
Abstract
The combination of cisplatin and pemetrexed remains the gold standard chemotherapy for malignant pleural mesothelioma (MPM), although resistance and poor response pose a significant challenge. Cytidine deaminase (CDA) is a key enzyme in the nucleotide salvage pathway and is involved in the adaptive stress response to chemotherapy. The cytidine analog capecitabine and its metabolite 5′-deoxy-5-fluorocytidine (5’-DFCR) are converted via CDA to 5-fluorouracil, which affects DNA and RNA metabolism. This study investigated a schedule-dependent treatment strategy, proposing that initial chemotherapy induces CDA expression, sensitizing cells to subsequent capecitabine treatment. Basal CDA protein expression was low in different mesothelioma cell lines but increased in the corresponding xenografts. Standard chemotherapy increased CDA protein levels in MPM cells in vitro and in vivo in a schedule-dependent manner. This was associated with epithelial-to-mesenchymal transition and with HIF-1alpha expression at the transcriptional level. In addition, pretreatment with cisplatin and pemetrexed in combination sensitized MPM xenografts to capecitabine. Analysis of a tissue microarray (TMA) consisting of samples from 98 human MPM patients revealed that most human MPM samples had negative CDA expression. While survival curves based on CDA expression in matched samples clearly separated, significance was not reached due to the limited sample size. In non-matched samples, CDA expression before but not after neoadjuvant therapy was significantly associated with worse overall survival. In conclusion, chemotherapy increases CDA expression in xenografts, which is consistent with our in vitro results in MPM and lung cancer. A subset of matched patient samples showed increased CDA expression after therapy, suggesting that a schedule-dependent treatment strategy based on chemotherapy and capecitabine may benefit a selected MPM patient population.
Similar content being viewed by others

Introduction
Malignant pleural mesothelioma (MPM) is a rare and deadly cancer originating from mesothelial cells of the pleura1. Currently, there are several treatment options for malignant mesothelioma, e.g., surgery, radiotherapy, chemo- and immunotherapy, and combinations thereof. In 2020, the FDA approved immunotherapy for treating treatment-naive unresectable MPM patients as a first-line therapy2. However, due to contraindications or side effects, not all patients with mesothelioma are eligible for immunotherapy. Since 2004, the standard first-line therapy for MPM has been combination therapy with cisplatin and pemetrexed (MTA)3. It has already been shown that schedule-dependent administration of various treatment regimens can increase the efficacy of standard therapy. In this context, in comparison to concurrent treatment, pretreatment with pemetrexed followed by chemotherapy results in reduced cancer cell growth due to persistent DNA damage and the induction of senescence not only in MPM4 but also in non-small cell lung cancer (NSCLC)5, which was also sensitized to subsequent radiotherapy by pretreatment with pemetrexed 6.
DNA replication is a prerequisite for uncontrolled cell proliferation and is a hallmark of cancer7. DNA replication is dependent on sufficiently high nucleotide levels8. Cancer cells can switch to the energy-efficient nucleotide salvage pathway to maintain DNA replication9,10. In detail, pyrimidine de novo synthesis is critically dependent on thymidylate synthase (TS), which is the principal target of MTA11. It has been shown that MTA treatment allows cancer cells to adapt their metabolism and increase their thymidine salvage pathway activity12. Pyrimidine salvage is fueled by recycling intracellular nucleic acids derived from DNA and RNA degradation or by the uptake of extracellular nucleosides and nucleobases13. Cytidine deaminase (CDA) and thymidine phosphorylase (TYMP) perform the first and second steps of the salvage pathway, e.g., the conversion of cytidine to uridine and uracil, respectively. Capecitabine (sold under the brand name Xeloda among others) is a prodrug processed in the liver to the cytidine analog 5′-deoxy-5-fluorocytidine (5’-DFCR). CDA and TYMP convert 5’-DFCR to 5-fluorouracil (5-FU), which inhibits TS. Previously, our group reported that a chemotherapy-resistant subpopulation of the NSCLC cell line A549 exhibited high CDA and TYMP expression14. Moreover, the expression of CDA and TYMP in NSCLC cells was associated with the induction of an epithelial-to-mesenchymal transition (EMT) and increased expression in response to standard chemotherapy, which further augmented the efficacy of subsequent treatment with 5’-DFCR in vitro15.
In this study, we aimed to demonstrate that standard chemotherapy also increases CDA expression in MPM in a schedule-dependent manner, thereby increasing sensitivity to treatment with 5’-DFCR and capecitabine in vitro and in vivo, respectively. Importantly, minimal toxicity of the schedule-dependent triplet regimen was observed in vivo. In addition, in a subset of MPM patient samples, CDA expression was increased after therapy, suggesting that a schedule-dependent treatment strategy based on chemotherapy and capecitabine may be a potentially beneficial treatment strategy for a selected MPM patient population.
Materials and methods
Cell culture and reagents
The MPM cell lines ACC-MESO-1, ACC-MESO-4, NCI-H28, NCI-H2052, NCI-H2452, MSTO-211H, and JL-1 (hereafter MESO-1, MESO-4, H28, H2052, H2452, MSTO, and JL-1, respectively) and the human NSCLC cell line NCI-H358 (hereafter H358) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). The cell lines were fingerprinted (Microsynth, Bern, Switzerland) and routinely tested for mycoplasma contamination using a Mycoplasma Detection Kit (Cat. # rep-mys-10; InvivoGen, San Diego, California, USA). The cell lines were cultured as recommended by the American Type Culture Board (ATCC) in Dulbecco’s modified Eagle’s medium nutrient mixture F12 Ham (Cat. #D6421; Sigma‒Aldrich, St. Louis, MO, USA) or RPMI-1640 (Cat. #11835030; Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (Cat. #10270-106; Life Technologies, Grand Island, NY, USA), Antibiotic-Antimycotic (1X) (Cat. #15240062; Thermo Fisher Scientific, Waltham, MA, USA) and 1% L-glutamine (Cat. #25030-024; Sigma‒Aldrich) at 37 °C in a humidified 5% CO2 incubator. The medium was changed every 3 days. Cells were passaged using TrypLE as indicated by the manufacturer (Cat. # A1217702, Thermo Fisher Scientific, Waltham, MA, USA).
