
Abstract
The interactions between CD40 and tumor necrosis factor receptor-associated factor 6 (TRAF6) are implicated in chronic inflammation and fibrosis. Given their poorly understood role in chronic transplant rejection, our study focused on investigating the CD40-TRAF6 interactions in murine models of cardiac transplantation, particularly in relation to cardiac allograft vasculopathy (CAV). We established murine heart transplantation models using BALB/C to C57BL/6 and H-2bm12 to C57BL/6 pairings. A specific antagonist for TRAF6 was administered post-transplantation, either alone or in combination with cyclosporin A (CsA). We analyzed cells infiltrating the cardiac allografts and splenic immune cells. Additionally, We explore the potential mechanistic effects of TRAF6 inhibition in CAV by bone marrow-derived macrophages (BMDMs) co-culture. The inhibition of CD40-TRAF6 interaction significantly prolonged the survival of cardiac allografts. When combined with CsA, this treatment induced long-term survival of the allografts. Specifically, in the H-2bm12 to C57BL/6 heart transplantation model, inhibiting TRAF6 mitigated the development of CAV. This blockade led to a decrease in CD11b + and CD4 + cells within the allografts. In vitro experiments showed that TRAF6 inhibition had limited effects on mixed lymphocyte culture responses and minimally affected the proliferation of naive CD4 + cells activated by CD3/CD28. Furthermore, BMDMs under CD40-TRAF6 inhibition were more likely to differentiate into an anti-inflammatory phenotype, and their migration capability was reduced. Our findings demonstrate that inhibiting the TRAF6 pathway can significantly ameliorate both acute and chronic allograft rejection. The combination with CsA appears to have a synergistic effect, suggesting that targeting the TRAF6 could be a beneficial co-strategy for managing alloimmune responses. Importantly, our results position TRAF6 as a promising complementary target for enhancing outcomes in CAV.
Similar content being viewed by others

Introduction
Chronic rejection is a prevalent issue following cardiac transplantation, often leading to the development of Cardiac Allograft Vasculopathy (CAV). In contrast, acute rejection has become less of a concern due to advancements in immunosuppressive therapies1. Despite the growing prevalence of CAV, retransplantation remains the most effective treatment available. The optimal approach involves preventing CAV development or diagnosing it early to mitigate its severity and frequency. CAV’s pathogenesis is driven by both immunologic and non-immunologic factors, with the prevailing view being that both humoral and cellular immunity play significant roles in its progression2.
The CD40/CD40L signaling pathway has been implicated in various inflammatory conditions, including rheumatoid arthritis, transplant rejection, and multiple sclerosis3,4,5. Specifically, the interaction between CD40 and tumor necrosis factor receptor-associated factor 6 (TRAF6) in myeloid cells, particularly macrophages6, is crucial. This interaction prompts macrophages to produce inflammatory cytokines. The CD40-TRAF6 signaling pathway is intricately regulated to balance protective and pathogenic inflammation7. It is essential for the infiltration of inflammatory cells, with TRAF6 acting as a pivotal regulator. Furthermore, CD40-TRAF6 signaling activates additional pathways, such as IKK–NFκB, directly influencing NFκB, IRF, MAPK, and PI3K pathways8.
Macrophages have been identified as key players in the development of CAV, with infiltrating macrophages being a common presence in CAV allografts. These macrophages exhibit a range of phenotypes, from pro-inflammatory to anti-inflammatory and fibrogenic, impacting transplant outcomes through various mechanisms. Particularly, activated macrophages contribute to interstitial fibrosis and the progression of CAV9,10. Small-molecule inhibitors targeting macrophages have shown promise in inhibiting activation and preventing foam cell formation11. This evidence supports the hypothesis that the CD40-TRAF6 interaction plays a role in CAV development. In this context, a small-molecule inhibitor, CD40-TRAF6 inhibitor 6,877,002 (CT6i for short), was explored as a specific antagonist in a murine cardiac transplantation model to evaluate its impact on cardiac transplantation outcomes.
Materials and methods
Animals
Male C57BL/6 mice (6–8 weeks and 20–25 g) were purchased and raised in pathogen-free conditions at the Animal Center of Tongji Medical College. B6.C-H-2bm12KhEg (H-2bm12) mice were kindly sponsored by Professor Fang Zheng (Department of Immunology, Basic Medical College, Huazhong University of Science and Technology, China). All mice were sacrificed by cervical dislocation after isoflurane gas anesthesia. All experiments were conducted according to the Guide of the Care and Use of Laboratory Animals of the Institutional Animal Care and Use Committee of Tongji Medical College, Huazhong University of Science and Technology (IACUC Number: 2358). All procedures for the animal experiments were approved by the Animal Care Committee of Hubei University of Medicine (Shiyan, China), and performed in accordance with the institutional guidelines (approval number 2022–022). All methods are reported in accordance with ARRIVE guidelines.
Abdominal ectopic heart transplantation in mice
The model was conducted as previously described12. Briefly, H-2bm12 or BALB/C mice as donors and C57BL/6 mice as recipients to conduct the abdominal ectopic heart transplantation model. The success of the model was confirmed by detecting that the cardiac allografts had strong and fast beats. Allografts and tissues were harvested at each time point. CT6i, DMSO, and CsA were all purchased from MCE MedChemExpress. After transplantation, the mice were injected with CT6i at a dose of 5 mg/kg/day respectively. In the control group, the mice were given CT6i at a dose of 5 mg/kg plus CsA at a dose of 5 mg/kg. The control group was given an equal volume of DMSO.
