
Abstract
D-allulose/D-psicose is a significant rare sugar with broad applications in the pharmaceutical, food, and other industries. In this study, we cloned the D-allulose 3-epimerase (DPEase) gene from Arthrobacter globiformis M30, using pET22b as the vector. The recombinant E. coli strain pET22b(+) was successfully constructed and expressed, providing an efficient whole-cell catalyst for converting inexpensive D-fructose into D-allulose. Subsequently, we optimized the induction and incubation conditions step by step using the single-factor method and used Lactobacillus plantarum(LAB) 217-8 to enhance the purity of D-allulose in the system. Ultimately, the BL21/pET22b(+)-E. coli strain achieved a conversion rate of up to 33.91% under optimal conditions, converting D-fructose to D-allulose. After purification, the purity of D-allulose reached 64.73%. Efficient production of D-allulose is a significant achievement, paving the way for future probiotic applications in its conversion.
Similar content being viewed by others
Introduction
D-allulose, a C-3 epimer of D-fructose, is a notable rare sugar and a novel low-calorie sweetener with the molecular formula C6H12O61. It occurs naturally in trace amounts, mainly in wheat, sugar cane, beet molasses, and some bacterial strains2. Studies characterize D-allulose as a white, odorless, crystalline powder, with only a tiny caloric content and 70% the sweetness of sucrose, positioning it as a promising alternative in the sweetener market3. Beyond its sweetness, D-allulose has physiological functions, with potential for preventing and treating hyperlipidemia, obesity, and diabetes4. It has also demonstrated anti-inflammatory and antioxidant properties5 and other beneficial effects6. In 2011, the U.S. Food and Drug Administration (FDA) recognized D-allulose as Generally Recognized As Safe (GRAS), paving the way for its inclusion in a variety of foods and dietary supplements7. Long-term consumption of D-allulose shows no adverse effects in humans, affirming its favorable safety profile8,9. This body of evidence underscores the potential of D-allulose as a safe and effective ingredient in the pharmaceutical, food, and dietary supplement industries.
At present, the main methods for synthesizing D-allulose are chemical synthesis and biological enzyme-catalyzed processes10,11. The chemical synthesis route for industrial D-allulose production is limited by its complex process, low yields, and substantial environmental impact. In contrast, the bio-enzymatic approach offers advantages such as economic viability, environmental sustainability, and straightforward product purification, making it a compelling choice.
The enzymatic synthesis of D-allulose mainly depends on D-allulose 3-epimerases (DPEase). Literature indicates that the equilibrium conversion rate for this process is typically between 24% and 33%12. Optimizing reaction conditions and selecting suitable host organisms for enzyme expression can increase conversion rates, thus enhancing the enzymatic production efficiency of D-allulose.
However, traditional enzymatic methods face challenges such as difficult enzyme purification and complex product separation, restricting their use in large-scale industrial production. Recently, whole-cell biocatalysts have garnered interest in producing target compounds, offering benefits such as cost-effectiveness, ease of use, straightforward product recovery, and consistent performance13.
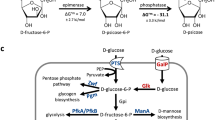
Researchers currently utilize a biological enzyme synthesis system that relies on phosphorylation-dephosphorylation to convert D-fructose to D-allulose. This process involves additional reactions, blocks the Embden-Meyerhof-Parnas (EMP) pathway, and incorporates an ATP regeneration system, enhancing D-allulose synthesis efficiency. However, despite improved synthesis efficiency, the process remains more complex and the production cycle is prolonged (as shown in Fig. 1, Pathways I and II)14.
This study primarily employs D-allulose 3-epimerase (DPEase) from microorganisms for in vitro D-allulose synthesis. DPEase is active in various microorganisms, including Agrobacterium tumefaciens15, Treponema primitia16, Ruminococcus sp17, etc. Arthrobacter globiformis M30, like D-allulose, is also classified as “GRAS”. Furthermore, the D-psicose 3-epimerase (DPEase) enzyme, isolated from the M30 strain, exhibits marked substrate specificity and robust catalytic efficiency, and these characteristics have informed our decision to source exogenous genes from the M30 strain18. Reports indicate that DPEase genes from diverse sources have been cloned and expressed in systems like Engineered yeast19 and Bacillus subtilis20. Escherichia coli(E. coli) is a commonly used host in cell factory development, known for its well-defined genetic background and short life cycle. It utilizes D-fructose exclusively as a carbon source and can convert it into D-allulose using methods like phosphorylation21. Studies have confirmed the successful introduction and expression of various foreign genes22. Ultimately, we heterologously expressed DPEase in E. coli(Fig. 1, Pathway III) and conducted single-factor optimizations of induction, incubation, and product purification processes. The goal is to develop a microbial fermentation process as an alternative to the traditional D-allulose synthesis method.
The study highlights the application of whole-cell biocatalysis in E. coli, capitalizing on its economic and operational benefits and efficient D-fructose utilization. The research aims to develop a sustainable, scalable method for D-allulose production, enhancing synthesis efficiency and streamlining the process. This approach could surpass the constraints of traditional chemical synthesis and conventional enzymatic methods, providing a viable solution for large-scale industrial manufacturing that ensures product quality and safety. Successful implementation of this method could transform D-allulose production, increasing accessibility and broadening its applications from sweeteners to therapeutics.

Metabolic pathway in E. coli cell factory for the production of D-allulose from D-fructose via phosphorylation-dephosphorylation and DPEase.