Pemetrexed/MTA (commercial name ‘ALIMTA’; Cat# VL7640) was purchased from Eli Lilly (Suisse) SA (Vernier/Geneva, Switzerland). Cisplatin (cisplatin Ebewe) was purchased from Sandoz Pharmaceuticals AG (Steinhausen/Cham, Switzerland). 5’-deoxy-5-fluorocytidine (5’-DFCR) was purchased from CaymanChem (Cat. #10010682).
Hypoxic conditions
Hypoxia was induced by supplying a humidified incubator with 1% O2, 5% CO2, and 94% N2. Mesothelioma cell lines were incubated at 37 °C. The culture medium was replaced every 2–3 days.
Cell viability assay
MESO-1 and JL-1 cells were seeded at a density of 1000 cells per well in 96-well plates. After overnight incubation, different concentrations of 5’-DFCR were added, and the cells were incubated for another 3 days. Cell viability was measured using a PrestoBlue Assay Kit16 according to the manufacturer’s protocol (Cat. #A13261, Thermo Fisher Scientific, Waltham, MA, USA). Changes in absorbance were recorded with an Infinite M1000 Tecan Reader.
Western blot analysis
MPM cells were seeded into 10 cm plates (0.05–0.5 × 106 cells, depending on the doubling time of the different cell lines). Twenty-four hours after seeding (on the morning of day 1 (D1)), 5 μM MTA and 10 μM cisplatin were added for 24 h. Forty-eight hours after seeding (on the morning of day 2 (D2)), the drugs were removed, the medium was changed, and the cells were collected at the indicated time points. Cells were lysed in RIPA buffer (Merck, R0278) containing 1 × protease and phosphatase inhibitor cocktail (Thermo Scientific, 78444). Protein concentrations were determined using a Pierce™ BCA Protein Assay Kit. Equal amounts of protein lysates (35–50 μg/lane) were resolved by SDS‒PAGE and blotted onto nitrocellulose membranes. The membranes were blocked in Intercept® Tris-buffered saline (TBS) blocking buffer for 1 h at room temperature and then incubated with specific primary antibodies (anti-CDA (atlasantibodies, HPA064202), anti-VIM (Novus Biologicals, NBP2-34809R), or anti-actin (Cell Signaling Technology, #3700)) at 4 °C overnight on an orbital shaker: TBS containing 0.2% Tween-20 was used to wash the membranes. IRDye 680LT-conjugated goat anti-mouse IgG and IRDye 800CW-conjugated goat anti-rabbit IgG secondary antibodies (Li-COR Biosciences) were used at a dilution of 1:5000. Finally, signals of the immobilized secondary antibodies were imaged and quantified using the Image Studio Lite System.
Public databases
The expression atlas (https://www.ebi.ac.uk/gxa/home) was used to collect RNA-seq data from human cancer cell lines to compare gene expression. Gene expression data from MPM patient samples were obtained from cBioPortal (https://www.cbioportal.org/). The cancer genome atlas (TCGA) was used for gene correlation analysis of MPM patient samples.
Use of experimental animals, and human participants
All experiments with live animals were (i) approved by the Veterinary Office of the Canton of Berne (approval number AB_BE49_2022), (ii) performed in accordance with the relevant guidelines and regulations, and (iii) reported according to the ARRIVE guidelines (https://arriveguidelines.org).
All experiments involving tissue samples from human participants were (i) approved by the Ethics Committee of the Canton of Bern, which waived the requirement for written informed consent (KEK-BE: 2016-01497), and (ii) conducted in accordance with the relevant guidelines and regulations.
In vivo mouse study
The experiments were performed in RAG mice, mixed in sex, aged 6–8 weeks. To establish tumor xenografts, suspensions of 0.5 × 106 MESO-1 tumor cells (in PBS) mixed 1:1 with BD Matrigel Basement Membrane Matrix (Cat. #356231; Corning, Inc., Corning, NY, USA) were subcutaneously inoculated on the left and right flanks. When the tumor volume reached 150–200 mm3, the mice were randomly assigned to one of the following treatment groups: (1) the control group; (2) the concomitant treatment group: cisplatin (4 mg/kg) plus pemetrexed (300 mg/kg) (i.p., 1 injection every second week) for six weeks; (3) the capecitabine/Xeloda treatment group (360 mg/kg, per os., 5 days/every second week) for six weeks; (4) the triple combination group: concomitant treatment + 1 week later capecitabine for 5 days. Each cycle lasted 2 weeks, and 3 treatment cycles were performed. Body weight and tumor size were measured every week. Tumor size was calculated as (length × width × width)/2. At the endpoint, tumor samples were collected for weight measurements and analysis by immunohistochemistry.
Tissue and immunohistochemistry
Immunohistochemistry (IHC) was performed at room temperature with the fully automated BOND RX® staining system (Leica Biosystems). CDA staining was conducted using a polyclonal antibody against human CDA (Atlasantibodies, HPA064202, Tris, 1:500). CDA expression on tumor cells was evaluated using the H-score, the product of staining intensity and frequency of positive tumor cells, where 0 = none, 1 = weak, 2 = intermediate, and 3 = strong17.
Statistical analysis
The data were analyzed using Prism 8 (GraphPad). In all the studies, the data represent biological replicates (n). The data are presented as the mean ± standard deviation (SD) or mean ± standard error of the mean (SEM), as indicated in the figure legends. Comparisons of mean values were conducted with two-tailed Student's t tests and one-way and two-way ANOVA with Tukey’s multiple comparisons tests, as indicated in the figure legends. In all analyses, p values less than 0.05 were considered to indicate statistical significance.