Bone marrow derived macrophages (BMDMs)
After anesthesia and disinfection of C57BL/6 mice, the femur and tibia were harvested. Bone marrow was isolated, flushed with cold PBS, lysed with red blood cell lysis buffer (BD Bioscience), and then cultured in RPMI medium (containing 10% fetal bovine serum). Then, 10 ng/mL murine M-CSF was added every 2 days. On day 6, the cells were with 100 ng/mL LPS and 20 ng/mL IFN-γ stimulated for 24 h for M1 polarization. Stimulated with 20 ng/mL IL-13 and 20 ng/mL IL-4 for M2 polarization. Cells were harvested after 24 h of polarization and assessed for the expression of macrophage markers.
Mixed lymphocyte culture
Splenocytes (1 × 106/mL) were isolated and purified from C57BL/6, inactivated splenocytes (1 × 106/mL) were isolated and purified from BALB/c mice then inactivated mitomycin C (MedChemExpress HY-13316). These cells were co-cultured in 96-well plates for 5 days. Proliferation was assessed by Cell Counting Kit-8 (Dojindo, Japan) on day 5. Cellular proliferation was expressed as the optical density (OD) at a wavelength of 450 nm.
BMDMs and CD4 + T cell co-culture
Splenocytes (1 × 106/mL) from C57BL/6 mice and CD4 + T cells were isolated and purified from spleens and lymph nodes via magnetic selection according to the instructions (Miltenyi Biotec). Then purified CD4 + cells were stained with CFSE or CTV (Invitrogen) and cultured in 24-well plates (1 × 106 cells/well) with anti-CD3 (2ug/mL) and anti-CD28 (2ug/mL) monoclonal antibodies. BMDMs and CD4 + T cells were cultured in a ratio of 1:1. Then, the CellTrace™ Violet Cell Proliferation Kit (Invitrogen, C34557) was applied according to the manufacturer’s instructions: Purified CD4 + cells were stained for 20 min at room temperature with gentle agitation. Then, the cells were cultured for 48 h, and proliferation was assessed by flow cytometry.
The antigen-presenting function of BMDMs was assessed as blow: Stimulate the C57BL/6 origin BMDMs with BALB/c skin tissue pieces. Incubate the co-culture of BMDMs and purify C57BL/6 origin CD4 + T cells for 72 h at 37 °C with 5% CO2. After the incubation, harvest the cells and analyze the activation status of CD4 + T cells by flow cytometry.
Flow cytometry
Spleens were surgically removed under anesthesia by isoflurane. Cells were isolated and purified from the spleens in RPMI with 1% FBS. The red blood cells were removed by Red Blood Cell Lysis Buffer (BD Biosciences, USA), and the cell suspension was filtered by a 40-µm nylon mesh. The following antibodies were used (BD Biosciences, San Diego, USA): FITC-anti-CD4, APC-anti-IFN-γ, PE-anti-IL-17A, APC-CY7-anti-Live/Dead, PE-CY7-anti-TNF-a, PE-anti-IL-2, PE-CY7-anti-Foxp3, APC-anti-Granzyme B, PE-anti-F4/80, APC-anti-CD206, and PE-CY7-anti-CD80. For intracellular cytokine staining, cells were stimulated by a cell activation cocktail containing brefedin A (2 μg/mL; BD Biosciences, USA) for 4 h, followed by a collection for intracellular staining. Marker expression was assessed by flow cytometry using a FACS Calibur Flow Cytometer (BD Biosciences, USA). Multicolor flow cytometry analysis was performed using the Flowjo V10 software (Tree Star, Ashland, USA).
Transwell migration assay
BMDMs were treated with 1% FBS DMEM for 24 h, and 2.5 × 105 cell suspension was prepared. PET Transwell (8 µm porous membrane) was placed on a 24-well plate, and 200μL cell suspension was added into the upper chamber. The cell suspension was cultured for 24 h. IFN-γ was added to adjust the final concentration to 1 ug/mL. The culture was continued for 24 h. Then, the culture medium was removed from the lower chamber, and PBS was slowly added for washing three times. Cells attached to the upper chamber were wiped before being wetted, and 500µL of 1% crystal violet staining solution was added to the lower chamber for dyeing for 15 min. The dye solution in the upper and lower chambers was removed, and PBS was slowly added to the lower chamber, which was rinsed three times. PBS was removed, the chamber was completely dried, and the cells were observed and counted using an inverted microscope.
Histological analysis of allografts
Mice were disinfected and anesthetized with isoflurane. The allografts and spleens were obtained at each time point. The tissue sections were under H&E and Masson’s trichrome stain. Immunohistochemical staining of Arg-1 + cells, iNOS + cells, CD4 + cells, and CD11b + cells were performed. The number of positive cells was averaged over five random 400 × fields.
Western blot
Cultured cells were lysed in sodium dodecyl sulfate (SDS) buffer and boiled for 10 min for denaturation. Proteins were resolved by 10% SDS-PAGE and transferred to a nitrocellulose membrane for immunoblotting. Proteins were visualized using an HRP-conjugated anti-mouse or anti-rabbit IgG and the ECL system (Amersham Biosciences). Total cell extracts were prepared in PBS containing protease inhibitors. Protein concentrations were determined using a protein assay kit (eBioscience).
