
Abstract
It has been believed that immunosenescence plays a crucial role in tumorigenesis and cancer therapy. Nevertheless, there is still a lack of understanding regarding its role in determining clinical outcomes and therapy selection for gastric cancer patients, due to the lack of a feasible immunosenescence signature. Therefore, this research aims to develop a gene signature based on immunosenescence, which is used for stratification of gastric cancer. By integrative analysis of bulk transcriptome and single-cell data, we uncovered immunosenescence features in gastric cancer. Random forest algorithm was used to select hub genes and multivariate Cox algorithm was applied to construct a scoring system to evaluate the prognosis and the response to immunotherapy and chemotherapy. The Cancer Genome Atlas of Stomach Adenocarcinoma (TCGA-STAD) cohort was implemented as the training cohort and two independent cohorts from the Gene Expression Omnibus (GEO) database were used for validation. The model was further tested by our Fudan cohort. In this study, immunosenescence was identified as a hallmark of gastric cancer that is linked with transcriptomic features, genomic variations, and distinctive tumor microenvironment (TME). Four immunosenescence genes, including APOD, ADIPOR2, BRAF, and C3, were screened out to construct a gene signature for risk stratification. Higher risk scores indicated strong predictive power for poorer overall survival. Notably, the risk score signature could reliably predict response to chemotherapy and immunotherapy, with patients with high scores benefiting from immunotherapy and patients with low scores responding to chemotherapy. We report immunosenescence as a hitherto unheralded hallmark of gastric cancer that affects prognosis and treatment efficiency.
Similar content being viewed by others

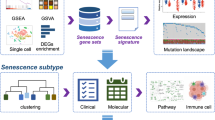

Introduction
Worldwide, gastric cancer (GC) ranks fifth among cancers and is third among cancer deaths1. Overall survival (OS) of GC patients is unsatisfactory and patients with advanced GC only have a median OS of around 1 year1. Chemotherapy based on fluoropyrimidine, platinum, taxane, and irinotecan is the basic and main treatment for GC. Meanwhile, immunotherapy represented by immune checkpoint inhibitors (ICIs) has elicited sustained antitumor reactions in a variety of solid cancers, including GC2. Considering it is a molecularly and phenotypically highly heterogeneous disease, not all patients respond to chemotherapy or immunotherapy, calling for biomarkers to perform better clinical management.
Immunosenescence plays an important role in the occurrence of tumors. The conception of immunosenescence is used to be defined as the immune system, which includes immune organs and cells, degenerates with age3. However, the concept is a little out-of-date these days, since immunosenescence is not age-dependent4. Although it is related to chronological age, the development of immunosenescence in individuals is sensitive to environmental clues, such as the history of immunology and infection. Stress, obesity and inflammation can be the causes of immunosenescence other than age4. More emphasis should be put on the tumor microenvironment (TME) rather than the whole immune system when considering immunosenescence. It also represents the crosstalk between immune and senescence in TME to some extent. It can bring thymatic diseases, decrease T cell population and antigenic expression capacity of dendritic cells, weaken phagocytic effects of macrophages, and increase cells myeloid-derived suppressor cells (MDSCs), which leads to deficiencies in cancer cell clearance and immune surveillance5. Deficiencies in immune monitoring can trigger tumor immunity escape, which directly promotes cancer proliferation, metastasis, and drug resistance. Several clinical trials or drugs related to immunosenescence have been implemented, indicating that immunosenescence is a potential drug target6.
Our research selected the immunosenescence-related genes (ISGs) from the public database and verified that they are associated with the features of immunosenescence in The Cancer Genome Atlas stomach adenocarcinoma (TCGA-STAD) cohort, which also served as the training set. In this study, we performed an extensive analysis of the immunosenescence landscape in precancerous lesions and GC, and further dissected its link with clinicopathologic and molecular characteristics, as well as signaling pathway activation and immune cell infiltration. Finally, we have established an immunosenescence scoring system of GC, which not only has the strong predictive ability of prognosis, but also the efficiency of immunotherapy and adjuvant chemotherapy.
Materials and methods
Data collection and processing
From UCSC Xena database (http://xena.ucsc.edu/), we achieved the transcriptional data with fragments per kilobase per million mapped reads (FPKM) format, corresponding clinical information, and somatic mutation data of TCGA-STAD cohort7. After excluding patients with incomplete clinical data, we finally adopted a total of 32 adjacent normal samples and 374 GC tissue ones for further research. The FPKM data was subsequently transformed to log2(transcripts per million + 1). The clinical information and microarray expression matrix of GSE62254, GSE15459, and GSE26899 were obtained from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/)8. As for GC immunotherapy cohort, we used Kim’s cohort (n = 45) since it was the only GC immunotherapy cohort whose transcriptomic and clinical data were available9. 56 GC patients diagnosed between 2007 and 2011 with necessary clinical data from Fudan University Shanghai Cancer Center were collected to further verify the prognostic value of genetic signatures. We selected cases on the basis of the following criteria: histologically confirmed adenocarcinoma of the stomach; surgical resection of primary GC; age ≥ 18 years; not treated with prior chemotherapy or radiotherapy. At least two expert gastrointestinal pathologists reviewed H&E-stained slides to select cases with estimated carcinoma content of at least 60%. Cases did not meet the above criteria were excluded. The statistical clinical information of TCGA-STAD and Fudan cohort is presented in Supplementary Table S1.
Identification of immunosenescence-related genes
Senescence-related genes (SRGs) were selected from the Molecular Signatures Database (MSigDB, https://www.gsea-msigdb.org/gsea/msigdb/)10. Immune-related genes (IRGs) were obtained from the Immunology Database and Analysis Portal (ImmPort) database (https://www.immport.org/)11, which is a useful and renowned immunology database and analysis portal system. Regarded as the main features of immunosenescence, senescence-associated secretory phenotype (SASP), cellular response to reactive oxygen species (ROS), mitochondrial biogenesis, and glycolysis gene sets were also achieved from MSigDB6. The intersections of SRGs and IRGs were regarded as ISGs which were confirmed by main hallmarks of immunosenescence afterwards. The mentioned gene sets are all included in Supplementary Table S2. In the STRING database, protein–protein interaction (PPI) networks were analyzed for ISGs12. The expression correlation network of ISGs was explored by “corrr” package.
Functional enrichment analysis
R package “clusterProfiler” was used for Kyoto Encyclopedia of Genes and Genomes (KEGG)13 and Gene Ontology (GO)14 enrichment analyses with a p-value < 0.05 and a q-value < 0.0515. To further investigate the important biological pathways, we conducted gene set enrichment analysis (GSEA)16 by “clusterProfiler” R package as well. Gene sets with adjusted p-value < 0.05 and false discovery rate (FDR) < 0.05 after performing 1,000 permutations were considered to be significantly enriched. The results were visualized using the R packages “enrichplot” , “GOplot” and “ggplot2”17.