Tissue microarray construction and analysis
The study cohort comprised 98 patients with consecutively resected MPM diagnosed at the Institute of Tissue Medicine and Pathology, University of Bern, Switzerland. All tumors were resected at the Department of General Thoracic Surgery, Inselspital, Bern University Hospital, University of Bern, Switzerland. The cohort was assembled according to the pathologic reports and validated using clinical files. The study was approved by the Cantonal Ethics Commission of the Canton of Bern, which waived the requirement for written informed consent (KEK-BE: 2016-01497).
A next-generation tissue microarray (TMA) was constructed from FFPE tissue blocks as described elsewhere using an automated tissue microarrayer (Grandmaster, 3DHistech, Budapest, Hungary)18,19. In brief, after review of the hematoxylin and eosin-stained slides, digitization, and annotation, tissue cores (diameter = 0.6 mm) were transferred to the TMA acceptor block. At least six punches from different tumor regions were included, which allowed us to evaluate intratumoral heterogeneity. Punches from the tumor center (n = 3) and the infiltration front (n = 3) were selected separately. Where adjacent lung tissue was present, additional punches of lung tissue infiltration were added (n = 3). The subsequent automated TMA construction was performed as described previously18,19.
R software (version 4.3.0, https://cran.r-project.org) was used for statistical analysis. To assess heterogeneity between different cores, different regions, or different time points (before neoadjuvant therapy vs. after neoadjuvant therapy), we used the Friedman test and the Wilcoxon signed rank test to determine Benjamini–Hochberg corrected p values. Bland‒Altman plots were generated for visualization. For patient or region-wise comparisons, the arithmetic average of the overall assessed cores was used for statistical analysis. Fisher’s exact test and the Wilcoxon rank-sum test were used to compare baseline characteristics according to CDA expression. For survival analysis, the survival (https://cran.r-project.org/web/packages/survival/index.html) and survminer packages (https://cran.r-project.org/web/packages/survminer/index.html) were used. We applied the log-rank test for univariate analysis, and Kaplan‒Meier plots were generated for visualization. Overall survival was defined as the time from the pathology-based diagnosis until death from any cause or loss to follow-up.
Ethics approval and consent to participate
This study was approved by the Cantonal Ethics Committee of Bern, Switzerland, and was performed in accordance with the Declaration of Helsinki.
Results
The basal levels of CDA expression in vitro and in patient samples and its association with EMT
CDA is a key enzyme of the nucleotide salvage pathway20 (Fig. 1A). Our in silico analysis of the gene expression data of the TCGA dataset revealed that CDA expression is relatively low in MPM patient samples compared to that in samples from other tumor entities (third lowest out of the 30 analyzed tumor entities; Fig. S1A). Furthermore, basal CDA protein expression was variable at low levels in the MPM cell lines tested, e.g., in MESO-1, MESO-4, H28, H2052, H2452, MSTO, and JL-1, and was relatively high in the NSCLC cell line H358 (Fig. 1B), as we previously reported15. The MPM cell line H2052 was an exception because it presented higher CDA protein expression compared with the other MPM cell lines. Our in silico analysis showed a similar pattern at the RNA level; e.g., the mRNA expression of CDA was greater in the MPM cell line H2052 and the NSCLC cell line H358 than in the other MPM cell lines tested (Fig. 1C).

CDA expression in MPM cell lines and patient samples. (A) Schematic representation of the role of CDA in capecitabine metabolism and its role in the pyrimidine de novo and salvage pathways. (B) Western blot analysis of basal CDA protein expression in the indicated MPM cell lines. The epithelial NSCLC cell line H358 was included as a positive control for CDA expression. (C) Graphical representation of CDA mRNA expression data from selected cell lines from the RNAseq Gene Expression Atlas. (D) In silico analysis of gene expression data from MPM tumor samples from patients in the TCGA database. (E) Correlation analysis of gene expression data from tumors of mesothelioma patients derived from the cBioPortal. (F) Analysis of MESO-1 cell viability normalized to that of the solvent-treated control; n = 3 independent experiments. (G) Analysis of the viability of JL-1 cells normalized to that of solvent-treated controls. The experiments were performed in triplicate and were replicated at least twice.
Our group has previously shown that CDA expression in NSCLC cells is associated with EMT and can be induced by treatment with chemotherapy or TGF-β, both of which are known to induce EMT15. Our in silico analysis of gene expression data from 82 patient-derived MPM tumors from The Cancer Genome Atlas (TCGA) revealed that high CDA expression significantly correlated with an increased EMT score (Fig. 1D). In detail, CDA expression was positively correlated with the mesenchymal transcription factors SNAI1, SNAI2, TWIST1, ZEB1, and ZEB2 and the mesenchymal markers FN1 and CDH2 and negatively correlated with the epithelial transcription factor TWIST2, the epithelial marker CDH1 encoding E-cadherin21 (Fig. 1E). In agreement with these findings, analysis of gene expression data from cBioportal (https://www.cbioportal.org) revealed that CDA expression is also significantly positively correlated with the mesenchymal markers CD90 (THY1) and vimentin (VIM) (Fig. S1B,C). Moreover, the mRNA expression of CDA in MPM patient samples was relatively low in epithelial tumors, slightly increased in biphasic tumors, and highest in sarcomatoid tumors (only two sarcomatoid samples were available in this dataset) (Fig. S1D). In conclusion, the basal expression of CDA in MPM cell lines and patient samples is relatively low, but CDA expression is positively correlated with EMT status.
Mesothelioma cell lines feature low basal sensitivity to 5’-DFCR treatment
Previously, we showed that sensitivity to 5’-DFCR was related to CDA expression and the EMT status of NSCLC cells. In detail, for the EMT-related subpopulations in the NSCLC cell line A549, the IC50 for 5’-DFCR was 0.3 mM for the mesenchymal-like paraclonal subpopulation. In contrast, the epithelial-like, holoclonal subpopulation was resistant to 5’-DFCR treatment15. The IC50s for 5’-DFCR in the epithelioid cell line MESO-1 and the sarcomatoid cell line JL-1 were 3.3 and 5.0 mM, respectively (Fig. 1F,G, respectively). Since the enzymatic activity of CDA is required to convert 5'-DFCR to 5-FU, the relatively high IC50 values for 5’-DFCR are consistent with the relatively low basal CDA expression levels in MPM cell lines.