RNA sequencing data processing
Raw count data of murine bone marrow derived macrophages (BMDM) cells before (n = 3) and after (n = 3) interferon-gamma stimulation (IFN-γ) were obtained from the GEO database identified by accession number GSE232827. Data normalization and differential expression analysis were conducted with the Bioconductor R package DESeq213. Genes displaying an absolute log2 fold change of at least 0.6 and p values less than 0.05 were considered differentially expressed genes (DEGs). Gene ontology (GO) enrichment analysis was carried out using the R package ClusterProfiler v4.0, using the enrichGO() function14.
Single-cell RNA sequencing data processing
Single-cell RNA sequencing (scRNA-seq) data from cardiac grafts and spleens in a murine heterotopic heart transplantation model were obtained from Gene Expression Omnibus (GEO) under accession numbers GSE14256415 and GSE17919716. The dataset included samples from four experimental groups: graft heart (day 0), allograft heart (day 5 post-transplant), isograft spleen, and allograft spleen. Raw count matrices were processed using the Seurat package (v4.3.0) in R17. Doublets and contamination were removed using scDblFinder18 (v1.14.0) and decontX (v1.14.0)19, respectively, retaining only singlets for downstream analysis. Quality control (QC) was performed to exclude low-quality cells and genes. Mitochondrial content (percent.mt) was calculated with PercentageFeatureSet (pattern: ^Mt-), and cells were filtered to retain those with 600 < nFeature_decontx_RNA < 5000 (number of detected genes per cell) and percent.mt < 10%. Genes expressed in fewer than five cells were excluded. Normalization was conducted using the LogNormalize method (NormalizeData), followed by identification of variable features (FindVariableFeatures). Dimensionality reduction was performed via principal component analysis (PCA) on variable genes, and batch effects were corrected using Harmony integration20 (IntegrateLayers, dims = 1:20). UMAP embeddings were generated on the integrated data (RunUMAP, dims = 1:20). Clustering was performed by identifying neighbors (FindNeighbors, dims = 1:20) and applying the shared nearest neighbor modularity optimization algorithm (FindClusters, resolution = 0.5). Cluster-specific markers were identified using FindAllMarkers (logfc.threshold = 0.25, only.pos = TRUE), and cell types were annotated using canonical markers (e.g., Cd3d for T cells, Cd79a for B cells, Nkg7 for NK cells). Gene expression patterns were visualized via dot plots (DotPlot) and violin plots (VlnPlot), while correlations between gene pairs (e.g., Traf6 and Cd4/Cd8a) were assessed using Pearman correlation. Differential expression analysis between groups (e.g., graft vs. allograft) was conducted using the Wilcoxon rank-sum test. Key parameters included Harmony integration (dims = 1:20, theta = default), clustering resolution = 0.5, and statistical thresholds of p < 0.05 with FDR correction.
Statistical analysis
Data are presented as the mean ± SD. Fisher’s LSD was used to assess mean graft survival, and comparisons between two groups using a two-tailed Student’s t-test. All data were analyzed using Prism 7 (GraphPad Software, CA, USA). Values of p < 0.05 were considered statistically significant.
Results
CD40-TRAF6 inhibition ameliorated allograft rejection and prolonged the survival of cardiac allografts
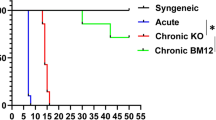
In the CT6i-treated group, the recipients were given CD40-TRAF6 specific inhibitor 6,877,002 (CT6i) 5 mg/kg by intraperitoneal injection (i.p.), and the control group was injected with the same amount of DMSO (n = 5) (Fig. 1A). We found that the CT6i group showed a much longer mean allograft survival time than the control group (Fig. 1B). The pathology showed that lymphocyte infiltration of allografts was less severe in the CT6i group (Fig. 1C), while Masson staining showed that myocardial fibrosis was much more progressed in the control group (Fig. 1D). Moreover, the CT6i group exhibited fewer infiltrated CD4 + and CD11b + cells than the control group (Fig. 1F,G). The control group, was characterized by widespread myocardial cell injury with interstitial inflammation, and higher parenchymal rejection (PR) scores (Fig. 1E). Altogether, CT6i markedly decreased the severity of acute rejection and significantly prolonged the mean survival time of cardiac allografts.

HYPERLINK "sps:id::fig1||locator::gr1||MediaObject::0" CD40-TRAF6 inhibition ameliorated acute rejection and prolonged the survival of cardiac allografts in the murine cardiac transplantation acute rejection model. (A) The murine cardiac heart transplantation model was performed using BALB/C mice as donors and C57BL/6 mice as recipients. CD40-TRAF6 specific antagonist 6,877,002 (CT6i for short) or DMSO was administered after transplantation. (B) Survival of cardiac allografts in each group.(p = 0.002). (C) HE staining of cardiac allografts 7 days after heart transplantation. DMSO vs CT6i. (D) Masson staining of cardiac allografts 7 days after heart transplantation. (E) Immunohistochemical assay of cardiac allografts infiltrated by CD4 + and CD11b + cells. (F) Parenchymal rejection scores of cardiac allografts were evaluated on day 7 after cardiac transplantation. The CT6i group exhibited lower PR scores than the DMSO group (n = 5 for each group)(p = 0.001).