The bulk transcriptomic and genomic analyses of the whole immunosenescence microenvironment in GC
Heatmap visualization was performed using R package “pheatmap”. The single sample gene set enrichment analysis (ssGSEA) score consisting of ISGs was calculated by “GSVA” and “GSEABase” package18. The median ssGSEA score was used as the cutoff value to divide the TCGA-STAD cohort into “down” and “up” groups. Survival analysis of OS was implemented as well. Gene set variation analyses (GSVA) of 2982 canonical pathways downloaded from MSigDB database were compared between the two groups with “GSVA” package. Microenvironment cell populations-counter (MCP-counter) method was used to compute two stromal and eight immune cell populations in immunosenescence-down and -up groups19. T cell dysfunction, exclusion and cytotoxicity scores were contrasted between them20. Single nucleotide variants (SNV) data of TCGA-STAD was handled with “maftools” package21 while the somatic copy number alterations (SCNAs) achieved by “TCGAbiolinks” package were further processed with Genomic Identification of Significant Targets in Cancer (GISTIC) 2.022.
Single-cell RNA sequencing data analyses
To analyze the expression level of ISGs at a single-cell level, we utilized human single-cell RNA (scRNA) sequencing data from DRscDB database23. However, DRscDB database lacked human stomach tissues. To further depict the dynamic immunosenescence microenvironment in GC development, we used previously published scRNA data of gastric tissues, which included non-atrophic gastritis (NAG), chronic atrophic gastritis (CAG), wild intestinal metaplasia (IMW), severe intestinal metaplasia (IMS) and early gastric cancer (EGC)24. We downloaded the raw data from GSE134520 which contained 3 NAG samples, 3 CAG samples, 2 IMW samples, 4 IMS samples, and one EGC sample. R package Seurat was used to further process raw count matrices of scRNA data25. Cells would be removed as poor quality cells if they reached a threshold for the number of expressed genes below 400 or above 7000. Genes would be ignored if they were expressed in less than 3 cells. The initial data was normalized using the “NormalizeData” function in the Seurat package and the “LogNormalize” normalization method was selected. The proportion of mitochondrial or ribosomal genes was controlled (Supplementary Figure S1). As a consequence, 20,685 genes and 48,657 cells were detected. Highly variable genes (HVGs) were explored by ‘FindVariableGenes’ function (nfeatures = 2000). The Venn diagram of HVGs was plotted by “venn” R package. Then, we implemented the ‘FindCluster’ function with resolution parameters 2 to recognize the number of cell clusters. Cell clusters were annotated based on gene expression by using the “FindAllMarkers” function to search for differentially expressed genes. “RunUMAP” and “DimPlot” functions were used for uniform manifold approximation and projection (UMAP) visualization26. Average GSVA scores of immunosenescence were calculated in different cell types. Hub genes used in the multivariate Cox model were studied in Tumor Immune Single-cell Hub (TISCH) database as well27.
Construction of an immunosenescence-related predictive and prognostic signature
To screen out certain immunosenescence genes related to GC immunotherapy response, we employed “random forest”, a machine learning algorithm, by R package “randomForest”28. Two thirds of Kim’s cohort’s patients were randomly selected as the training set and the others were used as the validation set for the random forest model. The immunotherapy-related genes were put into multivariate Cox regression analysis using “survival” R package. The RNA expression data of GC tissue samples in TCGA-STAD cohort was used as the training set for the multivariate Cox regression model. GSE62254 and GSE15459 were employed as validation sets. Fudan cohort was applied as the testing set. To maintain the comparability between different cohorts, we normalized every dataset and used z-score in the multivariate Cox regression model29. Each patient's risk score was calculated using the formula that risk score = coefficient × gene expression level. TCGA cohort samples were divided into high- and low-risk groups based on the median risk score. Both the validation cohorts and the testing cohort used the same cutoff value.
Exploring potential biological pathways in different risk populations
By using the "limma" R package, differentially expressed genes (DEGs) were identified between high- and low-risk groups30. There were two factors to consider, one was the thresholds of log2 fold change > 1 or < -1, the other was adjusted p-values (adj.p.val) < 0.05. Volcano plot and heatmap were used for the visualization of the DEGs by “ggplot2”, “ggrepel” and “pheatmap” package. GO bar plot, KEGG bubble plot and GSEA analyses were employed to research the potential biological pathways.
Validation of the model to effectively predict patient outcomes
Kaplan–Meier survival analyses and log-rank tests were performed in GSE62254, GSE15459 and Fudan cohort to assess the prophetical value of the model by using the R package “survival” and “survminer”. To determine the specificity and sensitivity of the multivariate Cox regression model by measuring the area under the curve (AUC). time-dependent receiver operating characteristic (ROC) curves were conducted with “survivalROC” R package31. Risk score correlation diagrams including risk score distributions, survival status distributions, and heatmaps of signature expression levels were plotted by R package “ggrisk”.
Correlation between the risk score and clinicopathological features
Univariate and multivariate Cox analyses were utilized to determine whether the risk score was an independent and reliable prognostic factor among other clinicopathological variables. Unknown clinical characteristics were determined as “X” status. The relationships between the risk score and other clinical characteristics such as age, gender, stage and so on were evaluated as well. The alluvial diagram was plotted by “ggalluvial” package to show the proportions of high and low risk groups in different clinical subsets.
Construction and evaluation of the signature-based nomogram
331 TCGA-STAD patients with complete data of risk score, age, gender and stage were taken into account when constructing a nomogram. With the "rms" R package, TCGA-STAD cohort generated a prognostic nomogram based on the risk score32. The nomogram model was calibrated for 1, 3 and 5 years to determine its predictive accuracy. The concordance index (C-index) and ROC curve were also utilized to evaluate the prediction accuracy of the model, and the time-dependent ROC curve was portrayed using “riskRegression” and “survival” R packages. By “ggDCA” R packages decision curve analyses (DCA) were performed to assess the net benefit of nomogram33.