Standard chemotherapy increases CDA expression in mesothelioma cells in an EMT- and schedule-dependent manner
We tested whether standard chemotherapy with cisplatin and MTA could increase CDA expression levels in mesothelioma cells, as we previously observed in various NSCLC cell lines15. As we described before, H358 cells were used as a control for positive CDA expression (Fig. 2A)15. After concomitant treatment with 10 μM cisplatin and 5 μM MTA for 24 h, the CDA expression levels increased over time in the MESO-1 cells compared to those in the untreated control (D0), reaching the highest expression levels on day 6 post-treatment, i.e., on recovery day 6 (RD6). The expression of the EMT marker VIM initially decreased and subsequently also increased in a schedule-dependent manner, slightly earlier than CDA expression did (Fig. 2A–C). These results suggest that delaying the additional administration of 5’-DFCR after the EMT-associated increase in CDA levels, e.g., after D6, might enhance anticancer efficacy compared with concomitant administration of standard chemotherapy.

Treatment with chemotherapy increased CDA expression in MPM cells in a schedule-dependent manner. (A) Western blot analysis of CDA protein expression in MESO-1 cells on recovery days (RD) after 24 h of cisplatin/MTA exposure. (B) Quantification of CDA protein expression, normalized to the expression of beta-actin (ACTIN), relative to the untreated control on day 0. (C) Quantification of VIM protein expression, normalized to the expression of beta-actin (ACTIN), relative to the untreated control on day 0.
CDA expression is associated with hypoxia, which differentially affects the growth of MPM histotypes
Previously reported in MPM, hypoxic conditions promote EMT and induce cisplatin resistance, which is associated with a more aggressive phenotype22. Thus, we tested whether hypoxic conditions affect CDA expression in mesothelioma cells in vitro. Indeed, compared with that in MESO-1 cells cultured under normoxic conditions, CDA protein expression increased over time after the initiation of hypoxia (1 and 20% O2, respectively), with a peak in expression occurring at D6 (Fig. 3A,B). To investigate whether the association of CDA expression with hypoxia is also relevant in the clinical setting, we analyzed 87 mesothelioma patient samples from a public database (www.cBioportal.org). Intriguingly, in those patient samples, CDA mRNA expression was significantly positively correlated with the expression of HIF1A, which encodes hypoxia-inducible transcription factor 1-alpha (HIF-1α) (Fig. S1E).

Hypoxia increases CDA expression in MESO-1 cells over time and differentially affects the growth of MPM cell lines. (A) Western blot analysis of CDA and beta-actin protein expression in MESO-1 cells exposed to hypoxia for the indicated time points (B) Quantification of the data in Fig. 3A, i.e., CDA protein expression, normalized to the expression of beta-actin (ACTIN) relative to the untreated control on day 0. (C–F) Relative cell growth of the indicated cell lines. After the cells were seeded into 96-well plates, the plates were incubated in parallel under normoxic and hypoxic conditions for 72 h, after which the medium was replaced after 48 h. Cell growth was determined in parallel for all plates by the Presto-Blue assay. The data are presented as the mean ± SEM (n = 12). Not significant (ns), *p < 0.05 and ****p < 0.0001 according to an unpaired t test.
Hypoxia has previously been shown to affect colony formation in several mesothelioma cell lines22. Compared to that of normoxic cells, the growth of the epithelial NSCLC cell line H358 was decreased under hypoxic conditions (Fig. 3C). Concerning the more epithelial-like MPM cell lines, hypoxia increased the growth of the MESO-4 and JL-1 cell lines (Fig. 3D), whereas the growth of the epithelial/fibroblast-like cell line H2452 was not affected, and the growth of the more fibroblast-like MESO1 cell line was decreased (Fig. 3E). Interestingly, the growth of the sarcomatoid MPM cell lines H2052 and H28 was significantly increased under hypoxic conditions (Fig. 3F). In summary, high CDA expression is associated with hypoxia and a sarcomatoid phenotype in MPM. Furthermore, the growth of sarcomatoid MPM cell lines increased under hypoxic conditions, whereas the growth of epithelial-like cell lines was more variable.
High CDA expression in tumor xenografts is further augmented by chemotherapy
It has been shown that MPM tumors contain areas of hypoxia23. Thus, we characterized CDA expression in vivo, i.e., during tumor development and after exposure to chemotherapy. As described above, the MESO-1 MPM cell line features relatively low basal CDA expression in vitro (Fig. 1B,C). However, 7 days after implantation, the MESO-1-derived tumors contained cells featuring high CDA expression (Fig. 4A). We also compared CDA expression in different parts of the tumor, as in the periphery, the estimated tissue oxygenation was increased compared to that in the tumor center24. Indeed, CDA expression was greater in the centers of tumor xenografts than in more peripheral regions (Fig. 4B).

MPM tumor xenografts feature high CDA expression and are more susceptible to schedule-dependent than concurrent therapy. (A) Representative image of CDA expression in a MESO-1 xenograft tumor. (B) In the periphery and center of each tumor, at least two regions with high and weak CDA intensities were selected for further H-score analysis. The tumor center was determined to be 1000 µm from the tumor edge. ***p < 0.001 according to an unpaired t test. (C) H-scores over time in untreated tumors (n = 2). (D) H-scores over time after treatment with one cycle of chemotherapy compared to those of the untreated control. The data are presented as the mean ± SEM (n = 4). **p < 0.01, comparison between groups by two-way ANOVA with Tukey’s multiple comparisons test. (E) Mouse body weight during the experiment shown in panel F. (F) Analysis of tumor growth over time in the MESO-1 xenograft tumors treated with the indicated regimens. The data were analyzed by two-way ANOVA with Tukey’s multiple comparisons test and are presented as the mean ± SEM (n = 4 or 5). **p < 0.01, ****p < 0.0001. (G) Tumor weight on day 100 after the start of the experiment. The data are presented as the mean ± SEM (n = 4 or 5). Not significant (ns), *p < 0.05, **p < 0.01, and ****p < 0.0001 according to unpaired t tests.