TRAF6 mediate acute allograft rejection through Th1/Th17 response
Single-cell RNA sequencing (scRNA-seq) of CD45-positive immune cells isolated from cardiac grafts and spleens in a murine heterotopic heart transplantation model revealed 11 major immune cell types, including B cells, macrophages, T cells, cytotoxic CD8 + T cells, plasma cells, monocytes, neutrophils, proliferating T cells, NK cells, monocyte-derived dendritic cells (monoDCs), and innate lymphoid cells (ILCs) (Fig. 2A). Cell type identification was confirmed by canonical marker gene expression, such as Cd3d for T cells, Cd19 for B cells, S100a8 for monocytes, and Nkg7 for NK cells (Fig. 2B). Analysis of cell composition across four groups—graft heart, allograft heart, isograft spleen, and allograft spleen—revealed significant shifts in immune populations following allogeneic transplantation. T cells exhibited a marked increase in proportion in allograft hearts compared to graft hearts (7.1% vs. 5.9%) and a more pronounced expansion in allograft spleens relative to isograft spleens (27% vs. 13.5%) (Fig. 2B). Further investigation of T cell–associated signaling pathways demonstrated that Traf6 (TNF receptor-associated factor 6) expression correlated strongly with Cd4 levels in T cells (Pearson’s, p < 0.05) but showed no significant correlation with Cd8a (Fig. 2D,E). Traf6 expression was significantly upregulated in allograft hearts compared to graft hearts (p < 0.005), suggesting its role in alloimmune responses. Additionally, Ifng (interferon-γ) expression was elevated in allograft hearts versus graft hearts (p < 0.001) and in allograft spleens compared to isograft spleens (p < 0.05). Conversely, Tbx21 (T-bet), a transcription factor linked to Th1 differentiation, was selectively upregulated in spleen tissues (p < 0.01), highlighting tissue-specific immune activation patterns. Further investigation revealed the phenotype and function of immune cells in the spleen of the recipient mice. Percentage of CD4 + T-bet + cells, CD4 + IFN-γ + cells, CD4 + RORγ-t + , CD4 + IL-17A + that were decreased in the CT6i group compared with the DMSO group (Fig. 2F–M). Percentage of CD4 + Foxp3 + cells that remained unchanged between the two groups (Fig. 2N,O).

TRAF6 mediates acute allograft rejection through Th1/Th17 response. (A) Uniform Manifold Approximation and Projection (UMAP) dimensionality reduction plot following Harmony batch-effect correction, displaying 11 distinct cell clusters colored by cell type: B_cells, Macrophage, T_cells, Cytotoxic_CD8_T, plasma, Monocyte, Neutrophil, Proliferating_T, NK_cells, monoDCs (monocyte-derived dendritic cells), and ILCs (innate lymphoid cells). (B) Stacked bar plot depicting relative proportions of the 11 cell types across four experimental groups: graft heart, allograft heart, isograft spleen, and allograft spleen. Colors correspond to cell types as in A; bars represent tissue- and treatment-specific immune composition. (C) Dot plot showing expression levels of canonical marker genes across cell types. Each dot represents a cell type, with color intensity indicating normalized gene expression (log scale) and dot size reflecting the percentage of cells expressing the gene. Key markers include Cd79a (B cells), CD68 (macrophages), Cd3d (T cells), Nkg7 (NK cells), Ly6c2 (monocytes), and Rora (ILCs). (D) Scatter plot illustrating correlations between Traf6 expression and Cd4 /Cd8a levels in T_cells. Each point represents an individual T cell; red and blue lines indicate linear regression fits for CD4 and CD8A, respectively. Pearson correlation coefficients (R) and significance values (p) are annotated. (E) Violin plots comparing expression distributions of Traf6, Ifng (interferon-γ), and Tbx21 (T-bet) in graft vs. allograft hearts (left) and isograft vs. allograft spleens (right). Statistical significance between groups was determined by Wilcoxon rank-sum test (p < 0.05, FDR-corrected). (F,G) FCM assay shows CT6i exhibited markedly reduced CD4 + Tbet + subsets. (H,I) FCM assay shows CT6i exhibited markedly reduced CD4 + IFN-γ + subsets. (J,K) CT6i exhibited markedly reduced CD4 + RORγt + subsets. (L,M) CT6i exhibited markedly reduced CD4 + IL17A + subsets. (N,O) CT6i exhibited markedly reduced CD4 + Foxp3 + subsets.
CD40-TRAF6 inhibition alleviated CAV in bm12 to C57BL/6 cardiac transplantation models
In the chronic rejection heart transplantation model, the H-2bm12 heart was transplanted into C57BL/6 recipients. One group was treated with CT6i (CT6i; n = 6), and another group was given the same amount of DMSO (control; n = 6) (Fig. 3A,B). Histological assessments of the cardiac allograft sections at 60 days after transplantation revealed that CAV was developed in the control group, while CAV was much less severe in the CT6i group (Fig. 3C,D). Infiltrated CD4 + cells were significantly decreased in the CT6i group compared with the control group (Fig. 3C,E). Treatment with CT6i also reduced splenic CD4 + IFN-γ and CD4 + IL-2 + cells compared with the control group (Fig. 3F,G). Flow cytometry analysis of splenic F4/80 + CD80 + and F4/80 + CD206 + cells (Fig. 3H,I) showed that the F4/80 + CD80 + cell percentage was decreased, while F4/80 + CD206 + cells were slightly increased in the CT6i group. CD80 is the T cellsThe histochemical assessment showed that the number of CD11b + cells infiltrated in the allografts of the CT6i group was greatly decreased compared with the control group (Fig. 3J). CD11b + iNOS + cells (Fig. 3K,L) were also decreased in the CT6i group. All these data indicate that CD40-TRAF6 inhibition mitigated the inflammatory environment after heart transplantation.