Materials and methods
Strains and plasmids
In this study, we used Arthrobacter globiformis M30 as the source of DPEase to construct the heterologous expression strain BL21/pET22b(+)-psi. The gene vector pET22b and the expression host E. coli BL21 (DE3) were utilized. Details of the strains and plasmids used are presented in Table 1.
Culture media and primary reagents
The media23,24 used to culture E. coli were MRS, LB, TB, SB, 2xYT, SOB, and the buffers required for the HPLC and protein purification stages were binding buffer, 10-fold volume Elution Washing Buffer, 5-fold volume Elution Buffer, and SDS-PAGE sample buffer25.
The reagents used in the experiment were analytically pure and purchased from China National Pharmaceutical Group Chemical Reagents Co., Ltd. (Shanghai, China). D-fructose and D-allulose were chromatographically pure. The genome extraction kit, product purification kit, and homologous recombinase were purchased from Vazyme Company (Nanjing, China), and the plasmid extraction kit was purchased from Tiangen Biotechnology Co., Ltd. (Beijing, China).
Vector construction
The DPEase gene, derived from Arthrobacter globiformis M30 (GI: 1111512028), was optimized and synthesized by Nanjing Paisennuo Company. Primers psi-F (5’-GTAACTTTGAAAGGAGCTTCCTCATGAAGATCGGTTGTCACGGTTT-3’) and psi-R (5’-GAACTAGTGGTACCGCATGCCTGCAGTTAGTGTAATTCGATAGTCT-3’) were designed to amplify the exogenous DPEase gene. The DPEase gene and the digested pET-22b plasmid with restriction endonuclease Xho I were purified using a kit from Vazyme (Nanjing, China), and the fragments were ligated with T4 DNA ligase. The ligated product was transformed into E. coli DE3, and recombinant strains were selected on LB agar with ampicillin. The recombinant plasmids were then extracted and sequenced by Sangon Bioengineering (Shanghai, China).
Target protein expression, production and purification
The successfully constructed recombinant plasmid pET22b(+)-psi was introduced into E. coli DE3 for protein expression, spread onto an LB agar plate containing ampicillin (50 µg/mL), and incubated overnight at 37 °C. Selected transformants were then inoculated into LB medium with ampicillin (50 µg/mL) and incubated at 37 °C at 220 rpm for 12 h. The culture was then transferred to LB liquid medium containing 2% (v/v) ampicillin. Once the OD600 reached 0.5 to 0.6, Isopropyl-β-D-thiogalactoside (IPTG) was added to a final concentration of 0.4 mM, and the culture continued to incubate at 37 °C. The induction was performed for 24 h. The experiments and the subsequent optimization of induction conditions were conducted in 500 mL conical flasks10,25.
E. coli cells were harvested by centrifuging for 5 min, washed twice with PBS, and resuspended in a 20 mM imidazole solution. Ultrasonication at 400 W with a 5-second on/off cycle for 99 pulses was used to lyse E. coli cells. The whole-cell lysate was centrifuged at 4 °C and 13, 400 ×g for 15 min, and the supernatant was filtered through a 0.22-µm membrane before Ni column purification. Purification involved sequentially adding solutions: binding buffer, filtered supernatant, washing buffer, and finally elution buffer. The eluted solution was collected, dialyzed against a pH 7.4 phosphate buffer overnight to remove imidazole, and then used to purify L-arabinose isomerase. SDS-PAGE was used to analyze whole-cell, intracellular, Ni-column flow-through, and protein samples. Samples were mixed with SDS-PAGE loading buffer and denatured at 5 min. SDS-PAGE, using a 50 g/L stacking gel and a 120 g/L separating gel, was performed to resolve protein bands, which were stained with Coomassie brilliant blue R-25015,25.
DEPease activity assay
To verify if the purified target protein from the engineered strain can catalyze D-allulose synthesis from D-fructose, we incubated a precise amount of D-allulose 3-epimerase with a D-fructose solution. After a 3-hour reaction, it was terminated using a boiling water bath. The mixture was then centrifuged at 11, 500 ×g for 20 min, and the supernatant was filtered through a 0.22-µm membrane to yield a liquid-phase sample.
The D-fructose substrate and D-allulose product concentrations were determined using an HPLC system equipped with a RID-10 A refractive index detector, LC-20AT pump, and SIL-20 A sample injector. The sample was chromatographed on a Sugar-Pak™ column (6.5 × 300 mm) at 83 °C, with a mobile phase of 50 mg/L calcium EDTA at a flow rate of 0.3 mL/min over 30 min26.
Optimization of fermentation conditions for recombinant E. Coli
Initially, cultivation is conducted using standard induction and incubation conditions. Activate the BL21/pET22b(+)-psi strain and gradually optimize fermentation conditions throughout its growth, focusing on single-factor optimization for induction conditions including: medium types (LB, TB, SB, 2×YT, SOB), induction temperatures (20, 25, 30, 37, 42 °C), inducers (IPTG and lactose at varying concentrations), metal ions (Zn2+, Fe3+, Mg2+, Mn2+, Ca2+), induction times (12, 18, 24, 30, 36 h); Incubation conditions, including temperatures (55, 60, 65, 70, 75 °C), substrate pH levels (5.0, 6.0, 7.0, 8.0, 9.0), substrate concentrations (50, 100, 150, 200, 250 g/L), bacterial contents (1, 3, 5, 7, 9 v/v), and incubation times (6, 12, 18, 24, 30 h), were optimized one variable at a time, and high-performance liquid chromatography (HPLC) was used to analyze the D-allulose produced by the system. Quantify and calculate the relative conversion rate to ultimately determine the optimal reaction conditions with the highest conversion rate.
