
Abstract
The genus Camellia, known for species such as Camellia japonica, is of significant agricultural and ecological importance. However, the genetic diversity and evolutionary relationships among Camellia species remain insufficiently explored. In this study, we successfully sequenced and assembled the complete chloroplast (cp) genomes of nine Camellia accessions, including the species Camellia petelotii, and eight varieties of C. Japonica (C. Japonica ‘Massee Lane’, C. Japonica ‘L.T.Dees’, C. Japonica ‘Songzi’, C. Japonica ‘Kagirohi’, C. Japonica ‘Sanyuecha’, C. Japonica ‘Xiameng Hualin’, C. Japonica ‘Xiameng Wenqing’, and C. Japonica ‘Xiameng Xiaoxuan’). These genomes exhibited conserved lengths (~ 156,580–157,002 bp), indicating minimal variation in genome size. They consistently predicted 87 protein-coding genes, although variations were observed in the rRNA and tRNA genes. Structural and evolutionary analyses revealed the highly conserved nature of these cp genomes, with no significant inversions or gene rearrangements detected. Consistent codon usage patterns were observed across these accessions. Five hypervariable regions (rpsbK, psbM, ndhJ, ndhF, and ndhD) were identified as potential molecular markers for species differentiation. Phylogenetic analysis of 82 accessions from the Camellia genus, along with outgroup accessions revealed close genetic relationships among certain C. japonica varieties, including Songzi, Sanyuecha, L.T.Dees, and Kagirohi, which formed sister groups. Massee Lane was located within Sect. Camellia. Moreover, Xiameng Hualin, Xiameng Wenqing, Xiameng Xiaoxuan, and C. petelotii demonstrated a strong genetic affinity. These findings provide valuable insights into the structural and evolutionary dynamics of Camellia cp genomes, contributing to species identification and conservation.
Similar content being viewed by others


Introduction
Camellia, the largest genus within the Theaceae family, comprises a diverse group of flowering plants with substantial ornamental, economic, and ecological significance1,2,3. Among the Camellia species, Camellia japonica stands out as one of the most widely cultivated, renowned for its significant floral beauty and ornamental value. Despite its popularity, the genetic diversity and evolutionary relationships within C. japonica, as well as among other species in the genus remain insufficiently understood. Currently, this genus is faced with various challenges, such as discrepancies in genetic relationships, difficulties in species identification, and ongoing debates regarding the parentage of numerous Camellia species. While Camellia species exhibit limited morphological diversity and have been extensively hybridized to develop new cultivars, they also display trait variability within the same species under different environmental conditions, which complicates morphological classification4. Therefore, relying solely on morphological features for species identification is inadequate. Accurate species identification requires a comprehensive phylogenetic analysis using whole-genome sequencing and molecular markers. Chloroplast (cp) genomes, which offer valuable insights into plant evolution, systematics, and phylogenetics, remain underexplored across the diverse varieties of Camellia. While previous studies have characterized the cp genomes of several Camellia species and provided insights into the phylogenetic relationships within the genus5,6,7,8, its phylogenetic reconstruction is not incomplete.
Cp genomes in plants are recognized for their highly conserved evolutionary nature and typically adopts a circular, double-stranded quadripartite structure. These genomes comprise a large single-copy (LSC) region, a small single-copy (SSC) region, and a pair of inverted repeat (IR) regions. The two IRs, which are identical in length and oriented in opposite directions, are separated by the LSC and SSC regions9. Cp genomes are generally conserved in structure and gene content across plant species. Their relatively simple structure and small genome size, coupled with features such as moderate evolutionary rates, low recombination and variation frequency, maternal inheritance, and independent inheritance patterns10,11, make cp genomes highly valuable for applications in molecular marker development, phylogenetic research, and genetic engineering in plant breeding12. Comparative cp genomics has therefore emerged as a powerful tool for exploring plant evolutionary relationships, facilitating species identification, and supporting conservation initiatives. Advances in next-generation sequencing technologies have further enabled the complete sequencing of cp genomes, allowing for detailed comparative analyses across species and cultivars. Based on whole-genome resequencing technology, this study focuses on the cp genomes of nine accessions from the Camellia genus, including Camellia petelotii, and eight varieties of the species C. japonica (C. Japonica ‘Massee Lane’, C. Japonica ‘L.T.Dees’, C. Japonica ‘Songzi’, C. Japonica ‘Kagirohi’, C. Japonica ‘Sanyuecha’, C. Japonica ‘Xiameng Hualin’, C. Japonica ‘Xiameng Wenqing’, and C. Japonica ‘Xiameng Xiaoxuan’). Our primary objectives were uncover structural variations and conserved features, and explore their evolutionary trajectories. Furthermore, we aimed to identify hypervariable regions within cp genomes that can serve as molecular markers for species identification and phylogenetic studies. A phylogenetic tree was constructed using cp genome data from 82 accessions from the Camellia genus, with Zea mays, Hordeum vulgare , Triticum aestivum , Brachypodium distachyon , Ficus microcarpa, four varieties of Arabidopsis thaliana, as well as four species of the Diospyros genus included as the outgroup. We seek to elucidate the genetic relationships and evolutionary patterns, with a particular focus on C. japonica and its closely related species. Our findings contribute valuable insights into the structural and evolutionary dynamics of cp genomes in the Camellia genus, laying a foundation for future research on genetic diversity, species identification, and conservation strategies.
Results
Complete cp genome overview of the nine Camellia accessions
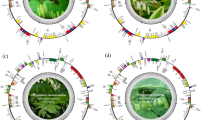
We first assembled and annotated the complete cp genomes of the nine Camellia accessions using whole genome sequencing. All nine genomes displayed a typical quadripartite structure, consisting of an LSC, an SSC, and two IRs (Fig. 1, Figure S1-8). They were predicted to annotate 87 coding sequences (CDSs) and 8 rRNAs each. Both Xiameng Hualin and C. petelotii were predicted to annotate 36 tRNAs, while the remaining seven accessions were annotated with 37 tRNAs. The nine cp genomes exhibited minimal length variation, with the genome of Xiameng Hualin being the longest at 157,059 bp, and that of Xiameng Xiaoxuan being the shortest at 156,580 bp. Moreover, the lengths of the LSC regions, SSC regions and IR regions ranged from 86,589 bp to 86,761 bp, 18,140 bp to 18,394 bp, and 25,953 bp to 26,134 bp, respectively, with the total GC content varying between 37.28 and 37.31%. Among these regions, the IR regions accounted for the highest GC content (42.96–43.03%), followed by the LSC regions (30.54–35.31%), while the SSC regions had the lowest GC content (30.54–30.6%) (Table S1). The higher GC content in the IR regions suggests that the Camellia cp genomes were stable and highly conserved.