CD40-TRAF6 inhibition alleviated CAV in bm12 to C57BL/6 cardiac transplantation models. (A,B) The murine cardiac heart transplantation model was performed using BM12 mice as donors and C57BL/6 as recipients. CD40-TRAF6 specific antagonist or DMSO was administered after transplantation (n = 5 for each group). (C) HE staining of cardiac allografts from recipient mice 60 days after heart transplantation. (D) Masson staining of cardiac allografts from recipient mice 60 days after heart transplantation. (E) Immunofluorescence assay of cardiac allografts infiltrated by CD4 + cells. (F,G) FCM assays of CD4 + IFN-γ + splenocytes(p = 0.001) and CD4 + IL-2 + splenocytes(p = 0.017). (H,I) FCM assays of F4/80 + CD80 + cells and F4/80 + CD206 + splenocytes. (J) Immunofluorescence assay of cardiac allografts infiltrated by CD11b + cells. (K,L) Immunohistochemical assay of cardiac allografts infiltrated by CD11b + cells, i-NOS + , and Arg-1cells.
Combined inhibition of TRAF6 and CsA induced allograft long-term survival in chronic cardiac transplantation rejection model
CsA and CT6i co-inhibition in a BALB/C to C67BL/6 murine cardiac transplantation model showed surprisingly that the survival of cardiac allografts was significantly prolonged (Fig. 4A,B). HE and Masson staining of the allograft sections showed that CAV was also markedly alleviated (Fig. 4C,D). Co-inhibition decreased the number of CD4 + and CD11b + cells infiltrated in cardiac allografts (Fig. 4E,F). Simultaneously, the flow cytometry results demonstrated that co-inhibition decreased the proportion of M1 macrophages and increased the proportion of M2 macrophages in the recipient (Fig. S1 A, B). T-cell activation in the recipient is also reduced (Fig. S1 C, D). Meanwhile, treatment of CT6i or CsA restrained the anti0antigen specific response of CD4 T cells (Fig. 4G,H, Fig. S2). Furthermore, while CT6i alone did not affect donor-specific antibody (DSA) levels, combined inhibition significantly suppressed DSA expression in recipient serum (Fig. S3). These results suggested that co-inhibition may have synergistic effects in the murine models of cardiac transplantation.

Combined inhibition of TRAF6 and CsA induced allograft long-term survival in chronic cardiac transplantation rejection model. (A) The murine cardiac heart transplantation model was performed using BALB/C mice as donors and C57BL/6 mice as recipients. CD40-TRAF6 specific antagonist (with or without cyclosporin A) was administered after transplantation (n = 5 for each group). (B) Mean survival times of cardiac allografts in different groups. CD40-TRAF6 inhibitor and CsA-induced long-term allograft survival. CsA vs CsA + CT6i (p = 0.002) (95% CI 0.3750 (0.1086–295) (C) HE staining of cardiac allografts from recipient mice 4 weeks after heart transplantation. (D) Masson staining of cardiac allografts from recipient mice 4 weeks after heart transplantation. (E,F) Infiltrated CD4 + (p = 0.001), CD11b + (p = 0.016) cells in the allografts 4 weeks after heart transplantation. (G,H) FCM assays of CD4 + CD154 + splenocytes (p = 0.001).
CD40-TRAF6 inhibition affected the immunostimulation function of BMDMs in vitro
The mixed lymphocyte reaction (MLR) was used to assess T-cell proliferation (Fig. 5A). Differences in the OD values between the two groups were detected only for a high CT6i concentration (10uM) (Fig. 5B). A naive CD4 + T cell isolation kit was used to purify CD4 + T cells. Then, on day 0, CD3 and CD28 antibodies were added to activate CFSE-labeled CD4 + T cells in different groups. After 48 h, no significant differences in the CD4 + T cell proliferation were observed between these two groups (Fig. 5C). BMDM-induced CD4 + T-cell proliferation was assessed by CD4 + T cells (labeled with CFSE) and BMDMs co-cultured in vitro. Significant differences in the CD4 + T cell proliferation were detected between these two groups (Fig. 5D). These results suggested that CD40-TRAF6 inhibition may not directly affect CD4 + T cells. It is more likely that CD40-TRAF6 inhibition hampered the immunostimulation function of BMDMs to alleviate CD4 + T cell-mediated alloimmune responses.

CD40-TRAF6 inhibition affected the immunostimulation function of BMDCs in vitro. (A) The mixed lymphocyte reaction (MLR) was used to assess T-cell proliferation. (B) CCK-8 proliferation analysis of the lymphocyte mixing assay: only high concentrations of CD40-TRAF6 inhibitor (10 uM) affected the OD value (p = 0.045). (C) CFSE-labeled CD4 + T cell (activated by CD3/CD28) proliferation was not hampered by CD40-TRAF6 inhibition (10 uM)(p = 0.287). (D) Proliferation of CFSE-labeled CD4 + T cells (co-cultured in BMDMs) was hampered by CD40-TRAF6 inhibition (10 uM) (p = 0.014).
