bristol Myers Squibb, Caris, CG Oncology, Daiichi Sankyo, Dendreon, Glytheryx, Invitae, Janssen, MDxhealth, Merck, Minomic, Novartis, Nusano, PhotoCure, Pfizer, Sumitomo, Telix, Tolmar, Tutelix and Urogen. T.B.P. reports honoraria: Astellas, AstraZeneca, Bristol Myers Squibb, Eisai, Elexis, Gilead, Incyte, Ipsen, Johnson & Johnson, Mashup, Merck Sharp & Dohme, Merck Serono, Novartis, Pfizer, Roche and Seagen; consulting or advisory role: Astellas, AstraZeneca, Bristol Myers Squibb, Eisai, Elexis, Gilead, Incyte, Ipsen, Johnson & Johnson, Mashup, Merck Sharp & Dohme, Merck Serono, Novartis, Pfizer, Roche and Seagen; research funding: Astellas, AstraZeneca, Bristol Myers Squibb, Eisai, Elexis, Incyte, Ipsen, Johnson & Johnson, Merck Sharp & Dohme, Merck Serono, Novartis, Pfizer, Roche and Seagen; and travel, accommodations or expenses: Astellas, AstraZeneca, Gilead, Ipsen, Merck Sharp & Dohme, Pfizer and Roche. J. Bedke reports financial interests from Astellas, AstraZeneca, Bristol Myers Squibb, Daiichi Sankyo, Eisai, Ipsen, Janssen, Merck Sorono, Merck Sharp & Dohme, Pfizer, Roche, Nektar, Novartis and Seagen and membership in the European Association of Urology and the Renal Cell Carcinoma Guidelines Panel (Vice-Chairman). M.D.G. reports stock and other ownership interests: Pfizer, Merck and Gilead Sciences; research funding: Merck, Bristol Myers Squibb, Mirati Therapeutics, Seagen, Alliance Foundation Trials, Alliance for Clinical Trials in Oncology, Clovis Oncology, Arvinas, ALX Oncology, Hoosier Cancer Research Network, Novartis, Acrivon Therapeutics, Astellas, Genentech, Accuray, PCCTC, G1 Therapeutics, OncoC4, Flare Therapeutics, Loxo/Lilly, Roche and Pfizer; other relationships: Elsevier, Medscape and Research to Practice; uncompensated relationships: G1 Therapeutics and Loxo/Lilly. J.P.R. reports advisory/consultancy roles for Arquer Diagnostics, Biotech, BTA Pharmaceuticals, Combat, Genomic Expression and Olympus; steering committee membership for AstraZeneca, Bristol Myers Squibb, Janssen and Pfizer; grant or research support from Arquer Diagnostics, Biotech, BTA Pharmaceuticals, Cepheid, TARIS Biomedical and Nucleix; and research funding from Pfizer. J.H.K. reports research funding from Pfizer. M.K. reports research funding from Pfizer. E.X. reports consulting or advisory role: Pfizer, Ferring and Boston Scientific and research funding: Ferring and Pfizer. B.A. reports honoraria: AstraZeneca, Astellas, Eisai, Janssen, Bayer, Merck Sharp & Dohme, Merck, Pfizer, Roche and Bristol Myers Squibb; consulting or advisory role: AstraZeneca, Astellas, Bayer, Bristol Myers Squibb, Janssen, Merck, Pfizer, Merck Sharp & Dohme, Roche and Eisai; speakers’ bureau: Janssen, Astellas, Pfizer, AstraZeneca, Bayer, Merck, Bristol Myers Squibb, Merck Sharp & Dohme, Eisai and Roche; research funding: AstraZeneca, Merck, Bayer, Astellas, Janssen, Bristol Myers Squibb, Pfizer, ICON Clinical Research, Eisai, Merck Sharp & Dohme and Roche; and travel, accommodations and expenses: AstraZeneca, Astellas, Bayer, Bristol Myers Squibb, Janssen, Merck Sharp & Dohme, Pfizer and Sanofi. D.Y. reports research funding from Pfizer. F.G.-R. reports research support/principal investigator: Johnson & Johnson, Pfizer, Taris, Bristol Myers Squibb, Roche, Seagen, AstraZeneca, Combat Medical, Cepheid, Fidia, Astellas, UroGen and Merck Sharp & Dohme; employment: SERMAS (Servicio Madrileño de Salud); consultancy: Johnson & Johnson, Pfizer, Merck, Roche, Taris, Combat Medical, AstraZeneca, Merck Sharp & Dohme and Bristol Myers Squibb; stockholder: CG Oncology; speakers’ bureau: Janssen, Nucleix, Merck Sharp & Dohme, Pfizer, Merck, Bristol Myers Squibb, AstraZeneca, Palex, Combat Medical, Johnson & Johnson and Recordati; travel: Pfizer, Recordati, Ipsen, Combat Medical, Alter, Salvat, Nucleix, AstraZeneca, Fidia and Johnson & Johnson; advisory board: AstraZeneca, Bristol Myers Squibb, Combat Medical, Johnson & Johnson, Nucleix, Pfizer, Taris, Roche and Merck Sharp & Dohme; and manuscript support: Pfizer, Janssen, Combat Medical, AstraZeneca, Johnson & Johnson and Bristol Myers Squibb. A.B. reports consulting or advisory role: Astellas, Janssen-Cilag, OPKO Health, MDxHealth, Ferring, Bayer, AstraZeneca, Hauora and Pfizer; speakers’ bureau: Astellas; and research funding: Sandoz-Novartis, Merck Sharp & Dohme and Pfizer. G.S.K. reports honoraria: AbbVie, Tersera, Bayer, Knight Pharmaceuticals, AstraZeneca Canada and PhotoCure; consulting or advisory role: Merck, Theralase, Janssen Oncology, Ferring, Verity Pharmaceuticals, Bristol Myers Squibb, EMD Serono, Pfizer, Novartis and enGene; research funding: Johnson & Johnson/Janssen and Pfizer. J. Brinkmann reports employment: Pfizer Pharma GmbH. A.