During the development of xenograft tumors, the area of hypoxia has been shown to increase24. Interestingly, our analysis of non-treated tumors revealed that CDA expression increased over time (Fig. 4C). Our in vitro experiments revealed that treatment with standard chemotherapy increased CDA expression in MPM cells in a time-dependent manner (see Fig. 2A). Compared to that in the untreated control tumors, standard chemotherapy further increased CDA expression in tumors in a time-dependent manner; i.e., CDA expression was increased 24 h after treatment with chemotherapy and further increased up to day 13, with elevated expression persisting throughout the experiment (Fig. 4D). Thus, in comparison to in vitro cell culture conditions, CDA expression in MPM cells is higher in vivo and further increased after chemotherapy.
Schedule-dependent therapy in vivo
We have already shown in vitro that the efficacy of capecitabine treatment depends on high CDA levels15. Thus, we hypothesized that treatment of MPM tumors with chemotherapy combined with delayed capecitabine treatment should have increased anticancer activity compared to concomitant treatment. To test this hypothesis, we compared several treatment strategies in vivo (Fig. S2A). Body weight was not significantly affected by any of the tested treatment regimens (Fig. 4E). During the treatment phase (after 7–48 days), all the treatment regimens effectively suppressed tumor growth compared with that in the vehicle-treated control group (Fig. 4F). Additionally, immediately after the treatment phase, e.g., between days 48 and 62, the tumor volume did not significantly increase after treatment with the four treatment regimens. However, after day 62, tumor growth was significantly accelerated after concomitant chemotherapy and capecitabine treatment. The accelerated growth phase was only reached after day 84 with concomitant triple combination therapy. Interestingly, with schedule-dependent combination therapy, e.g., combined treatment with pemetrexed and cisplatin on day 1 followed by treatment with capecitabine on days 8–12 of a 14-day treatment cycle, this accelerated growth phase was not reached until the end of the experiment, i.e., on day 100 (Fig. 4F and Fig. S2B). Consistent with these findings, tumor weight was reduced on day 100 after concurrent triplet therapy compared to after chemotherapy or capecitabine alone and was further reduced after schedule-dependent triplet therapy (Fig. 4G). Thus, sequential treatment combining chemotherapy with delayed capecitabine administration demonstrated increased anticancer efficacy against MPM tumors compared to concomitant treatment, as evidenced by suppressed tumor growth and reduced tumor weight on day 100, suggesting a schedule-dependent benefit.
Increased CDA expression in patient MPM samples before neoadjuvant therapy is significantly associated with worse overall survival
Our previous study revealed that chemotherapy elicits patient-specific changes in CDA expression in NSCLC tumor tissue samples15. To investigate the clinical significance of our findings in MPM, we examined CDA expression levels in patient samples before and after chemotherapy. For this purpose, we generated a tissue microarray (TMA) from 98 MPM patients, consisting of 867 TMA cores, 327 before and 540 after neoadjuvant therapy (Fig. S3A). Our cohort had a median age of 67 years, the disease stage was mainly stage I or III, and the predominant histotype was epithelial (baseline characteristics; see Tables 1, 2). Our xenograft experiments revealed that CDA expression was increased in the tumor center compared to the periphery (Fig. 4A,B). Therefore, we included three punches from three different tumor regions, namely, the infiltration zone and the infiltration region toward the lung and the tumor center. It was not always possible to assess all the cores; thus, not the same number of cores was available for all patients. However, for most patients, at least six cores were assessed. Only 14.4% of the samples had positive CDA staining, the intensity of which varied considerably (Figs. 5A,B and S3A,B). No intercore or interregional heterogeneity was detected in the samples before or after neoadjuvant therapy. For 70 patients, samples from either time point were available and could be compared via an unpaired test. There was no significant difference in the H-scores between these patients (p = 0.394, n = 70). This finding held when comparing each of the different regions (infiltration zone; p = 0.724, n = 69; infiltration toward the lung: p = 0.703, n = 55; tumor center: p = 0.496, n = 70). For 28 patients, samples from two time points were available and could be compared via paired procedures. There was no significant difference in the H-scores between these patients (p = 0.185, n = 28; Fig. S3C). However, when considering only the tumor centers, samples had significantly higher CDA expression after neoadjuvant therapy (p = 0.035, n = 26; Fig. 5A,B; Fig. S3D).

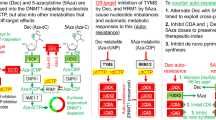
CDA expression in MPM patient samples before neoadjuvant therapy was significantly associated with worse overall survival. (A) Example of a tumor center with a low basal CDA staining intensity that increased significantly after neoadjuvant therapy (sample 7522045; see also Fig. S3D). (B) Example of a tumor center with high basal CDA staining intensity that increased significantly after neoadjuvant therapy (sample 12678090; see also Fig. S3D). (C) Correlation analysis of CDA expression and survival in non-matched MPM samples before neoadjuvant therapy (p = 0.029, n = 57). (D) Correlation analysis of the relative changes in the expression of CDA and survival time in paired MPM tissue samples before and after therapy (p = 0.18, n = 28). (E) Schematic illustration of the concept of schedule-dependent, capecitabine-based therapy. In detail, tumors are composed of various cellular subsets, including cancer cells with shared genetic alterations. These cancer cells can be further divided into subpopulations, each characterized by distinct epigenetic programs, resulting in different states along the EMT spectrum. Standard chemotherapy, specifically the combination of cisplatin and MTA for lung cancer treatment, primarily eradicates cancer cells with an epithelial phenotype. In contrast, cancer cells with a more mesenchymal phenotype tend to be more resistant to this treatment. Additionally, chemotherapy induces EMT in a subset of cancer cells, causing some cells to newly acquire a resistant state. This mesenchymal/resistant state is associated with increased CDA expression, rendering these cells sensitive to capecitabine treatment.