CD40-TRAF6 inhibitor-induced BMDMs developed an anti-inflammatory phenotype through NF-κB pathway
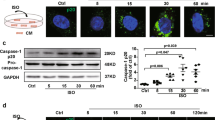
To further assess the effect of CD40-TRAF6 inhibition in transplantation, BMDMs were cultured in 100 ng/mL LPS and 20 ng/mL IFN- γ for M1 polarization or stimulated with 20 ng/mL IL-4 and 20 ng/mL IL-13 for M2 polarization(Fig. 6A). CD40-TRAF6 inhibition decreased the immunofluorescence of CD68 + Arg-1 + cells less significantly than that of CD68 + iNOS + cells (Fig. 6B). Flow cytometry analysis showed that the number of CD11b + CD86 + and CD11b + CD163 + cells was decreased by CT6i (Fig. 6D,E). The transwell migration assay indicated that the number of migrated BMDMs in the CT6i group was decreased compared with that in the control group (Fig. 6C). Consistently, RNAseq of BMDM before and after stimulation of IFN- γ revealed genes including Nfkbia, Cd40, Rela and Traf6 were significantly up-regulated (Fig. 6F). Gene ontology enriched by the up-regulated DEGs were involved in response to interferon-gamma, cytokine mediated signaling pathway, I-kappaB kinase/NF-kappaB signaling and cellular response to interferon gamma (Fig. 6G). NF-κB is a major transcription factor that regulates genes that are involved in both adaptive and innate immunity. CD40-TRAF6 inhibition hampered BMDMs’ NF-κB signal-associated phosphorylation of proteins (Fig. 6H,I, Fig. S4).

TRAF6 inhibition induced BMDCs developed a less pro-inflammatory phenotype via NF-κB pathway. (A) Macrophages were activated by stimulation in vitro. (B) Immunofluorescence assay of CD68 + Arg-1 + BMDMs. (C) Immunofluorescence assay of CD68 + i-NOS + BMDMs. (D) CT6i decreased CD11b + CD86 + BMDMs in M1 polarization (p = 0.001). Slightly decreased CD11b + CD163 + BMDMs in M2 polarization. (p = 0.002) (E) Transwell assay showing that the migration capacity of BMDMs was hampered by CT6i. (F,G) scRNAseq shows the expression increased pathway in macrophage activation. (H,I) Western blot assay of NF-κB pathway proteins showed that the phosphorylation level of IκBα was decreased.
Discussion
The rejection response to heart grafts involves a complex immune response, encompassing both humoral and cellular rejection, with acute and chronic rejection reactions intertwined. At different stages of graft rejection, acute rejection and chronic rejection play major roles respectively. In this study, we demonstrated that CD40-TRAF6 inhibition by a small-molecule inhibitor ameliorated murine cardiac allograft rejection in both acute and chronic cardiac transplantation models. Blocking CD40-TRAF6 combined with CsA induced long-term tolerance of cardiac allografts.
Several studies showed that CsA, which was commonly used in post-transplant patients, was effective in inhibiting CD40 ligand expression in T lymphocytes21,22. We investigated the effects of CD40-TRAF6 and CD40L co-inhibition. Therefore, we used CT6i and CsA in BALB/C to C57BL/6 murine cardiac transplantation models. The co-inhibition significantly prolonged the survival time of cardiac allografts compared with CT6i or CsA alone. It suggested that there might be a synergistic effect of co-inhibition in maintaining alloimmune responses.
CAV is the leading cause of long-term graft failure in cardiac transplant recipients23. Although numerous kinds of cells were found to be involved in the development of CAV, it is still unknown which kind of cells mainly contribute to the pathology. Macrophages were reported to account for 38–60% of graft-infiltrating cells in human transplant biopsies in acute allograft rejection and contribute to graft damage through multiple mechanisms24,25. Macrophages are involved in both acute and chronic rejection, contributing to both graft rejection and tolerance9. In our study, intragraft macrophages were decreased due to CD40-TRAF6 inhibition. Once macrophages infiltrated into the allograft, they secreted inflammatory cytokines and acquired a pro-inflammatory phenotype. Activated macrophages could induce colony-stimulating factor-1 (CSF-1), promote cytotoxic T cells, and activate endothelial cells26. These pro-inflammatory macrophages were the main source of reactive nitrogen species (RNS) and reactive oxygen (ROS)27. Both can directly injure the allograft and cause the following rejection. Macrophages are abundant in CAV lesions and capable of producing growth factors implicated in neointimal proliferation. Therefore, they are leading end-effector candidates28. Although the specific role of macrophages in CAV is not well understood, depleting host macrophages with carrageenan could suppress the development of CAV in a heterotopic cardiac transplant system28. M1 cells are potent pro-inflammatory cells: they release pro-inflammatory cytokines (IL-1, IL-6, and TNF-α) and ROS, which play a critical role in cell-mediated immunity. Patients with CAV had significantly lower M2 macrophage production and were less able to induce the production of functional M2 macrophages than patients without CAV(26). In our study, the number of iNOS + cells that infiltrated into the cardiac allografts was decreased by CT6i.
Macrophages that are recruited to the intima secrete several kinds of pro-inflammatory cytokines and growth factors that cause smooth muscle cell migration to the intima, proliferation, and extracellular matrix deposition29, which are important to the pathogenesis of CAV. With CD40 engagement and alloantigen challenge, macrophages acquired some degree of alloantigen specificity. They sensitized and rejected allogeneic cells independent of any other adaptive immune cells30.
The relationship between T cells and macrophages is complex, and macrophages are APCs that are critical in the activation and differentiation of CD4 + T cells. CD40-CD40L engagement is also important in T-cell-mediated macrophage activation31. This raised the question of whether the results of CD40-TRAF6 inhibition of macrophages or CD40-TRAF6 inhibition have a direct effect on T cells. Previous studies had revealed that pharmacological inhibition of TRAF6 had a strong effect on macrophages but only minor effects on the activation status of T and B cells11.