Evaluation of tumor immune microenvironment and the ability to predict immunotherapeutic response
CIBERSORT was utilized to estimate the fractions of 22 tumor-infiltrating immune cells in different risk groups34. Estimates of immune infiltration to characterize the cellular composition from TCGA-STAD gene expression matrix were performed using Tumor Immune Estimation Resource (TIMER)35, CIBERSORT-ABS, xCELL36, and Estimating the Proportions of Immune and Cancer cells (EPIC)37. Pearson correlation analysis by “ggcorrplot” package was conducted to clarify the correlation between hub genes used in the model and immune infiltration cells. The stromal, immune and ESTIMATE scores of different subgroups were assessed with the ESTIMATE algorithm38. We then evaluated the association between the two risk groups and programmed cell death protein 1 (PD-1), programmed cell death ligand 1 (PD-L1), and cytotoxic T-lymphocyte antigen 4 (CTLA-4) expression. The microsatellite instable (MSI) status of the TCGA-STAD cohort was obtained from https://tcia.at/39. Kim’s cohort was used for testing the ability of the multivariate Cox model to predict the response to immunotherapy.
Prediction of the sensitivity to chemotherapy in clinical cohorts and cell lines
299 patients from GSE62254 and 92 patients from GSE26899 with chemotherapy and survival information were selected into research. To study the clinical performance of chemotherapy drugs in patients, we calculated semi-inhibitory concentration (IC50) values for 198 drugs in Genomics of Drug Sensitivity in Cancer (GDSC) database with the “oncoPredict” package40,41. We analyzed the correlation between IC50 and risk score and the relationship between IC50 and risk groups.
Statistical analysis
All of the R packages in this study were executed in the R software (version 4.1.2, https://www.r-project.org). The distribution of the variables was tested via the Shapiro–Wilk test. In order to compare the two groups, unpaired Student’s t test or the Wilcoxon rank-sum test was used. If there were more than two groups, the Kruskal–Wallis test was applied to evaluate significant differences. Correlations coefficients were calculated by Spearman at the transcriptomic level. To compare the statistical differences in survival, the survival curves of prognosis analysis were generated and the Kaplan–Meier method and the log-rank test were used. Entire heatmaps were generated by functions of the pheatmap R package. The p-values were two-sided and less than 0.05 was considered statistically significant.
Results
Identification of the immunosenescence-related genes
579 senescence-related genes were selected from MSigDB database and 1793 immune-related genes were obtained from ImmPort. 99 common genes among them were recognized as the potential ISGs (Supplementary Figure S2A). With STRING online database, the PPI analysis of the potential ISGs was conducted and the PPI enrichment p-value was < 1.0e-16 (Supplementary Figure S2B). We also employed k-means algorithm on the PPI result and the 99 ISGs were generally classified into two groups. Functional enrichment analysis indicated that the green genes were more related to senescence while the red genes were more related to immune. The PPI network showed close crosstalk between senescence and immune. Most of the expressions of the 99 ISGs revealed a positive correlation in TCGA-STAD (Supplementary Figure S2C). GO biological process (GO-BP) and KEGG analyses of ISGs proved that these genes were connected with immune and senescence pathways (Supplementary Figure S2D, E). GSEA showed that those biological pathways indicating immunosenescence, including SASP, mitochondrial biogenesis, glycolysis, and ROS, were significantly enriched in GC than in normal tissues (Supplementary Figure S2F).
Confirmation of immunosenescence as a hallmark of GC at the transcriptomic and genomic level
To investigate the general expression condition of immunosenescence in GC, a heatmap of the expression levels of the ISGs in TCGA-STAD was plotted, showing that immunosenescence was major significantly enriched in tumor tissues compared to adjacent normal ones (Fig. 1A). The ssGSEA was utilized to compute the enrichment level of the ISGs among the samples from the TCGA-STAD. Samples whose ssGSEA scores were more than the median level were considered as the immunosenescence up-regulation ones and had worse OS (p = 0.025), implying that immunosenescence was significantly linked with the prognosis of GC (Fig. 1B). Top 10 variant canonical pathways were exhibited, indicating that immunosenescence up-regulation was positively connected with adhesion and diapedesis of lymphocytes and granulocytes, and local acute inflammatory response (Supplementary Figure S3A). The standardized scores of eight immune and two stromal cells calculated by MCP-counter method implied that all these 10 cells were highly expressed in immunosenescence up-regulation group (Supplementary Figure S3B). Although having a higher number of cancer-associated fibroblasts (CAFs) and a higher score of T cell dysfunction, immunosenescence-up patients tended to have fewer MDSCs and the M2 subtype of tumor-associated macrophages (TAMs). They also had a lower T cell exclusion score and a higher cytotoxicity score (Fig. 1C, D). Furthermore, the waterfall plots of mutations revealed that the up-group had higher tumor mutational burden (TMB) (Fig. 1E), and the six genes, namely TP53, TTN, ARID1A, LRP1B, MUC16, and SYNE1 exhibited the top 6 highest mutation frequency (Supplementary Figure S3C). Differentially mutated genes were identified and HKDC1, MTA1, TNFRSF19 only mutated in ssGSEA-up set (Supplementary Figure S3D). Based on GISTIC 2.0 tool, immunosenescence ssGSEA-down group exhibited relatively higher G-scores (G = Frequency × Amplitude), implying it had more SCNAs (Fig. 1F). Heatmaps of the raw copy number profiles had proven this point of view (Supplementary Figure S3E).

Confirmation of senescence as a hallmark of GC at transcriptomic and genomic level. (A) Heatmap of the expression levels of the ISGs in TCGA-STAD. (B) Survival analysis of different immunosenescence ssGSEA scores. (C) T cell dysfunction and exclusion status of different ssGSEA classes. (D) cytotoxicity scores comparison. (E) SNV and (F) SCNA G-score analyses. PPI, Protein–protein interaction; ISGs, immunosenescence-related genes; TCGA-STAD, The Cancer Genome Atlas stomach adenocarcinoma; ssGSEA, single-sample gene set enrichment analysis; SNV, single nucleotide variant; SCNA, somatic copy number alteration. ns p ≥ 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Single-cell profiling of immunosenescence within TME in GC
To further analyze the immunosenescence features of the level of single-cell, we first researched DRscDB database (Supplementary Figure S4A). ISGs were enriched in all kinds of cells such as macrophages and microglia. Considering that DRscDB database lacked human stomach tissues, we downloaded the raw data of GSE134520 which contained normal and cancerous gastric tissues to research. After essential quality control, 20,685 genes and 48,657 cells (NAG 5639, CAG 19,837, IMW 4165, IMS 15,382, EGC 3634) were identified. 2000 HVGs were identified as well (Supplementary Figure S4B). The Venn diagram of HVGs showed that the 5 tissues had 541 common HVGs while each of them had its own unique HVGs (Fig. 2A). 17 cell types were recognized according to known marker genes (Supplementary Figure S4C). UMAP of the 17 cell types demonstrated that the cells could be classified into 3 clusters, malignant cells (cancer cells, stem cells, enterocytes), normal stomach cells (mucous cells, parietal cells), and other cells, including immune and stromal cells (Fig. 2B). Immunosenescence GSVA score was calculated in each kind of tissue to portray the dynamic immunosenescence microenvironment in GC development. Interestingly, lollipop diagram, UMAP and heatmap of the GSVA score showed that certain cell types consisting of fibroblasts and macrophages, but not the cancer cells, stem cells, and proliferative cells (PC), were highly enriched in immunosenescence signature (Fig. 2C–E). With the development of the disease, D cells, G cells, B cells, T cells, and endothelial cells gained more and more immunosenescence GSVA scores. The result suggested that fibroblasts and macrophages, rather than the cancer cells or stem cells, primarily contributed to the immunosenescence features in GC.