Since most samples exhibited negative CDA staining, we checked whether CDA expression was associated with overall survival separately before and after neoadjuvant therapy. In non-matched samples, CDA expression after neoadjuvant therapy was not significantly associated with improved overall survival (p = 0.71, n = 69). However, CDA expression before neoadjuvant therapy was significantly associated with worse overall survival (p = 0.029, n = 57; Fig. 5C). Finally, in the available paired samples, an increase in CDA expression was not significantly associated with worse survival; however, the survival curves were separate, and it would be interesting to investigate this effect with an increased sample size (p = 0.18, n = 28; Fig. 5D).
In summary, in vitro, the basal level of CDA protein expression in MPM cells is increased by hypoxia and treatment with chemotherapy. The CDA protein level in MPM cells increases upon subcutaneous xenograft transplantation, increases over time during tumor development, and can be further increased by chemotherapy in a schedule-dependent manner. Our in vivo experiments revealed the superiority of a schedule-dependent regimen, e.g., the inhibition of tumor growth by cisplatin and pemetrexed combination therapy was further augmented by schedule-dependent treatment with capecitabine compared to concomitant triple therapy (Fig. 5E). Finally, before neoadjuvant therapy, CDA expression is detectable in a subset of MPM patient samples and is associated with worse overall survival, indicating that CDA expression might be relevant for managing MPM in the clinical setting.
Discussion
We have previously shown that chemotherapy-induced CDA expression renders NSCLC cancer cells sensitive to subsequent capecitabine treatment15. To our knowledge, such a treatment approach has never been tested in MPM. Extensive studies with various cancer cell lines revealed that CDA expression is downregulated in approximately 60% of cancer cell lines and that low CDA levels correlate with CDA promoter methylation25. Changes in promoter methylation are described as a hallmark of EMT26. Indeed, we found that CDA expression in MPM strongly correlates with EMT status both in patient samples and in vitro (Figs. 1, 2, respectively), which agrees with our previous findings in NSCLC15. However, recent single-cell sequencing data revealed significant differences between EMT trajectories, including a so-called hybrid-E/M state27, characterized by the simultaneous expression of both epithelial and mesenchymal markers14. The hybrid E/M state is associated with hypoxia28. Hypoxia contributes to the development of neoplastic disease, the initiation of an aggressive phenotype, and chemotherapy resistance in various cancers, including mesothelioma29. We found that CDA expression correlated strongly with HIF-1-alpha expression in patient samples (Fig. S1E) and was highest in sarcomatoid and biphasic MPM samples (Fig. S1D), which have a more aggressive phenotype and are more resistant to therapy. Our experiments demonstrated that CDA expression was only modestly elevated in response to exposure to 1% O2, which simulates physiological hypoxia in tumors30. However, the actual oxygen levels during treatment and tumor progression are likely to be considerably lower. Thus, it would be of interest to investigate how CDA expression changes at oxygen levels below 1%. Hypoxia is intrinsically linked with angiogenesis, which is crucial for MPM tumor growth31. Therefore, further studies at the single-cell level are needed to determine the molecular mechanisms underlying the association between CDA expression and EMT status, hypoxia, and angiogenesis. Although these features constitute a complex network, identifying molecular markers associated with increased CDA activity may serve as predictive and prognostic markers for capecitabine-based combination therapies.
Concerning capecitabine-based therapies, this study revealed that mesothelioma cell lines are relatively resistant to a single treatment with 5’-DFCR in vitro (Fig. 1F,G). However, treatment of MPM cells in vitro with chemotherapy led to a schedule-dependent increase in CDA expression (Fig. 2). This peak CDA expression was even greater than the basal CDA expression in the NSCLC cell line H358 (Fig. 2), which was used as a positive control and was shown in our previous study to be sensitive to 5’-DFCR treatment15. These results suggest that a schedule-dependent therapy for MPM is feasible: a first phase consisting of standard chemotherapy to increase CDA expression, which is exploited in a second phase by treatment with capecitabine. Intriguingly, CDA expression was readily detectable in vivo, e.g., in subcutaneous xenograft tumors generated from MESO-1 cells (Fig. 4). Consequently, the differences in CDA expression between our in vitro and in vivo experiments suggest that either in vivo orthotopic models or ex vivo patient samples should be used to investigate the underlying molecular mechanisms in subsequent studies.
Finally, our in vivo experiments revealed that cisplatin-pemetrexed combination therapy, the current gold-standard chemotherapy for treating MPM, effectively inhibited tumor growth only during the treatment phase (Fig. 4E). As soon as the standard therapy was discontinued, the tumors resumed growth and almost reached the size of the untreated control group at the end of the experiment. Interestingly, treatment with capecitabine per se suppressed tumor growth as efficiently as standard chemotherapy did (Fig. 4E). In the clinical setting, a phase II multicenter study revealed that the antitumor activity of capecitabine is insufficient to warrant further exploration in patients with malignant mesothelioma32. Since we observed differences in the anticancer effect of capecitabine against different MPM cell lines in vitro (Fig. 1F,G) and capecitabine reduced the growth of MESO-1 xenografts in vivo (Fig. 4), we conclude that capecitabine may be effective against a subset of MPM tumors that can be identified by molecular markers related to CDA activity. However, once treatment was discontinued, tumors treated with capecitabine per se also exhibited a high proliferative capacity. Only the concomitant triple combination showed efficacy after the treatment was stopped, possibly due to the accumulation of persistent DNA damage, as we previously showed in NSCLC for schedule-dependent triple combination therapy15. Importantly, standard chemotherapy combined with the delayed administration of capecitabine had the greatest efficacy compared to all the other regimens and did not cause toxicity; i.e., the body weights of the animals remained stable throughout the experiment (Fig. 4E).
A limitation of our study is that we did not monitor tumor growth for the group receiving standard chemotherapy combined with the delayed administration of capecitabine beyond 100 days, i.e., the time point where the maximum tolerable tumor size was reached for the control group (Fig. 4F). Indeed, a slight increase in tumor size was detectable after day 84, suggesting that also this group may have benefited from an additional treatment cycle or an alternative treatment regimen in the adjuvant setting. Further, using an immuno-competent and orthotopic MPM animal model would better evaluate the impact of capecitabine-based combination therapy and its potential synergy with immune checkpoint inhibitors (ICIs). Indeed, in the clinical setting for the treatment of MPM, ICIs demonstrated superiority over standard chemotherapy and with improved overall survival in the first and later-line settings33. However, a significant proportion of MPM patients still do not derive benefit from ICIs, highlighting the need for new treatment strategies and predictive biomarkers of response.