Preliminary purification of D-allulose
Fructophilic Lactic Acid Bacteria(FLAB) can use D-fructose as a carbon source for growth27. In this study, the substrate D-fructose was not completely consumed. When D-fructose and D-allulose levels in the reaction system stabilized, Lactobacillus plantarum 217-8 was added at 2% (v/v) and incubated at 37 °C for 30 h. Samples of the bacterial solution were collected every 8 h to prepare liquid-phase samples for detecting D-fructose content. The detection method is detailed in “DEPease activity assay” section. The definition of purity for allulose is as follows:
A: Content of D-allulose; B: Content of D-fructose.
Data analysis
This experiment characterizes results using relative activity. Definition of Relative Activity: The maximum enzyme activity, recorded under optimal single-factor conditions for whole-cell catalysis, is set at 100%. The relative activity is calculated as the ratio of activities under various conditions to this maximum. Each experiment was conducted three times, and the results are presented as mean ± standard deviation after analysis with Prism 9. Error bars in tables and figures indicate the standard deviation from three independent experiments.
Results
Construction and PCR identification of plasmid containing DPEase
In this study, we constructed the inducible plasmid pET22b(+)-psi(6283 bp, Fig. 2), which contains the DPEase gene, and obtained the recombinant strain BL21/pET22b(+)-psi, screened with 100 µg/mL ampicillin. The plasmid was extracted using a commercial kit, and amplified by PCR with primers psi-F and psi-R. The PCR product’s length was 870 bp, confirming the expected size (Fig. 3).

Schematic representation of pET22b(+)-E. coli.

Plasmin PCR identification; Note: M: Marker; 1, 2, 3: Target electrophoretic bands.
Expression, isolation, and purification of D-allulose 3-epimerase in E. Coli
The recombinant strain BL21/pET22b(+)-psi was induced for protein expression, as shown in the electropherogram in Fig. 4 post-cell lysis. As depicted in Fig. 4, E. coli expressed a protein of approximately 32 kDa, matching the expected size of DPEase, suggesting successful expression; Furthermore, the purified protein’s single band on the gel confirms a molecular weight of approximately 32 kDa, demonstrating effective separation from other proteins and impurities during purification.

Expression of D-allulose-epimerase Note: M: Marker, 1: Unpurified protein, 2: Purified target protein.
D-allulose 3-epimerase activity detection
The recombinant strain was incubated in a medium containing D-fructose as the substrate, and D-allulose production was detected using HPLC. Figure 5a is a liquid-phase chromatogram showing the separate detection of substrates. As shown in Fig. 5b, the presence of D-allulose at 25 min indicates that DPEase is functional in E. coli. It expresses active DPEase, which catalyzes the synthesis of D-allulose.

HPLC detection of recombinant DPEase. (a) The peak of D-allulose; (b) peak diagram of D-fructose catalyzed by whole cell.
Optimization of induction conditions
E. coli is one of the most used expression hosts for producing and expressing recombinant proteins. However, for various reasons, improved efficiency and yield are often required. Therefore, optimizing the E. coli expression system conditions is crucial for efficiently expressing target proteins28.
The effect of culture medium on enzyme activity
Ineffective media use can be prevented by assessing the effectiveness of various media throughout the production process29. As illustrated in Fig. 6a, the evaluation of the relative enzyme activity of DPEase in E. coli revealed that the different compositions of the medium exerted a considerable influence on the enzyme activity. The highest relative enzyme activity of DPEase was observed in TB medium, at 100% composition, followed by SOB medium (59.30%) and LB medium (37.21%). Conversely, the relative enzyme activities in 2×YT and SB media were lower, at 34.40% and 31.01%, respectively.
The effect of induction temperature on enzyme activity
The induction temperature is crucial for enzyme secretion; lower temperatures facilitate proper protein folding into soluble forms but can marginally decrease overall expression levels30. In exploring the effect of induction temperature on the relative enzyme activity of DPEase, it was observed that the enzyme activity showed a specific pattern of change with increasing induction temperature (Fig. 6b). Upon reaching the temperature threshold of 30 °C, the enzymatic activity of DPEase within E. coli cultures achieved a zenith, attaining a relative activity of 100%. In contrast, at the reduced temperatures of 20 °C and 25 °C, the relative enzymatic activities were markedly diminished, recording values of 25.63% and 31.85%, respectively. Upon further elevation of the thermal gradient to 37 °C and 42 °C, a decrement in enzymatic activity was observed, with relative activities plummeting to 54.30% and 39.35%, respectively.
The effect of induction metal ion on enzyme activity
Metal ions are crucial for the conversion of D-fructose to D-allulose, as they stabilize the D-fructose molecule. The DPEase enzymes exhibit significant variation in their dependence on metal ions like Mn2+, Co2+, and Mg2+31,32. In this study, Fig. 6c illustrates that both Fe3+ and Zn2+ significantly enhance DPEase activity, suggesting that this action may play an essential role in regulating DPEase conformation; in contrast, the activities of Ca2+ and Mg2+ are just 15% of the Fe3+ activity (p < 0.0001).