Single-cell profiling of immunosenescence within TME in GC. (A) The Venn diagram of HVGs. (B) UMAP of the 17 cell types. (C) Average immunosenescence GSVA scores of different cell types from total and 5 tissues. (D) UMAP of immunosenescence GSVA scores in EGC. (E) Heatmap of the dynamic immunosenescence microenvironment in GC development. TME, tumor microenvironment; HVG, highly variable gene; UMAP, uniform manifold approximation and projection; GSVA, gene set variation analysis; EGC, early gastric cancer; GC, gastric cancer.
Construction of the predictive and prognostic signature
After screening out genes absent in GC immunotherapy cohort (Kim’s cohort), 92 ISGs were reserved to find ISGs related to programmed cell death 1 (PD-1) targeted therapy in GC by random forest algorithm. Four hub genes were selected as "ADIPOR2", "BRAF", "APOD", and "C3" (Supplementary Figure S5A). 45 patients in Kim’s cohort were randomly divided into training set (n = 30) and validation set (n = 15) to construct the random forest model based on the hub genes. The model had reached stabilization when the number of trees (ntree) = 600 (Supplementary Figure S6A). The ROC curve was plotted to investigate the specificity and sensitivity of the random forest model by “pROC” package (Supplementary Figure S6B). The AUC spectacularly attained one, indicating that the 4 hub genes were significantly connected with the response of GC immunotherapy. Then, the 4 hub genes were utilized for constructing a multivariate Cox regression model to predict OS of GC patients. TCGA-STAD cohort was used as the training cohort. To ensure the comparability between different datasets, we normalized every cohort and used z-score in the multivariate Cox regression model. The risk score was calculated as follows: risk score = 0.303*APOD + 0.064*ADIPOR2-0.038*BRAF-0.011*C3. The model was statistically related to prognosis (p = 0.02). According to the median cut-off value which was equal to -0.004, all the patients were divided into the high- and low-risk subgroups. To further research the expression of the hub genes at a single-cell level, TISCH database was used to display the enrichment level of different cell types (Supplementary Figure S6C). APOD was especially enriched in fibroblasts. ADIPOR2 was slightly enriched in myofibroblasts and mast cells while BRAF was slightly enriched in CD8 + T cells. Last but not least, C3 was enriched in fibroblasts and dendritic cells (DCs).
Exploration of the biological features of the two risk groups
We used the limma R software package to obtain DEGs between high risk groups and low risk groups. The adjusted p-values of 9196 genes were < 0.05, among which 380 genes were identified that the absolute values of log2 fold change were > 1. Volcano plot identifying DEGs among two groups was exhibited and APOD was the most significant one (Supplementary Figure S5B). GO and KEGG enrichment analyses indicated that intercellular communication, cell adhesion and growth, Calcium signaling pathway, cGMP − PKG signaling pathway, and Apelin signaling pathway were observed in the high-risk group of GC (Supplementary Figure S5C, D). Overexpression of apelin and apelin receptor could result in increased migration and invasion abilities by stimulating migratory protrusion formation and proteolytic activity by PI3K/AKT and MAPK signaling pathways42. Although cGMP − PKG signaling pathway could work as a protective signature to intercept proliferation and tumor angiogenesis via inhibiting β-catenin/TCF and SOX9 signaling43, it was the negative pathways that played the dominating role in high-risk group. GSEA analysis of “hallmark gene sets” demonstrated that adipogenesis, epithelial-mesenchymal transition, hypoxia, and KRAS signaling pathway were enriched in the high-risk group (Supplementary Figure S5E). These signaling pathways play an important role in gastric cancer.
Validation of risk model’s prognostic value
The survival analysis of TCGA-STAD revealed that the high immunosenescence risk score was associated with the poorer OS (p = 0.00022, Fig. 3A), which was opposite to the result based on senescence risk score assessed by Zhou et al.44. This finding cast light on the importance of immune for senescence and the crosstalk between them. The time-dependent ROC curve analysis demonstrated a significant predicting ability for OS (1-year AUC = 0.617, 3-year AUC = 0.602, 5-year AUC = 0.631, Fig. 3B). In order to further explore the impact of risk scores on the prognosis of GC, we drew a scatter plot of hub gene expression and the corresponding survival time in different TCGA-STAD samples (Fig. 3C). To confirm the robustness of the risk score in different people, we further explored GSE62254 and GSE15459 datasets with complete clinical information. Similar results of survival analysis were observed that a higher immunosenescence risk score was related to a worse OS of GC patients (p < 0.0001, p = 0.017, Fig. 3D, G). In the GSE62254 cohort, the AUCs of 1-, 3-, and 5-year ROC were 0.600, 0.651, and 0.655 (Fig. 3E), while in GSE15459, the 1-, 3-, and 5-year AUCs were 0.565, 0.580, and 0.625, respectively (Fig. 3H), showing pleasant prognostic discrimination of the model based on hub genes. The distribution of the risk score, the survival status, and heatmaps exhibiting the expression of the hub genes in GSE62254 and GSE15459 were presented as well (Fig. 3F, I). Furthermore, Fudan cohort with 56 patients was explored as the test set. Encouragingly, the prognostic value of the risk model was validated (p = 0.019, Fig. 3J). The AUCs of 1-, 3-, and 5-year ROC were all > 0.6 (Fig. 3K). Risk score correlation diagram of Fudan cohort was consistent with the result of TCGA (Fig. 3L).