-M.C. reports employment: Pfizer SRL. R.C. reports employment: Pfizer SRL. A.E. reports employment: Pfizer Ltd. E.M. reports employment: Pfizer Inc. J.V. reports employment: Pfizer Inc. C.W. reports employment: Pfizer Inc. G.D.S. reports advisory/consultancy roles: Heat Biologics, CG Oncology, PhotoCure, Merck, Roche/Genentech, Ciclomed, Taris Biomedical (now Janssen), MDxHealth, Fidia Farmaceutici, UroGen, Ferring, Aduro, Boston Scientific, Bristol Myers Squibb, AstraZeneca, Pfizer, Janssen, Epivax Therapeutics, Natera, FKD, EnGene Bio, Sesen Bio, BioCanCell (now Archiano), Nucleix, Ipsen, Combat Medical, Astellas, FerGene, Dendreon, AbbVie, Seagen, Verity Pharmaceuticals, Regeneron, STIMIT, Vyriad, Protara, xCures, Nonagen, Nanology and Imvax; clinical trial protocol committee membership for Bristol Myers Squibb, CG Oncology, Fidia, Janssen, Merck, Pfizer, PhotoCure, Protara and Seagen; stock/shares in CG Oncology, EnGene Bio, EpiVax Therapeutics and UroGen; and research funding from Pfizer.
Peer review
Peer review information
Nature Medicine thanks Bernhard Eigl, Jorg R. Oddens and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Ulrike Harjes and Saheli Sadanand, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Analysis of event-free survival in the patients with carcinoma in situ at randomization (with or without papillary tumors) for sasanlimab + BCG-I+M vs BCG-I+M.
For EFS, an event was defined as the first of recurrence of high-grade disease, progression of disease, persistence of CIS (for patients with CIS at randomization), or death due to any cause. Kaplan-Meier estimates of EFS according to treatment arm. The dashed line indicates EFS at 36 months. BCG-I+M, induction and maintenance regimen of intravesical Bacillus Calmette-Guérin; CIS, carcinoma in situ; EFS, event-free survival.
Extended Data Fig. 2 Analysis of event-free survival in the patients with T1 tumor (with or without carcinoma in situ) for sasanlimab + BCG-I+M vs BCG-I+M.
For EFS, an event was defined as the first of recurrence of high-grade disease, progression of disease, persistence of CIS (for patients with CIS at randomization), or death due to any cause. Kaplan-Meier estimates of EFS according to treatment arm. The dashed line indicates EFS at 36 months. BCG-I, Bacillus Calmette-Guérin induction regimen of intravesical Bacillus Calmette-Guérin; BCG-I+M, induction and maintenance regimen of intravesical Bacillus Calmette-Guérin; CIS, carcinoma in situ; EFS, event-free survival.
Extended Data Fig. 3 Forest plot of the analysis of event-free survival in prespecified subgroups for sasanlimab + BCG-I vs BCG-I+M.
The hazard ratio for EFS in all patients was calculated on the basis of an analysis stratified by the presence of carcinoma in situ at randomization (yes or no) and geography (US, Western Europe and Canada, or rest of world). In each subgroup, the hazard ratio for EFS was estimated with the use of unstratified Cox proportional-hazards models. Data are presented as hazard ratio (center) with 95% confidence intervals (error bars). BCG-I+M, induction and maintenance regimen of intravesical Bacillus Calmette-Guérin; CIS, carcinoma in situ; CRF, case report form; EFS, event-free survival.
Extended Data Fig. 4 Probability of remaining in Complete Response for patients with CIS at randomization who achieved Complete Response for sasanlimab + BCG-I+M vs BCG-I+M.
Kaplan-Meier estimates for duration of complete response for sasanlimab + BCG-I+M vs BCG-I+M in for patients with CIS at randomization who achieved CR. The dashed lines indicate the probability of remaining in complete response at 12, 24 and 36 months. BCG-I+M, induction and maintenance regimen of intravesical Bacillus Calmette-Guérin; CIS, carcinoma in situ.
Extended Data Fig. 5 Plot of mean change from baseline over time for EORTC QLQ-C30 global health status by visit.
BCG-I+M, induction and maintenance regimen of intravesical Bacillus Calmette-Guérin; C, cycle; D, day.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shore, N.D., Powles, T.B., Bedke, J. et al. Sasanlimab plus BCG in BCG-naive, high-risk non-muscle invasive bladder cancer: the randomized phase 3 CREST trial. Nat Med (2025). https://doi.org/10.1038/s41591-025-03738-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41591-025-03738-z