Optimization of plasmid PET22b-E. coli induced conditions (Calculated with a conversion rate of 100% under optimal single factor conditions. (a) The effect of culture medium on enzyme activity. (b) The effect of induction temperature on enzyme activity. (c) The effect of induction Metal ion on enzyme activity. (d) The effect of induction OD600 enzyme activity. (e) The effect of inducer concentration on enzyme activity. (f) The effect of induction time on enzyme activity. ns, no significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Data are expressed as the mean ± standard deviation (SD) of 3 independent experiments.
The effect of induction OD600 on enzyme activity
The amount of inoculum significantly affects the enzyme activity: too little leads to insufficient activity, too much to metabolic competition17. Figure 6d shows the relative enzyme activities of E. coli when induced by the addition of inducers at different cell densities. The activity was 47.75% at OD600 of 0.1, which increased to 61.55% at 0.3 and peaked at 100% at 0.5. Subsequently, the activity decreased to 75.23% at 0.7 and 0.9 and further decreased to 67.31% at 0.9. This trend indicates that the enzyme activity is not proportional to the cell density, with a maximum observed at an OD600 of 0.5, followed by a slight decrease at higher densities.
The effect of inducer concentration on enzyme activity
This study utilized pET22b as an inducible promoter, requiring the addition of an inducer to the recombinant strain for protein expression. IPTG, a commonly used inducer, effectively induces protein expression but is relatively expensive and has specific toxicity to humans33,34. Therefore, we selected lactose as an alternative to IPTG. Figure 6e demonstrates that lactose achieves optimal induction effects at a concentration of 2.0 g/L. However, subsequent experimental results showed that higher concentrations of lactose did not improve the enzyme activity yield when the lactose concentration exceeded 2.0 g/L. The relative enzyme activity was 78.20% at a lactose concentration of 2.5 g/L, and plummeted to 51.70% when the concentration increased to 3.0 g/L.
The effect of induction time on enzyme activity
Induction duration is directly proportional to protein yield, up to an optimal point. Prolonged induction enhances DPEase expression but may elicit negative feedback, reducing secretion once protein levels reach their peak. This study investigates the impact of induction time on conversion rates35. As shown in Fig. 6f, at an induction temperature of 30 °C, the relative conversion rate increased with the increase of induction time within 24 h. The optimal equilibrium of protein yield and activity was reached at the 24th hour, and its enzyme activity also reached its peak. Subsequently, the relative enzyme activity gradually decreased, and the relative conversion rate dropped to 65.23% at 30 h of induction time.
Optimization of incubation conditions
After inducing the expression of the recombinant strain with the inducible plasmid pET22b(+)-psi under the previously mentioned optimal conditions, we added a substrate to provide a suitable environment for DPEase activity. The DPEase family is characterized by highly conserved sequences, consistent structural homology, and substrate specificity. These characteristics include the optimal reaction pH range of 7.0–9.0 and temperature range of 40–70 °C, critical for enzyme activity and stability36. A thorough understanding of DPEase’s intrinsic characteristics is essential for strategically designing reaction conditions to fully harness the enzyme’s potential. Therefore, we have designed the fermentation conditions for DPEase, focusing on reaction pH, temperature, and substrate concentration. This targeted approach maximizes the conversion rate while ensuring the quality and consistency of the final product, crucial for subsequent large-scale industrial applications.
Effect of permeating agent on enzyme activity
The E. coli system, a leading expression vector, encounters issues with rare codons and complex proteins even under optimal expression conditions. Permeating agent enhances protein solubility, facilitates bacterial material exchange, and promotes proper folding, thus enabling the extraction of heterologous proteins from inclusion bodies37. Figure 7a shows that the relative enzyme activities of E. coli in the presence of osmotic agents were 83.7% for N, N-Dimethylformamide (DMF), 100% for Tween-80, 76.50% for glycine (Gly), and 65.06% for dimethyl sulfoxide (DMSO), with the most pronounced promotion by Tween-80, compared to no osmotic agent, Tween 80 and Gly increased the relative conversion rate compared to no addition, while DMSO inhibited the enzyme activity.
Effect of incubation temperature on enzyme activity
The thermostability of recombinant enzymes is attributed to numerous hydrophobic interactions and a rigid amino acid sequence configuration12. The reported optimal temperature range for DPEase in catalysis is 40 °C to 70 °C. The optimal temperature for the enzymatic reaction was found to be 65 °C. The catalytic efficiency declines rapidly if temperatures deviate from 65 °C. At 60 °C, the conversion efficiency drops to 55.4% of the optimum, and at 70 °C, it is 66.92%, showing that this DPEase is highly temperature-sensitive. Strictly control the temperature during the reaction (as shown in Fig. 7b).
Effect of incubation pH on enzyme activity
In the isomerization reaction of D-fructose to D-allulose, the optimal pH range is generally 7.0 to 9.0. A limited number of DPEase sources, such as Dorea sp, are adapted to weakly acidic conditions38. Extremes in pH can lead to the denaturation and inactivation of the enzyme. Consequently, this study discusses the impact of substrate pH on enzyme activity. Figure 7c shows that the relative enzyme activity of DPEase showed fluctuations in the range of pH 5 to 9. The highest enzyme activity of 100% was reached at neutral pH 7. As the pH deviated from neutral, the enzyme activity decreased, especially at pH 5 and 9 to 33.64% and 52.23%, respectively.