Validation of risk model’s prognostic value. Survival analyses, ROC curves, and risk score correlation diagrams were conducted in training set (A-C) TCGA-STAD, validation sets (D-F) GSE62254 and (G-I) GSE15459, and test set (J-L) Fudan cohort, respectively. ROC, receiver operating characteristic; TCGA-STAD, The Cancer Genome Atlas stomach adenocarcinoma. ns p ≥ 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
What’s more, higher immunosenescence risk score was also related to the worse progression free interval (PFI) (p = 0.00049) and disease specific survival (DSS) (p < 0.0001) in TCGA-STAD cohort (Supplementary Figure S7A). The time-dependent ROC curve analysis demonstrated a significant predicting ability for PFI (1-year AUC = 0.649, 3-year AUC = 0.631, 5-year AUC = 0.601) and DSS (1-year AUC = 0.676, 3-year AUC = 0.634, 5-year AUC = 0.670) as well (Supplementary Figure S7B). Risk score correlation diagrams of PFI and DSS were similar (Supplementary Figure S7C). Although high risk score could predict poorer PFI and DSS in TCGA-STAD, disease free survival (DFS) was not related to the risk score in GSE62254 and Fudan cohorts (Supplementary Figure S8).
Correlation between the risk score and clinicopathological characteristics
Univariate and multivariate Cox analyzes were employed to demonstrate that the risk score was an independent and robust prognostic factor. Several clinicopathological features such as gender, age, stage, histology, molecular subtypes, and immune subtypes of TCGA-STAD were taken into account. Among them, risk score, age, stage III and IV were independent prognosis factors of GC (P < 0.05, HR > 1, Fig. 4A). Moreover, significant differences in the risk scores were observed among clinicopathological subtypes in TCGA cohort. Patients older than 60 were defined as “old”. The risk score was found to be related to old, stage, CpG island methylator phenotype (CIMP)45, immune subtype46, molecular subtype45, and MSI status (Fig. 4B–G). Interestingly, younger GC patients tended to have a higher immunosenescence risk score. Recent studies have proved that immunosenescence is not an age-dependent process, but the dysfunction of the immune system and should include some characteristics of low-level chronic inflammation4. Although some aspects of immunosenescence may be related to aging, there is evidence that the response to stress (whether infectious or not) and obesity are the strong regulators of this process4. To better illustrate the characteristics of the risk score, an overview of the links among stage, methylation, histology, molecular subtype, immune subtype, old, and individual patient risk in the TCGA cohort was represented via the alluvial plot. (Fig. 4H).

Correlation between the risk score and clinicopathological features. (A) Univariate and multivariate Cox analyses. The relationships between the risk score and (B) old, (C) stage, (D) hypermethylation, (E) immune subtypes, (F) molecular subtypes, and (G) MSI status. (H) The alluvial diagram showed the proportions of high- and low-risk groups in different clinical subgroups. MSI, microsatellite instable. ns p ≥ 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Construction and verification of signature-based nomograms for OS and PFI
To make this model more suitable for clinical use, we next constructed a nomogram, which integrated age, sex, stage, and risk score to optimize current metrics for long-term OS prediction by multivariate Cox regression in the TCGA-STAD cohort (Fig. 5A). The nomogram was used to predict 1-, 3-, and 5-year OS in combination with clinical features and immunosenescence signature. Each patient was assigned a unique score based on the built nomogram, with higher scores indicating poorer prognosis. The distinction, consistency, and clinical utility of the nomogram were quantified by nomogram calibration curves, time-dependent ROC curves, and DCA.

Construction and validation of the signature-based nomogram for OS. (A) Nomogram was developed in the TCGA-STAD cohort based on the risk score, age, gender, and TNM stage to predict 1-year, 3-year, and 5-year outcomes. (B) Calibration curves of the nomogram for 1-, 3-, and 5-year OS prediction in TCGA-STAD. (C) Time-dependent ROC curves of the nomogram for 1-, 3-, and 5-year OS prediction in TCGA-STAD. (D-F) Time-dependent ROC curves of the nomogram, age, gender, TNM stage, risk score, and model without risk score for 1-, 3-, and 5-year OS prediction. (G) DCA curves were performed to evaluate the net benefit of the nomogram, age, gender, pathological characteristics, and risk score. TCGA-STAD, The Cancer Genome Atlas stomach adenocarcinoma; OS, overall survival; ROC, receiver operating characteristic; DCA, decision curve analysis.
For the nomogram in the training cohort, the C-index was 0.673. Nomogram calibration curves demonstrated good agreement between nomogram-predicted OS and actual observed 1-, 3-, and 5-year OS, further demonstrating the accuracy of the nomogram (Fig. 5B). The AUCs of 1-, 3- and 5-year ROC were 0.700, 0.661 and 0.659, respectively (Fig. 5C). Combined nomograms showed the largest 1-, 3-, and 5-year OS prediction AUCs compared to age, sex, stage, and models without risk scores, suggesting that incorporating risk scores into the Cox model improved the model's discriminative power (Fig. 5D–F). DCA curves showed that the nomogram produced a better net benefit compared to the individual predictors and the model without risk score, indicating that the conjunct nomogram could guide better prediction of OS in GC patients (Fig. 5G).
As for PFI, we applied the same method to construct a nomogram in the TCGA-STAD cohort (Supplementary Figure S9A). The C-index of the PFI nomogram model was 0.649. The calibration curves further demonstrated the accuracy of the PFI nomogram (Supplementary Figure S9B). The AUCs of 1-, 3- and 5-year ROC were 0.682, 0.647 and 0.657, respectively (Supplementary Figure S9C). The combined nomograms also showed the best AUCs compared to age, sex, stage, and models without risk scores in 1-, 3-, and 5-year PFI predictions (Supplementary Figure S9D-F). DCA curves also showed that the nomogram produced a better net benefit compared to other models (Supplementary Figure S9G). The similar results indicated that incorporating risk scores into the Cox model improved the model's discriminative power of both OS and PFI.
Evaluation of immune cell infiltration and the capacity of predicting immunotherapy response
In order to characterize the immune landscape related to ISGs, infiltrating immune cell populations were estimated by 4 algorithms (TIMER, CIBERSORT-ABS, xCELL, and EPIC) (Fig. 6A). Both risk score and hub genes were taken into account. Correlation analysis showed that a higher risk score indicated increased abundance of myeloid DCs, endothelial cells, eosinophils, CAFs, granulocyte monocyte progenitors, and hematopoietic stem cells. APOD was positively connected with more T-helper 1 (Th1) cells and less activated mast cells. ADIPOR2 and BRAF had a low positive correlation with most of the immune cells. C3 was positively connected with more CD8 T cells and fewer CD4 T cells and B cells. The CIBERSORT algorithm was employed to evaluate the correlation between risk score and 22 immune cells abundance (Fig. 6B). The risk score was positively related with the infiltration of naive B cells, regulatory T cells (Tregs), resting mast cells, resting DCs, and monocytes, while the contrary performance was revealed in relation to risk score and activated memory CD4 T cells, neutrophils, activated dendritic cells, resting NK cells, activated mast cells. Although high-risk patients had relatively lower PD-L1 expression, they tended to have higher stromal score, immune score, and ESTIMATE score, indicating a better immunotherapeutic response (Fig. 6C). More responders were observed in the high-risk group of Kim’s cohort which received anti-PD-1 therapy (Fig. 6D).