The construction of the TMA and the subsequent analysis revealed that 11 out of 57 (14%) MPM patient samples expressed CDA at various levels before neoadjuvant therapy (Fig. 5). Since increased CDA expression is associated with a mesenchymal phenotype, which is linked to resistance to cisplatin and pemetrexed treatment, positive CDA staining could be used to identify patients who might benefit from capecitabine-based chemotherapy. In this context, it was recently shown that ALK inhibitor-resistant NSCLC cells exhibit increased CDA expression and a mesenchymal phenotype and that targeting CDA expression reduces proliferation and reverses the EMT phenotype34. Interestingly, in 2009, a clinical case of NSCLC was reported in which standard chemotherapy was used in combination with capecitabine35. The patient achieved a partial response to capecitabine after several cycles of chemotherapy. In this context, capecitabine-based treatment regimen are well established for treating several solid tumors, including colorectal cancer36. Intriguingly, significant antitumor effects were achieved with an anti-VEGF drug followed by long-term, low-dose capecitabine therapy37, demonstrating that the efficacy of a capecitabine-based sequential treatment regimen is not limited to pretreatment with chemotherapy. Our results are consistent with these findings and provide further evidence that a schedule-dependent, capecitabine-based therapy may be a promising and affordable therapeutic option for lung cancer, MPM and other rare cancers.
Conclusions
In summary, for the first time, we demonstrated that exploiting the increase in pyrimidine salvage pathway activity upon chemotherapy might be a feasible treatment strategy for MPM. Our study also identified hypoxia and EMT status as potential prognostic and predictive markers for combination therapy based on increased CDA activity. Additional studies will be necessary to unravel the molecular pathways associated with regulating CDA expression not only in MPM cancer cells but also in tumor-associated immune and stromal cells. Nevertheless, our current study extends our previous findings in NSCLC and lays the foundation for developing a schedule-dependent therapy based on CDA activity for treating MPM. Approving novel therapeutics for cancer patients is highly time-consuming and costly. From a pragmatic perspective, the regulatory hurdles for a novel treatment strategy based on the combination of clinically approved drugs are lower than those for other agents, so its implementation in clinical practice could be more straightforward, which would greatly benefit MPM patients.
Data availability
This published article and its supplementary information files include all the data generated or analyzed during this study.
Abbreviations
- 5’-DFCR :
-
5’-Deoxy-5-fluoro-cytidine
- CDA:
-
Cytidine deaminase
- cisPt :
-
Cisplatin
- EMT :
-
Epithelial-to-mesenchymal transition
- HIF-1-alpha :
-
Hypoxia-inducible factor 1-alpha
- MPM :
-
Malignant pleural mesothelioma
- MTA :
-
Pemetrexed
- NSCLC :
-
Non-small cell lung cancer
- TMA :
-
Tissue microarray
References
Yang, H., Testa, J. R. & Carbone, M. Mesothelioma epidemiology, carcinogenesis, and pathogenesis. Curr. Treat. Opt. Oncol. 9(2–3), 147–157 (2008).
Kulkarni, A. A. & Kratzke, R. A. A turning point for mesothelioma therapy. JCO Oncol. Pract. 18(2), 125–126 (2022).
de Gooijer, C. J., Baas, P. & Burgers, J. A. Current chemotherapy strategies in malignant pleural mesothelioma. Transl. Lung Cancer Res. 7(5), 574–583 (2018).
Karatkevich, D. et al. Schedule-dependent treatment increases chemotherapy efficacy in malignant pleural mesothelioma. Int. J. Mol. Sci. 23(19), 11949 (2022).
Tieche, C. C. et al. Prolonged pemetrexed pretreatment augments persistence of cisplatin-induced DNA damage and eliminates resistant lung cancer stem-like cells associated with EMT. BMC Cancer 16(1), 125 (2016).
Dorn, P. et al. Schedule-dependent increased efficiency of pemetrexed-ionizing radiation combination therapy elicits a differential DNA damage response in lung cancer cells. Cancer Cell Int. 16(1), 66 (2016).
Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100(1), 57–70 (2000).
Gao, Y. et al. Cisplatin-resistant A549 non-small cell lung cancer cells can be identified by increased mitochondrial mass and are sensitive to pemetrexed treatment. Cancer Cell Int. https://doi.org/10.1186/s12935-019-1037-1 (2019).
Vander Heiden, M. G. et al. Metabolic pathway alterations that support cell proliferation. Cold Spring Harb. Symp. Quant. Biol. 76, 325–334 (2011).
Lumeau, A. et al. Cytidine Deaminase Protects Pancreatic Cancer Cells from Replicative Stress and Drives Resistance to DNA-Targeting Drugs (Cold Spring Harbor Laboratory, 2021).
Lam, S. W., Guchelaar, H. J. & Boven, E. The role of pharmacogenetics in capecitabine efficacy and toxicity. Cancer Treat. Rev. 50, 9–22 (2016).
Chen, X., Yang, Y. & Katz, S. Early detection of thymidylate synthase resistance in non-small cell lung cancer with FLT-PET imaging. Oncotarget 8(47), 82705–82713 (2017).
Walter, M. & Herr, P. Re-discovery of pyrimidine salvage as target in cancer therapy. Cells 11(4), 739 (2022).
Tieche, C. C. et al. Tumor initiation capacity and therapy resistance are differential features of EMT-related subpopulations in the NSCLC cell line A549. Neoplasia 21(2), 185–196 (2019).
Gao, Y. et al. Chemotherapy-induced CDA expression renders resistant non-small cell lung cancer cells sensitive to 5’-deoxy-5-fluorocytidine (5’-DFCR). J. Exp. Clin. Cancer Res. 40(1), 138 (2021).