Effect of substrate concentration on enzyme activity
Based on substrate differences, isomerases for synthesizing D-allulose are categorized into distinct families, including D-labeled enzyme-3-epimerases (DTEase) and D-allulose-3-epimerases (DPEase). Members of the DPEase family exhibit strong substrate specificity39. We used a gradient concentration of D-fructose as the substrate. Figure 7d illustrates a negative correlation between substrate concentration and enzyme activity found in this concentration range. The enzyme activity peaked at the lowest concentration of 50 g/L, recorded as 100%. When the substrate concentration was increased to 100 g/L, the relative activity decreased to 48.39%. Further increase in concentration to 150 g/L and 200 g/L resulted in 36.68% and 28.05% enzyme activity, respectively. At the peak concentration of 250 g/L of substrate, the enzyme activity decreased abruptly to 26.06%.
Effect of cell concentration on enzyme activity
The essence of whole-cell catalysis lies in intracellular enzyme action, with varying bacterial concentrations indicating different levels of enzyme addition40. The relative enzyme activity of the reaction system determined at different initial inoculum ratios showed a significant difference, as depicted in Fig. 7e. The relative enzyme activity of E. coli cultures showed a significant increase with the increase of the initial inoculum ratio. At the lowest inoculum ratio of 1%, the enzyme activity was 58.47%, reflecting a weak catalytic ability. When the inoculation ratio was increased to 3%, a significant increase in enzyme activity was observed to 94.51%. When the inoculum ratio was further increased to 5% and 7%, the activity reached 95.43% and 100%, respectively, indicating that the enzyme expression reached the optimal range. Interestingly, at the highest inoculum ratio of 9%, a slight decrease in activity was observed at 98.03%.
Effect of incubation time on relative enzyme activity
Incubation time refers to the duration of substrate-enzyme contact, during which substrate conversion rates are expected to increase theoretically. However, for reversible reactions such as DPEase-catalyzed conversion of D-fructose to D-allulose, equilibrium restricts additional product formation and may lead to a decrease41. As depicted in Fig. 7f, the relative enzyme activity of E. coli showed some differences at different incubation times. At 6 h incubation time, the enzyme activity was 58.47%, and as the incubation time was extended to 12 h, a significant increase in the enzyme activity was observed up to 94.51%. After extending the incubation time to 18 h, the activity increased slightly to 95.43%. The peak activity of 100% was reached at 24 h incubation time, which indicates that the enzyme expression was at its optimum. At the most extended incubation of 30 h, the activity slightly decreased to 98.03%.

Optimization of plasmid PET22b-E. coli in conditions (Calculated with a conversion rate of 100% under optimal single-factor conditions). (a) Effect of penetrant on enzyme activity. (b) Effect of Incubation temperature on enzyme activity. (c) Effect of Incubation pH on enzyme activity. (d) Effect of substrate concentration on enzyme activity. (e) Effect of bacterial concentration on enzyme activity. (f) Effect of incubation time on enzyme activity. ns, no significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Data are expressed as the mean ± standard deviation (SD) of 3 independent experiments.
D-allulose preliminary purification
After gradually optimizing the fermentation conditions, we achieved a conversion rate of 33.91%, with an expected D-allulose concentration of 16.95 g/L. However, due to LAB’s consumption of D-allulose as a carbon source, the final concentration is 15.53 g/L after accounting for losses. After a 16-hour reaction (Fig. 8), the purity reached 64.73%, with a D-allulose content of 13.28 g/L. By the 24th hour, the purity had increased to 75.60%, yet the D-allulose content was down to 11.34 g/L, This indicates that LAB, similar to E. coli, consumes a portion of D-allulose, hindering the accumulation of the final product. Consequently, the purification was halted at 16 h.

D-fructose content in the system and purity of D-allulose.
Discussion
Currently, D-allulose 3-epimerase (DPEase) activity has been found in various microorganisms, with enzymes from different sources often showing similar characteristics. Despite these similarities, the natural yield of DPEase from microorganisms is too low for industrial applications due to limited secretion. Consequently, developing engineered strains is essential for advancing research and industrial utilization of DPEase.
This study uses a whole-cell biocatalytic approach for D-allulose synthesis, providing several advantages over traditional enzymatic methods. The whole-cell method simplifies the synthesis process by eliminating the need for enzyme purification, reducing costs, and streamlining the procedure.
The experimental results demonstrate that optimizing incubation and induction conditions significantly affects DPEase activity. For instance, optimizing induction conditions revealed that the LB medium’s enzyme activity was just 37.20% of the TB medium’s, a phosphate-rich medium containing 20% more tryptophan and 380% more yeast extract (p < 0.0001). The higher phosphate in the TB medium likely contributes to the stability and enzyme protection; Conversely, at an induction temperature of 25 °C, enzyme activity dropped to 31.85% of the level at 30 °C (p < 0.0001).
Furthermore, the reversibility of DPEase, which converts D-fructose to D-allulose, can limit process efficiency. Considering the high cost of D-fructose, achieving higher D-allulose yields economically is a significant industrial challenge. Our exploration of substrate concentrations revealed that 50 g/L is optimal, yielding twice the conversion of 100 g/L and significantly reducing costs. Additionally, incubation temperature and duration significantly affect enzyme activity. By gradually optimizing incubation and induction conditions, we established optimal fermentation parameters that greatly enhanced D-allulose production efficiency. However, using E. coli as a host organism presents challenges, as its multiple metabolic pathways for fructose can consume the substrate, competing with the D-allulose synthesis pathway42,43.