The developed signature could predict immunotherapy response and antineoplastic drug sensitivity. (A) Immune infiltration estimations were performed using TIMER, CIBERSORT-ABS, xCELL, and EPIC. (B) CIBERSORT was utilized to estimate the fractions of 22 tumor-infiltrating immune cells in different risk groups. (C) PD-1, PD-L1, CTLA-4 expression and TME scores of the two risk subgroups. (D) The proportion of patients with the therapy response to immunotherapy in the high- or low-risk groups. (E) Survival analyses of patients from GSE62254 and GSE26899 with different risk and chemotherapy statuses. (F) The IC50 values of 6 representative drugs in the two risk groups. IC50, the half-maximal inhibitory concentration. TIMER, Tumor Immune Estimation Resource; CIBERSORT, cell type identification by estimating relative subsets of RNA transcripts; ABS, absolute; EPIC, Estimate the Proportion of Immune and Cancer cells; PD-1, programmed cell death protein 1; PD-L1, programmed cell death ligand 1; CTLA-4, cytotoxic T—lymphocyte antigen 4; ESTIMATE, Estimation of Stromal and Immune cells in Malignant Tumours using Expression data. ns p ≥ 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
The developed signature could predict the outcome of chemotherapy
In GSE62254 and GSE26899 datasets with information on adjuvant chemotherapy, patients were divided into high-risk and low-risk groups (Fig. 6E). Low-risk patients treated with adjuvant chemotherapy had a best OS rate. To determine the efficacy of risk scores as a biomarker to predict the treatment reactions in GC cell lines, we estimated the IC50 values of 198 drugs with available from GDSC database. The IC50 values of 122 drugs were found related to the risk classification (Supplementary Table S3), and most of them were positively connected with risk scores (Supplementary Figure S10). Representative drugs were docetaxel, 5-fluorouracil, paclitaxel, erlotinib, gefitinib, and oxaliplatin (Fig. 6F). The result indicated that high-risk patients were less sensitive to chemotherapy than low-risk ones. Since chemotherapy was still the first-line therapy for the majority of GC patients, the result was consistent with the survival analyses.
Discussion
In the current study, immunosenescence was identified as a hallmark of gastric cancer that is linked with transcriptomic features, genomic variations, and distinctive TME. Immunosenescence and the risk score model based on it have been proved connected with prognosis and the effects of immunotherapy and chemotherapy. A GC patient classified as high-risk will probably have a poor prognosis and an unsatisfied chemotherapeutic outcome, but will have a better immunotherapy response, indicating that high-risk patients are more suitable for immunotherapy and low-risk patients are recommended chemotherapy.
Although there are available immune or senescence-related gene signatures, immunosenescence signature is still lacking in GC44,47,48. Our study portrayed the general immunosenescence microenvironment in GC and constructed a prognostic and predictive model. Our immunosenescence signature shares similar characteristics with these immune or senescence signatures, but it still has its own features, considering immunosenescence is a unique phenotype different from both immune and senescence. With the rapid development of next generation sequencing technology, several molecular subtypes of GC have been widely used. For example, the molecular subtypes established by TCGA include MSI, genome stable (GS), chromosome instability (CIN), Epstein-Barr virus (EBV)-related and hypermutated-single-nucleotide variants (HM-SNVs)45, and by the Asian Cancer Research Group (ACRG) comprise microsatellite stable (MSS)/epithelial–mesenchymal transition (EMT), MSI, MSS/p53 + , and MSS/p53 −49. However, these definitions have encountered huge barriers in the limitations of clinical translation. Our classifications based on immunosenescence are related to prognosis and response to both chemotherapy and immunotherapy. Since the signature only contains 4 hub genes, it is easy for the clinical translation.
Multiple studies have confirmed the relationships between hub genes and immunosenescence. As a protein-coding gene related to lipoprotein metabolism, APOD is significantly and consistently overexpressed with age50. However, APOD seems to be in response to the stress of the body's exemption of oxidation. In fact, the expression of APOD extends the life span while lack of APOD expression results in premature senility51,52. APOD can also be identified as a novel molecular marker of human Th17 cells which mediate inflammation under certain conditions53. As the main effective cytokine of Th17, Interleukin (IL)-17 stimulates and promotes fibroblasts, epithelial cells, and endotheliocytes54. Similar to APOD, ADIPOR2 is continuously overexpressed with age in animal models55. It also works as a protective factor, activating adiponectin/AdipoR signaling pathways in favor of longevity56. The pathways expand the Treg population and reduce the Th17 cell population, which is opposite to the role of APOD in Th1757. BRAF mutations, most commonly the V600E mutation, are the most frequently identified oncogenic mutations, but BRAF mutations in GC are not as frequent as in other kinds of tumors such as melanoma and colorectal cancer. BRAF is associated with immunotherapy outcomes in advanced melanoma58. Considering BRAF-targeted therapy is able to modulate the immune microenvironment, combination treatment strategies with BRAF-targeted therapy and immune checkpoint inhibitors may result in a better response59. Via activation of MAPK ERK1/2 signaling, BRAF leads to effector T cell senescence60. Accumulating evidence suggests that BRAF gene mutations are not only associated with poor prognosis but also with less benefit from anti-epidermal growth factor receptor antibody therapy61. As an inseparable part of complement system, complement C3 generated by gene C3 drives local inflammatory tissue priming by metabolic reprogramming of fibroblasts62. Complement C3 deficiency protects against age-associated diseases, indicating that C3 promotes senescence to some extent63,64,65.
As for immunotherapy, ICIs have significantly prolonged OS in younger and older melanoma patients, and anti-PD-1 drugs are more effective in elder patients66. The aging T cells have low proliferation capabilities, while maintaining highly cytotoxicity and inflammatory cytokine production agents67. In our study, immunosenescence up-regulation group had a higher cytotoxicity score and was more sensitive to anti-PD-1 immunotherapy. This is not absolute, however. Efficacy data of ICIs in elderly NSCLC patients from phase III trials is conflict68. Only 45% high-risk GC patients responded to immunotherapy in our research. For the other high-risk patients, drugs targeting immunosenescence might be helpful.