In our study, we found that CD40-TRAF6 inhibition alleviated the development of CAV. Decreased intra-graft macrophages may be responsible for the invasive activity of activated macrophages or reduced macrophage activation. Inhibition of CD40-TRAF6 could lead to an impaired pro-fibrosis function of macrophages by reducing the secretion of pro-inflammatory factors. Meanwhile, CT6i inhibits the antigen-presenting function of macrophages and enhances the differentiation of M2 macrophages, both of which affect the activation and inflammatory function of T cells. Suzanne et al. investigated 6,877,002 in an autoimmune encephalomyelitis model. The inhibitor reduced the trans-endothelial migration of human monocytes and reduced neuroinflammation32. Our findings have extended previous studies by demonstrating that the inhibition of CD40-TRAF6 exerted effects on cardiac transplantation. In vitro experiments showed that blocking TRAF6 reduced phosphorylation levels of the canonical NF-κB pathway, which may be effective in containing macrophage migration and activation11. TRAF6 contributes to the CD40-mediated activation of NF-κB, as well as activation of stress-activated protein kinases and perhaps other signaling molecules. NF-κB is a major transcription factor that regulates the genes that are responsible for both the innate and adaptive immune response33.
Based on the above results, we can conclude that CD40-TRAF6 inhibition could exert effects on macrophages, and CsA hampers the expression of CD40L in T cells. Therefore, co-inhibition not only inhibited the activation of CD4 T cells but also affected the function of macrophages, thus leading to long-term cardiac allograft survival. The combined use of CT6i and CsA prolonged the survival of cardiac allografts synergistically. Our results may provide a clue for further clinical research. Big animal experiments, pre-clinical research, and clinical research are needed before this strategy can be applied in the clinic.
CD40-TRAF6 inhibition in vivo has a systematic influence according to previous studies. We focused on the alloimmune responses after allografts transplantation. Our results confirmed that the inhibition of CD40-TRAF6 contributes to alleviating CAV. However, it has not been clarified whether CD40-TRAF6 inhibition exerts effects through other types of APCs or even other cells. One study investigated the effects of LysMCre Traf6fl/fl and LysMCre Mtorfl/fl recipients in the prevention of CAV, and only LysMCre Mtorfl/fl recipients treated with CTLA-4Ig alleviated CAV9. Our study mainly explored the effect of CD40-TRAF6 inhibition in a cardiac transplantation model and demonstrated its application prospects.
In conclusion, we gave evidence that CD40-TRAF6 inhibition by a small-molecule inhibitor significantly alleviated the development of CAV and prolonged the survival of cardiac allografts in a murine cardiac transplantation model. CD40-TRAF6 inhibition by CsA induced long-term survival of cardiac allografts. Taken together, these data advanced our understanding of how CD40-TRAF6 impacts allograft rejection, especially in the setting of CAV.
Data availability
Sequence data that support the findings of this study have been deposited in the GEO database with the primary accession code GSE23282 and GSE179197. The datasets during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Abbreviations
- CAV:
-
Cardiac allograft vasculopathy
- BMDMs:
-
Bone marrow derived macrophages
- TRAF6:
-
Tumor necrosis factor receptor-associated factor 6
- CT6i:
-
CD40-TRAF6 inhibitor
- CTLA4-Ig:
-
Cytotoxic tlymphocyte antigen 4 immunoglobulin
- H-2bm12:
-
B6.C-H-2bm12KhEg
- DMSO:
-
Dimethylsulfoxide
- CsA:
-
Cyclosporin A
- APCs:
-
Antigen presenting cells
- CFSE:
-
Carboxylfluorescein diacetate succinimide ester
- TRAFs:
-
Tumor necrosis factor-related factors
- PR:
-
Parenchymal rejection
- CD40L:
-
CD40 Ligand
References
Arkonac, B., and J. Hosenpud. Pathogenesis of cardiac allograft vasculopathy (chronic rejection). The Transplantation and Replacement of Thoracic Organs: The Present Status of Biological and Mechanical Replacement of the Heart and Lungs: 321–331 (1996).
Weis, M. & von Scheidt, W. Cardiac allograft vasculopathy: A review. Circulation 96, 2069–2077 (1997).
Gerritse, K. et al. CD40-CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc. Natl. Acad. Sci. 93, 2499–2504 (1996).
Law, C.-L., and I. S. Grewal. Therapeutic interventions targeting CD40L (CD154) and CD40: the opportunities and challenges. Therapeutic Targets of the TNF Superfamily 8–36 (2009).
Sarantopoulos, A. et al. YI-845 CD40/CD40L in dyslipidemic patients with rheumatoid arthritis (RA). The immunomodulating effect of simvastatin. Atherosclerosis (Supplements) (Component) 1, 224 (2007).
Subauste, C. S., Andrade, R. M. & Wessendarp, M. CD40-TRAF6 and autophagy-dependant anti-microbial activity in macrophages. Autophagy 3, 245–248 (2007).
Walsh, M. C., Lee, J. & Choi, Y. Tumor necrosis factor receptor-associated factor 6 (TRAF 6) regulation of development, function, and homeostasis of the immune system. Immunol. Rev. 266, 72–92 (2015).
Donners, M. M. et al. The CD40-TRAF6 axis is the key regulator of the CD40/CD40L system in neointima formation and arterial remodeling. Blood J. Am. Soc. Hematol. 111, 4596–4604 (2008).
Salehi, S. & Reed, E. F. The divergent roles of macrophages in solid organ transplantation. Curr. Opin. Organ Tran. 20, 446–453 (2015).
Zhao, Y. et al. Macrophage subpopulations and their impact on chronic allograft rejection versus graft acceptance in a mouse heart transplant model. Am. J. Transplant. 18, 604–616 (2018).