Scientists have now improved the catalytic activity and thermal stability of the DPEase family of enzymes through molecular modification44. The researchers have successfully developed an ATP regeneration system coupled to a polyphosphate kinase to improve the efficiency of the enzymatic process. After optimization, 99% conversion was achieved in a reaction system with 2 mM ATP, 5 mM polyphosphate, 20 mM Mg2+, 50 °C, and pH 7.5. Compared with the system without ATP regeneration, the use of ATP can be reduced to 10% of the theoretical amount. However, its substrate concentration is only 20 mM, much lower than our substrate concentration (278 mM, 50 g/L). Although the concentration of the reaction substrate is low, the biosynthesis system provides a promising platform for converting D-fructose into D-allulose at a low cost of ATP. In additional research, wild-type DPEase from Arthrobacter globiformis M30, once purified, showed maximal activity and thermostability in the presence of Mg2+. The enzyme showed optimal activity between pH 7.0 and 8.0 and at 70 °C, surpassing the 65 °C optimal temperature for the engineered strain in our study, indicating superior high-temperature tolerance45. The high-temperature resistance property enhances the activity and stability of the enzyme under high-temperature environment, which not only speeds up the reaction rate and reduces microbial contamination but also improves its resistance to unfavorable environments and facilitates daily storage. In our experiments, we found that Fe3+ is critical to the catalytic efficiency of enzymes. In an experiment46 using various metal ions such as Li+, Na+, K+, Co2+, Mn2+, Ni2+, Fe3+. And Mg2+ as cofactors, the researchers found that the involvement of Mn2+ significantly increased the activity of the DPEase, with a conversion rate of 22%, and Mg2+ as a cofactor, and found that the involvement of Mn2+ significantly increased the activity of the DPEase, with a conversion rate of 22% at a substrate concentration of 5%. and Mg2+ as cofactors, it was found that the involvement of Mn2+ could significantly increase the activity of DPEase with a conversion rate of 22% at a substrate concentration of 5%. At a substrate concentration of 25% (w/v), the conversion rate was 22.42%, which was lower than ours (33.91%), and this is also in agreement with the results of other studies that emphasize the importance of metal ions in the catalysis of DPEase. This study is in agreement with other studies emphasizing the importance of metal ions in the catalysis of DPEase. However, there are some differences in the enhancement of enzyme activity by metal ions from different sources, which need to be demonstrated by more studies47.
This preference discrepancy for metal cofactors may result from genetic engineering modifications, suggesting potential alternative pathways or mechanisms for improving DPEase enzyme performance. In summary, although progress has been made in enhancing DPEase enzyme performance, improvements in scalability and temperature tolerance are still needed. Developing an ATP regeneration system, optimizing reaction conditions, and exploring alternative metal cofactors are promising directions for future research.
To address the aforementioned challenges, future research on D-allulose should explore various strategies, including the dual enzyme strategy44,45, multi-product strategy48,49, coupled ATP regeneration system50, Site-specific mutagenesis51, to enhance product yields.
Conclusion
This article focuses on optimizing fermentation conditions for E. coli-mediated D-allulose production. An inducible plasmid pET22b-psi, containing the DPEase gene sequence, was constructed and introduced into E. coli DE3 using the heat shock method for induced expression. The induction and incubation conditions for E. coli were optimized. Finally, HPLC was used to detect D-allulose content, determining the optimal fermentation conditions:
Under these conditions, the E. coli conversion yield of D-fructose to D-allulose was 33.91%. Once the reaction reaches a stable stage, the D-allulose purity in the system is 64.73%, with a content of 13.28 g/L.
This study enhanced D-allulose production, offering valuable insights for future research on the biosynthesis of rare sugars.
Data availability
Data Availability Statemet: The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Baek, J. & Lee, M. G. Oxidative stress and antioxidant strategies in dermatology. J. Redox Rep. 21, 164–169 (2016).
Zhang, L. et al. Characterization of D-tagatose-3-epimerase from Rhodobacter sphaeroides that converts D-fructose into D-psicose. J. Biotechnol. Lett. 31, 857–862 (2009).
Chen, Z., Gao, X. D. & Li, Z. Recent advances regarding the physiological functions and biosynthesis of D-allulose. J. Front. Microbiol. 13, 881037 (2022).
Han, Y. et al. Alteration of microbiome profile by D-allulose in amelioration of high-fat-diet-induced obesity in mice. J. Nutrients 12, 1 (2020).
Japar., S. et al. A pilot study on the effect of D-allulose on postprandial glucose levels in patients with type 2 diabetes mellitus during Ramadan fasting. J. Diabetol. Metab. Syndr. 14, 86 (2022).
Gou, Y. et al. d-Allulose ameliorates skeletal muscle insulin resistance in high-fat diet-fed rats. J. Molecules 26(20), 6310 (2021).
Zhang, W., Zhang, T. & Jiang, B. Mu. Enzymatic approaches to rare sugar production. J. Biotechnol. Adv. 35, 267–274 (2017).
Iida, T. et al. Acute D-psicose administration decreases the glycemic responses to an oral maltodextrin tolerance test in normal adults. J. Nutr. Sci. Vitaminol. 54, 511–514 (2008).
Hayashi, N. et al. Study on the postprandial blood glucose suppression effect of D-psicose in borderline diabetes and the safety of long-term ingestion by normal human subjects. J. Biosci. Biotechnol. Biochem. 74, 510–519 (2010).