Additional clinical data and functional experiments with cell lines and animal models are needed to further investigate the idiographic effect of immunosenescence in GC. Our data of 122 chemotherapy drugs in cell lines has reference value for in-depth research. More GC immunotherapy outcome data is necessary to further validate the predictive efficiency of the model. Interestingly, single-cell data has improved that stromal and immune cells such as fibroblasts and macrophages in TME are more important than cancer cells in the process of immunosenescence, so it may be better if we target both noncancerous and cancerous cells.
Our research is retrospective, and our Fudan cohort is lack of therapy response data, so that confounding and bias can be introduced. Therefore, further large-scale and prospective clinical trials are necessary to reconfirm the findings. We need to include abundant cases before they are treated, and collect complete pathological, surgical, treatment, and follow-up data to further assess the risk model’s ability to predict prognosis, chemotherapy, targeted-therapy and immunotherapy response. Prospective studies require a lot of manpower and resources, and we have not yet completed them. It is planned to be carried out in a follow-up study. These efforts will enable us to confirm the clinical relevance of our findings and develop improved treatment strategies for gastric cancer patients.
Conclusion
This study identifies and characterizes the phenotypic features of immunosenescence in gastric cancer. Using data from the MSigDB and ImmPort databases, we identified 99 ISGs, which showed significant enrichment in tumor tissues compared to adjacent normal tissues. Pathway enrichment analysis revealed that biological pathways associated with immunosenescence, including SASP, mitochondrial biogenesis, glycolysis, and ROS, are significantly enriched in GC tissues. High immunosenescence scores were found to correlate with worse OS, PFI, and disease-specific DSS. Genomic and transcriptomic analyses indicated that high immunosenescence scores are associated with a higher TMB and specific gene mutations, such as TP53, TTN, and ARID1A, highlighting the interplay between senescence and immune pathways. Single-cell profiling revealed that fibroblasts and macrophages, rather than cancer cells, primarily contribute to the immunosenescence features in GC. Risk models based on ISGs demonstrated prognostic and predictive value, indicating that high-risk patients, identified by immunosenescence scores, are likely to have poorer outcomes with chemotherapy but better responses to immunotherapy. The clinical relevance of immunosenescence was further supported by its significant association with age, stage, and molecular subtypes.
To sum up, the verification we discovered in the extensive patient cohorts, and the discovery that immunosenescence features reflect both biotic and clinical traits related with prognosis and treatment responses, will improve the development of optimal treatment recommendations and the optimization of personalized cancer therapy.
Data availability
The data that support findings of this study are available in the GEO (https://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE62254, GSE15459, and GSE26899, as well as TCGA (https://portal.gdc.cancer.gov) database. Other original contributions including Fudan cohort presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
References
Smyth, E. C., Nilsson, M., Grabsch, H. I., van Grieken, N. C. & Lordick, F. Gastric cancer. Lancet 396, 635–648 (2020).
Bagchi, S., Yuan, R. & Engleman, E. G. Immune checkpoint inhibitors for the treatment of cancer: Clinical impact and mechanisms of response and resistance. Annu. Rev. Pathol. 16, 223–249 (2021).
Pawelec, G. Age and immunity: What is “immunosenescence”?. Exp. Gerontol. 105, 4–9 (2018).
Xu, W., Wong, G., Hwang, Y. Y. & Larbi, A. The untwining of immunosenescence and aging. Semin. Immunopathol. 42, 559–572 (2020).
Santoro, A., Bientinesi, E. & Monti, D. Immunosenescence and inflammaging in the aging process: Age-related diseases or longevity?. Ageing Res. Rev. 71, 101422 (2021).
Lian, J., Yue, Y., Yu, W. & Zhang, Y. Immunosenescence: A key player in cancer development. J. Hematol. Oncol. 13, 151 (2020).
Goldman, M. J. et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 38, 675–678 (2020).
Barrett, T. et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 41, D991–D995 (2013).
Kim, S. T. et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat. Med. 24, 1449–1458 (2018).
Liberzon, A. et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Bhattacharya, S. et al. ImmPort: disseminating data to the public for the future of immunology. Immunol. Res. 58, 234–239 (2014).
Szklarczyk, D. et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49, D605–D612 (2021).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
The Gene Ontology C. The gene ontology resource: 20 years and still going strong. Nucleic Acids Res. 2019; 47: D330-D8.
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS. 16, 284–287 (2012).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 102, 15545–15550 (2005).
Walter, W., Sanchez-Cabo, F. & Ricote, M. GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics. 31, 2912–2914 (2015).
Hanzelmann, S., Castelo, R. & Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 14, 7 (2013).
Becht, E. et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 17, 218 (2016).
Jiang, P. et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 24, 1550–1558 (2018).
Mayakonda, A., Lin, D. C., Assenov, Y., Plass, C. & Koeffler, H. P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 28, 1747–1756 (2018).
Mermel, C. H. et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, 41 (2011).
Hu, Y. et al. DRscDB: A single-cell RNA-seq resource for data mining and data comparison across species. Comput. Struct. Biotechnol. J. 19, 2018–2026 (2021).
Zhang, P. et al. Dissecting the single-cell transcriptome network underlying gastric premalignant lesions and early gastric cancer. Cell Rep. 27(1934–47), e5 (2019).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell. 177(1888–902), e21 (2019).
Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2018.
Sun, D. et al. TISCH: a comprehensive web resource enabling interactive single-cell transcriptome visualization of tumor microenvironment. Nucleic Acids Res. 49, D1420–D1430 (2021).
Liu, Y. & Zhao, H. Variable importance-weighted random forests. Quant. Biol. 5, 338–351 (2017).
Cheadle, C., Cho-Chung, Y. S., Becker, K. G. & Vawter, M. P. Application of z-score transformation to Affymetrix data. Appl. Bioinformatics. 2, 209–217 (2003).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Kamarudin, A. N., Cox, T. & Kolamunnage-Dona, R. Time-dependent ROC curve analysis in medical research: Current methods and applications. BMC Med. Res. Methodol. 17, 53 (2017).
Iasonos, A., Schrag, D., Raj, G. V. & Panageas, K. S. How to build and interpret a nomogram for cancer prognosis. J. Clin. Oncol. 26, 1364–1370 (2008).
Van Calster, B. et al. Reporting and interpreting decision curve analysis: A guide for investigators. Eur. Urol. 74, 796–804 (2018).
Chen, B., Khodadoust, M. S., Liu, C. L., Newman, A. M. & Alizadeh, A. A. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol. Biol. 1711, 243–259 (2018).
Li, B. et al. Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol. 17, 174 (2016).
Aran, D., Hu, Z. & Butte, A. J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 18, 220 (2017).
Racle J, de Jonge K, Baumgaertner P, Speiser DE, Gfeller D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife. 2017; 6.