Seijkens, T. T. et al. Targeting CD40-induced TRAF6 signaling in macrophages reduces atherosclerosis. J. Am. Coll. Cardiol. 71, 527–542 (2018).
Huang, Y. et al. Inhibition of S-adenosyl-L-homocysteine hydrolase alleviates alloimmune response by down-regulating CD4(+) T-cell activation in a mouse heart transplantation model. Ann. Transl. Med. 8, 1582 (2020).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb.) 2, 100141 (2021).
Hua, X. et al. Single-cell RNA sequencing to dissect the immunological network of autoimmune myocarditis. Circulation. 142(4), 384–400. https://doi.org/10.1161/CIRCULATIONAHA.119.043545 (2020).
Chen, Z. et al. Single-Cell RNA sequencing reveals immune cell dynamics and local intercellular communication in acute murine cardiac allograft rejection. Theranostics 12, 6242–6257 (2022).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Germain, P. L., Lun, A., Garcia Meixide, C., Macnair, W. & Robinson, M. D. Doublet identification in single-cell sequencing data using scDblFinder. F1000Res. 10, 979. https://doi.org/10.12688/f1000research.73600.2 (2021).
Yang, S. et al. Decontamination of ambient RNA in single-cell RNA-seq with DecontX. Genome Biol. 21(1). https://doi.org/10.1186/s13059-020-1950-6 (2020).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Daoussis, D., Antonopoulos, I., Andonopoulos, A. P. & Liossis, S. N. Increased expression of CD154 (CD40L) on stimulated T-cells from patients with psoriatic arthritis. Rheumatology (Oxford) 46, 227–231 (2007).
Shoker, A., George, D., Yang, H. & Baltzan, M. Heightened CD40 ligand gene expression in peripheral CD4+ T cells from patients with kidney allograft rejection. Transplantation 70, 497–505 (2000).
Yun, J. J., Fischbein, M. P., Laks, H. & Ardehali, A. Chronic rejection of transplanted hearts. Curr. Opin. Organ Tran. 6, 205–210 (2001).
Alexander, D., Pearson, T., Hendrix, R., Ritchie, S. & Larsen, C. Analysis of effector mechanisms in murine cardiac allograft rejection. Transpl. Immunol. 4, 46–48 (1996).
Schaefer, N. et al. Role of resident macrophages in the immunologic response and smooth muscle dysfunction during acute allograft rejection after intestinal transplantation. Transpl. Int. 21, 778–791 (2008).
Liu, Y., Kloc, M. & Li, X. C. Macrophages as effectors of acute and chronic allograft injury. Curr. Transpl. Rep. 3, 303–312 (2016).
Di Meo, S., T. T. Reed, P. Venditti, and V. M. Victor. Role of ROS and RNS sources in physiological and pathological conditions. Oxidative medicine and cellular longevity 2016. (2016).
Kitchens, W. et al. Macrophage depletion suppresses cardiac allograft vasculopathy in mice. Am. J. Transplant. 7, 2675–2682 (2007).
Holzhauser, L. et al. Cardiac allograft vasculopathy is linked to impaired generation of regulatory M2 macrophages. J. Heart Lung Transplant. 38, S42 (2019).
Liu, W., Xiao, X., Demirci, G., Madsen, J. & Li, X. C. Innate NK cells and macrophages recognize and reject allogeneic nonself in vivo via different mechanisms. J. Immunol. 188, 2703–2711 (2012).
Stout, R. D., Suttles, J., Xu, J., Grewal, I. S. & Flavell, R. A. Impaired T cell-mediated macrophage activation in CD40 ligand-deficient mice. J. Immunol. (Baltimore, Md.: 1950) 156, 8–11 (1996).
Aarts, S. A. et al. Inhibition of CD40-TRAF6 interactions by the small molecule inhibitor 6877002 reduces neuroinflammation. J. Neuroinflammation 14, 1–14 (2017).
Hayden, M. S., West, A. P. & Ghosh, S. NF-kappaB and the immune response. Oncogene 25, 6758–6780 (2006).
Acknowledgements
B6.C-H-2bm12KhEg (H-2bm12) mice were kindly sponsored by Professor Fang Zheng (Department of Immunology, Basic Medical College, Huazhong University of Science and Technology, China).
Funding
The work was supported by the National Natural Science Foundation of China (Grant Number 81671578).
Author information
Authors and Affiliations
Contributions
Yajun Huang, Junlin Lai and Xing Chen contributed equally to this work. Yajun Huang, Junlin Lai, Xing Chen, Yuqi Chen, Xiangchao Ding and Guohua Wang participated in the research design. Chenghao Li, Wai Yen Yim, Yixuan Wang, Tian Xia and Guohua Wang participated in the performance of the research. Yajun Huang, Junlin Lai, Xing Chen, Jinfeng Yang and Guohua Wang participated in the writing of the article. Yajun Huang, Junlin Lai, Xing Chen, Jinfeng Yang, Xiangchao Ding,Yijie luo and Guohua Wang participated in the data analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
All experiments were conducted according to the Guide of the Care and Use of Laboratory Animals of the Laboratory Animal Ethical Commission of Tongji Medical College, Huazhong University of Science and Technology (IACUC Number: 2358).
Consent for publication
All authors approved the final manuscript and the submission to this journal.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, Y., Lai, J., Chen, X. et al. Mitigating cardiac allograft vasculopathy in a murine model via CD40-TRAF6 blockade and cyclosporin A synergy. Sci Rep 15, 22327 (2025). https://doi.org/10.1038/s41598-025-08315-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-08315-5