Tang, X. et al. The characterization of a novel D-allulose 3-epimerase from Blautia produca and its application in D-allulose production. J. Foods 11, 1 (2022).
Laksmi, F. A., Nirwantono, R., Nuryana, I. & Agustriana, E. Expression and characterization of thermostable D-allulose 3-epimerase from Arthrobacter psychrolactophilus(ap DAEase) with potential catalytic activity for bioconversion of D-allulose from d-fructose. J. Int. J. Biol. Macromol. 214, 426–438 (2022).
Jiang, S. et al. Review on D-allulose: in vivo metabolism, catalytic mechanism, engineering strain construction, bio-production technology. J. Front. Bioeng. Biotechnol. 8, 26 (2020).
Wachtmeister, J. & Rother, D. Recent advances in whole cell biocatalysis techniques bridging from investigative to industrial scale. J. Curr. Opin. Biotechnol. 42, 169–177 (2016).
Guo, Q. et al. Metabolically engineered Escherichia coli for conversion of D-fructose to D-allulose via phosphorylation-dephosphorylation. J. Front. Bioeng. Biotechnol. 10, 947469 (2022).
Kim, H. J., Hyun, E. K., Kim, Y. S. & Lee, Y. J. Oh. Characterization of an Agrobacterium tumefaciens D-psicose 3-epimerase that converts D-fructose to D-psicose. J. Appl. Environ. Microbiol. 72, 981–985 (2006).
Zhang, W., Zhang, T., Jiang, B. & Mu, W. Biochemical characterization of a D-psicose 3-epimerase from Treponema primitia ZAS-1 and its application on enzymatic production of D-psicose. J. Sci. Food Agric. 96, 49–56 (2016).
Zhu, Y. et al. Overexpression of D-psicose 3-epimerase from Ruminococcus sp. in Escherichia coli and its potential application in D-psicose production. J. Biotechnol. Lett. 34, 1901–1906 (2012).
Juneja, A., Zhang, G., Jin, Y. S. & Singh, V. Bioprocessing and technoeconomic feasibility analysis of simultaneous production of d-psicose and ethanol using engineered yeast strain KAM-2GD. Bioresour. Technol. 275, 27–34 (2019).
Chen, J. et al. High-level intra- and extra-cellular production of D-psicose 3-epimerase via a modified xylose-inducible expression system in Bacillus subtilis. J. Ind. Microbiol. Biotechnol. 43, 1577–1591 (2016).
Aristidou, A. A., San, K. Y. & Bennett, G. N. Improvement of biomass yield and recombinant gene expression in Escherichia coli by using fructose as the primary carbon source. Biotechnol. Prog. 15, 140–145 (1999).
Mu, W. et al. Cloning, expression, and characterization of a D-psicose 3-epimerase from Clostridium cellulolyticum H10. J. Agric. Food Chem. 59, 7785–7792 (2011).
Feliciello, I. et al. Regulation of ssb gene expression in Escherichia coli. J. Int. J. Mol. Sci. 23, 1 (2022).
Gao, Y. et al. Response surface methodology-based optimization of Inonotus Hispidus’ liquid fermentation medium and evaluation of its exopolysaccharide activities. J. Front. Microbiol. 15, 1456461 (2024).
Khalilvand, A. B. et al. Media optimization for SHuffle T7 Escherichia coli expressing SUMO-Lispro proinsulin by response surface methodology. J. BMC Biotechnol. 22(1), 1 (2022).
Ruiting, Z. et al. Preparation of sweet milk and yogurt containing d-tagatose by the l-arabinose isomerase derived from Lactobacillus rhamnosus. J. LWT 187, 1 (2023).
Su, L., Sun, F., Liu, Z., Zhang, K. & Wu, J. Highly efficient production of Clostridium cellulolyticum H10 D-psicose 3-epimerase in Bacillus subtilis and use of these cells to produce D-psicose. J. Microb. Cell. Fact. 17, 188 (2018).
Endo, A., Futagawa-Endo, Y. & Dicks, L. M. Isolation and characterization of fructophilic lactic acid bacteria from fructose-rich niches. J. Syst. Appl. Microbiol. 32, 593–600 (2009).
Gileadi, O. Recombinant protein expression in E. Coli: a historical perspective. J. Methods Mol. Biol. 1586, 3–10 (2017).
Wu, J., Du, G., Zhou, J. & Chen, J. Metabolic engineering of Escherichia coli for (2S)-pinocembrin production from glucose by a modular metabolic strategy. J. Metab. Eng. 16, 48–55 (2013).
Porowińska, D., Wujak, M., Roszek, K. & Komoszyński, M. Prokaryotic expression systems. J. Postepy High. Med. Dosw 67, 119–129 (2013).
Kim, H. J. et al. Mutational analysis of the active site residues of a D: -psicose 3-epimerase from Agrobacterium tumefaciens. J. Biotechnol. Lett. 32, 261–268 (2010).
Yang, J. et al. Development of food-grade expression system for d-allulose 3-epimerase preparation with tandem isoenzyme genes in Corynebacterium glutamicum and its application in conversion of cane molasses to D-allulose. J. Biotechnol. Bioeng. 116, 745–756 (2019).
Carneiro, S., Ferreira, E. C. & Rocha, I. Metabolic responses to recombinant bioprocesses in Escherichia coli. J. Biotechnol. 164, 396–408 (2013).