Yoshihara, K. et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612 (2013).
Charoentong, P. et al. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 18, 248–262 (2017).
Yang, W. et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 41, D955–D961 (2013).
Maeser D, Gruener RF, Huang RS. oncoPredict: an R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief Bioinform. 2021; 22.
PodgOrska, M., Pietraszek-Gremplewicz, K., OlszaNska, J. & Nowak, D. The role of Apelin and Apelin receptor expression in migration and invasiveness of colon cancer cells. Anticancer Res. 41, 151–161 (2021).
Browning, D. D., Kwon, I. K. & Wang, R. cGMP-dependent protein kinases as potential targets for colon cancer prevention and treatment. Fut. Med. Chem. 2, 65–80 (2010).
Zhou, L. et al. Senescence as a dictator of patient outcomes and therapeutic efficacies in human gastric cancer. Cell Death Discov. 8, 13 (2022).
Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014; 513: 202–9.
Thorsson, V. et al. The immune landscape of cancer. Immunity. 48(812–30), e14 (2018).
Jiang, Y. et al. ImmunoScore signature: A prognostic and predictive tool in gastric cancer. Ann. Surg. 267, 504–513 (2018).
Lin, Y. et al. Immune cell infiltration signatures identified molecular subtypes and underlying mechanisms in gastric cancer. NPJ. Genom. Med. 6, 83 (2021).
Cristescu, R. et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 21, 449–456 (2015).
de Magalhaes, J. P., Curado, J. & Church, G. M. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics. 25, 875–881 (2009).
Sanchez, D. et al. Aging without Apolipoprotein D: Molecular and cellular modifications in the hippocampus and cortex. Exp. Gerontol. 67, 19–47 (2015).
Muffat, J., Walker, D. W. & Benzer, S. Human ApoD, an apolipoprotein up-regulated in neurodegenerative diseases, extends lifespan and increases stress resistance in Drosophila. Proc. Natl. Acad. Sci. U S A. 105, 7088–7093 (2008).
Salkowska A, Karas K, Karwaciak I, Walczak-Drzewiecka A, Krawczyk M, Sobalska-Kwapis M, et al. Identification of novel molecular markers of human Th17 cells. Cells. 2020; 9.
Chang, S. H. T helper 17 (Th17) cells and interleukin-17 (IL-17) in cancer. Arch. Pharm. Res. 42, 549–559 (2019).
Feyzi, M., Tabandeh, M. R., Shariati, M. & Edalatmanesh, M. A. Age associated changes in transcription of adiponectin, AdipoR1 and AdipoR2 genes in pancreas of rats. Cell J. 22, 61–67 (2020).
Iwabu, M., Okada-Iwabu, M., Yamauchi, T. & Kadowaki, T. Adiponectin/adiponectin receptor in disease and aging. NPJ Aging Mech. Dis. 1, 15013 (2015).
Li, W., Geng, L., Liu, X., Gui, W. & Qi, H. Recombinant adiponectin alleviates abortion in mice by regulating Th17/Treg imbalance via p38MAPK-STAT5 pathway. Biol. Reprod. 100, 1008–1017 (2019).
Joshi, K., Atwal, D., Ravilla, R., Pandey, Y., Yarlagadda, N., Kakadia, S., et al. Immunotherapy outcomes in advanced melanoma in relation to age. Perm J. 2020; 24.
Kelley, M. C. Immune responses to BRAF-targeted therapy in melanoma: Is targeted therapy immunotherapy?. Crit. Rev. Oncog. 21, 83–91 (2016).
Gao, A., Liu, X., Lin, W., Wang, J., Wang, S., Si, F., et al. Tumor-derived ILT4 induces T cell senescence and suppresses tumor immunity. J. Immunother. Cancer. 2021; 9.
Sanz-Garcia, E., Argiles, G., Elez, E. & Tabernero, J. BRAF mutant colorectal cancer: Prognosis, treatment, and new perspectives. Ann. Oncol. 28, 2648–2657 (2017).
Friscic, J. et al. The complement system drives local inflammatory tissue priming by metabolic reprogramming of synovial fibroblasts. Immunity. 54(1002–21), e10 (2021).
Shi, Q. et al. Complement C3-deficient mice fail to display age-related hippocampal decline. J. Neurosci. 35, 13029–13042 (2015).
Wu, X. et al. Complement C3 deficiency ameliorates aging related changes in the kidney. Life Sci. 260, 118370 (2020).
Shi, Q., Chowdhury, S., Ma, R., Le, K.X., Hong, S., Caldarone, B.J., et al. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci Transl Med. 2017; 9.
Li, P. et al. The impact of immunosenescence on the efficacy of immune checkpoint inhibitors in melanoma patients: a meta-analysis. Onco. Targets Ther. 11, 7521–7527 (2018).
Rodriguez, J.E., Naigeon, M., Goldschmidt, V., Roulleaux Dugage, M., Seknazi, L., Danlos, F.X., et al. Immunosenescence, inflammaging, and cancer immunotherapy efficacy. Expert Rev. Anticancer Ther. 2022: 1–12.
Ferrara, R., Mezquita, L., Auclin, E., Chaput, N. & Besse, B. Immunosenescence and immunecheckpoint inhibitors in non-small cell lung cancer patients: Does age really matter?. Cancer Treat. Rev. 60, 60–68 (2017).
Acknowledgements
We would like to acknowledge the TCGA and GEO for providing data.
Funding
This work was funded by National Natural Science Foundation of China (82272681).
Author information
Authors and Affiliations
Contributions
R.Z.: Conceptualization (lead); data curation (equal); formal analysis (equal); writing – original draft (lead). X.J.: Conceptualization (supporting); data curation (equal); formal analysis (equal); writing – original draft (supporting). Z.L.: Data curation (supporting); formal analysis (supporting); writing – original draft (supporting); writing – review and editing (supporting). S.H.: Validation (lead); project administration (equal); writing – review and editing (equal). W.F.: Resources (lead). X.Z.: Funding acquisition (lead); project administration (equal); writing – review and editing (equal).
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, R., Jia, X., Li, Z. et al. Identifying an immunosenescence-associated gene signature in gastric cancer by integrating bulk and single-cell sequencing data. Sci Rep 14, 17055 (2024). https://doi.org/10.1038/s41598-024-68054-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-68054-x
Keywords
This article is cited by
-
Correlation between dyslipidaemia and gastric cancer: pathogenesis to prevention and treatment strategies
Lipids in Health and Disease (2025)
-
Integrating multiomics analysis and machine learning to refine the molecular subtyping and prognostic analysis of stomach adenocarcinoma
Scientific Reports (2025)