Xu, M., McCanna, D. J. & Sivak, J. G. Use of the viability reagent PrestoBlue in comparison with alamarBlue and MTT to assess the viability of human corneal epithelial cells. J. Pharmacol. Toxicol. Methods 71, 1–7 (2015).
Fedchenko, N. & Reifenrath, J. Different approaches for interpretation and reporting of immunohistochemistry analysis results in the bone tissue—A review. Diagn. Pathol. 9, 221–221 (2014).
Zlobec, I. et al. Next-generation tissue microarray (ngTMA) increases the quality of biomarker studies: an example using CD3, CD8, and CD45RO in the tumor microenvironment of six different solid tumor types. J. Transl. Med. 11(1), 104 (2013).
Zlobec, A. U. et al. A next-generation tissue microarray (ngTMA) protocol for biomarker studies. JoVE https://doi.org/10.3791/51893 (2014).
Frances, A. & Cordelier, P. The emerging role of cytidine deaminase in human diseases: A new opportunity for therapy?. Mol. Ther. 28(2), 357–366 (2020).
Deshmukh, A. P. et al. Identification of EMT signaling cross-talk and gene regulatory networks by single-cell RNA sequencing. Proc. Natl. Acad. Sci. 118(19), e2102050118 (2021).
Kim, M. C. et al. Hypoxia promotes acquisition of aggressive phenotypes in human malignant mesothelioma. BMC Cancer 18(1), 819 (2018).
Francis, R. J. et al. Characterization of hypoxia in malignant pleural mesothelioma with FMISO PET-CT. Lung Cancer 90(1), 55–60 (2015).
Zhang, M. et al. Induction of peroxiredoxin 1 by hypoxia regulates heme oxygenase-1 via NF-κB in oral cancer. PLoS ONE 9(8), e105994 (2014).
Mameri, H. et al. Cytidine deaminase deficiency reveals new therapeutic opportunities against cancer. Clin. Cancer Res. 23(8), 2116–2126 (2017).
Easwaran, H., Tsai, H. C. & Baylin, S. B. Cancer epigenetics: Tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell 54(5), 716–727 (2014).
Karacosta, L. G. et al. Mapping lung cancer epithelial-mesenchymal transition states and trajectories with single-cell resolution. Nat. Commun. 10(1), 5587 (2019).
Saxena, K., Jolly, M. K. & Balamurugan, K. Hypoxia, partial EMT and collective migration: Emerging culprits in metastasis. Transl. Oncol. 13(11), 100845 (2020).
Tirpe, A. A. et al. Hypoxia: Overview on hypoxia-mediated mechanisms with a focus on the role of HIF genes. Int. J. Mol. Sci. 20(24), 6140 (2019).
McKeown, S. R. Defining normoxia, physoxia and hypoxia in tumours—Implications for treatment response. Br. J. Radiol. 87(1035), 20130676 (2014).
Cantini, L. et al. Emerging treatments for malignant pleural mesothelioma: Where are we heading?. Front. Oncol. https://doi.org/10.3389/fonc.2020.00343 (2020).
Otterson, G. A. et al. Capecitabine in malignant mesothelioma: A phase II trial by the cancer and leukemia group B (39807). Lung Cancer 44(2), 251–259 (2004).
Borgeaud, M. et al. The evolving role of immune-checkpoint inhibitors in malignant pleural mesothelioma. J. Clin. Med. 12(5), 1757 (2023).
Heo, H. et al. DNA methylome and single-cell transcriptome analyses reveal CDA as a potential druggable target for ALK inhibitor–resistant lung cancer therapy. Exp. Mol. Med. 54(8), 1236–1249 (2022).
Mendiola, C. & Vaz, M. A. Is capecitabine a new choice of treatment for lung adenocarcinoma? A case report involving partial response in second line of treatment and hypothesis of the biological basis. Clin. Transl. Oncol. 11(8), 554–557 (2009).
Cura, Y. et al. Influence of single-nucleotide polymorphisms on clinical outcomes of capecitabine-based chemotherapy in colorectal cancer patients: A systematic review. Cancers (Basel) 15(6), 1821 (2023).
Zhang, Y. et al. Maintenance of antiangiogenic and antitumor effects by orally active low-dose capecitabine for long-term cancer therapy. Proc. Natl. Acad. Sci. 114(26), E5226–E5235 (2017).
Acknowledgements
We would like to express our gratitude to the Graduate School for Cellular and Biomedical Sciences for organizing the study process and providing constant support for the students. We thank the Lung Cancer Center, Bern University Hospital, the Tissue Bank Bern, Institute of Pathology, University of Bern, the Translational Research Unit (TRU) platform, University of Bern, and the ngTMA platform, Institute of Tissue Medicine and Pathology, University of Bern, for acquiring patient tissues, constructing the TMA and performing the IHC staining.
Funding
This work was supported by the Swiss Cancer Research (KFS-5405-08-2021) and the Swiss National Science Foundation (310030_2127661/1) to TMM. The research contribution of RWP was funded by the Swiss Cancer Research Foundation (KFS-4851-08-2019) and the Swiss National Science Foundation (310030_192648). This work was supported by the Stiftung zur Krebsbekämpfung (SKB503/AHW/de) to PD. The funding bodies were not involved in the study design or collection or analysis; data interpretation; or manuscript writing.
Author information
Authors and Affiliations
Contributions
DK, HD, YG, WW, RP, RAS, PD, and TMM substantially contributed to the conception and design of this study. DK, YG, CC, PD, and TMM conducted the molecular biology studies and acquired the data. DK, HD, WW, RP, RAS, PD, and TMM participated substantially in interpreting the data and critically drafted/revised the manuscript for important intellectual content. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Karatkevich, D., Losmanova, T., Zens, P. et al. Chemotherapy increases CDA expression and sensitizes malignant pleural mesothelioma cells to capecitabine treatment. Sci Rep 14, 18206 (2024). https://doi.org/10.1038/s41598-024-69347-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-69347-x