Jia, B. & Jeon, C. O. High-throughput recombinant protein expression in Escherichia coli: current status and future perspectives. J. Open. Biol. 6, 1 (2016).
Wei, Y. et al. Construction and immobilization of recombinant Bacillus subtilis with D-allulose 3-epimerase. J. Sheng Wu Gong. Cheng Xue Bao 37, 4303–4313 (2021).
Mu, W. et al. Characterization of a D-psicose-producing enzyme, D-psicose 3-epimerase, from Clostridium sp. J. Biotechnol. Lett. 35, 1481–1486 (2013).
Kaur, J., Kumar, A. & Kaur, J. Strategies for optimization of heterologous protein expression in E. coli: roadblocks and reinforcements. J. Int. J. Biol. Macromol. 106, 803–822 (2018).
Wang, Y. et al. Biocatalytic synthesis of D-allulose using novel D-tagatose 3-epimerase from Christensenella minuta. J. Front. Chem. 8, 622325 (2020).
Xia, Y. et al. Research advances of d-allulose: an overview of physiological functions, enzymatic biotransformation technologies, and production processes. J. Food 10, 1 (2021).
Elbing, K. L. & Brent, R. Growth of E. coli in liquid medium. J. Curr. Protoc. Mol. Biol. 125, e81 (2019).
Chan., H. C. et al. Crystal structures of D-psicose 3-epimerase from Clostridium cellulolyticum H10 and its complex with ketohexose sugars. J. Protein Cell 3, 123–131 (2012).
Endo, A. et al. Fructophilic lactic acid bacteria, a unique group of fructose-fermenting microbes. J. Appl. Environ. Microbiol. 84, 1 (2018).
Kornberg, H. L. Routes for fructose utilization by Escherichia coli. J. Mol. Microbiol. Biotechnol. 3, 355–359 (2001).
Xiao, Q. et al. High conversion of D-fructose into D-allulose by enzymes coupling with an ATP regeneration system. J. Mol. Biotechnol. 61, 432–441 (2019).
Yoshihara, A. et al. Purification and characterization of d-allulose 3-epimerase derived from Arthrobacter globiformis M30, a GRAS microorganism. J. Biosci. Bioeng. 123(2), 170–176 (2017).
Kim, H. J. et al. Characterization of an Agrobacterium tumefaciens D-psicose 3-epimerase that converts D-fructose to D-psicose. J. Appl. Environ. Microbiol. 72, 981–985 (2006).
Jia, M. et al. A D-psicose 3-epimerase with neutral pH optimum from Clostridium bolteae for D-psicose production: cloning, expression, purification, and characterization. J. Appl. Microbiol. Biotechnol. 98, 717–725 (2014).
Wen, X. et al. Optimization for allitol production from D-glucose by using immobilized glucose isomerase and recombinant E. coli expressing D-psicose-3-epimerase, ribitol dehydrogenase and formate dehydrogenase. J. Biotechnol. Lett. 42, 2135–2145 (2020).
Men, Y. et al. The development of low-calorie sugar and functional jujube food using biological transformation and fermentation coupling technology. J. Food Sci. Nutr. 7, 1302–1310 (2019).
Chen, X. et al. Production of d-psicose from d-glucose by co-expression of d-psicose 3-epimerase and xylose isomerase. J. Enzyme Microb. Technol. 105, 18–23 (2017).
Chen, Z. et al. Improving thermostability and catalytic behavior of l-rhamnose isomerase from Caldicellulosiruptor obsidiansis OB47 toward d-allulose by site-directed mutagenesis. J. Agric. Food Chem. 66, 12017–12024 (2018).
Funding
This work was financially supported by (1) Key innovation Project of Qilu University of Technology (Shandong Academy of Sciences) (Grant No. 2022JBZ01-06), (2) Natural Science foundation of Shandong Province (Grant No. ZR2022MC059, Grant No. ZR2023MC087), (3) The Science and Technology Support Plan for Young People in Colleges and Universities of Shandong Province (Grant No. 2020KJE005), (4) The Basic Research of Pilot Project for the Integration of Science, Education and Industry, Qilu University of Technology, Shandong Academy of Sciences (Grant No. 2022PY074). (5) The Natural Science Foundation of Shandong Province for Youth (Grant No. ZR 2020QC235).
Author information
Authors and Affiliations
Contributions
Author Contributions: 1.Haoran Liu and Kang Xu: writing—original draft preparation, writing—review and editing, validation; 2.Shuqi Sun: methodology; software;3.Yinbiao Wan and BoJia Zhang: formal analysis; methodology;4.Yang Song: methodology; software;4.Chuanzhuang Guo: methodology; software; funding acquisition5.Songsen Sui: methodology;6.Ruiming Wang: methodology; funding acquisition 7.Piwu Li:methodology; software8.Junqing Wang:methodology;software9.Xu Zhenshang: Conceptualization, validation, writing—review and editing, supervision, funding acquisition.10.Wang Ting: Conceptualization, validation, writing—review and editing, supervision, funding acquisition; All authors reviewed the manuscript.In addition, all the pictures and tables in the article were drawn by Liu Haoran.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, H., Xu, K., Sun, S. et al. Optimization of fermentation conditions for whole cell catalytic synthesis of D-allulose by engineering Escherichia coli. Sci Rep 14, 30771 (2024). https://doi.org/10.1038/s41598-024-80561-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-80561-5