Circular map showing the complete chloroplast (cp) genome of Camellia japonica ‘L.T. Dees’. This map includes key features of a cp genome, including the large single-copy (LSC) region, small single-copy (SSC) region, and a pair of inverted repeat (IR) regions. The genome is composed of 87 genes, with a total length of 157,002 bp and a GC content of 37%.
Codon usage analysis
Codon usage analysis was conducted on the CDSs from the complete cp genomes of the nine Camellia accessions. A total of 20 amino acids were encoded, each with 1–6 synonymous codons in the nine accessions. Differences were observed in the codons encoding leucine (Leu). Specifically, Kagirohi, L.T.Dees, Sanyuecha, Songzi, and Xiameng Wenqing each used four synonymous codons for Leu (CTT, CTC, CTA, CTG), while C. petelotii, Massee Lane, Xiameng Hualin, and Xiameng Xiaoxuan employed only two synonymous codons for Leu (TTA, TTG). The majority of amino acids exhibited a pronounced codon preference, with the notable exception of methionine (Met) and tryptophan (Trp). Each accession contained 29 codons with an RSCU value greater than 1. Moreover, most of these codons ended in A/T bases, indicating a strong preference for A/T usage (Fig. 2, Figure S9, Table S2). This A/T preference represents a common phenomenon observed in the cp genomes of higher plants. These findings highlight the high degree of evolutionary conservation in the cp genomes of the nine Camellia accessions.

Codon usage analysis of coding sequences (CDSs) from the cp genomes of C. petelotii.
Analyses of simple sequence repeats (SSRs) and dispersed repeats
Sequence analysis of the cp genomes of nine Camellia accessions revealed the distribution of dinucleotides to hexanucleotides. Specifically, a total of 16 SSRs were detected in each of the following accessions: Sanyuecha (J100), L.T.Dees (J49), Massee Lane (J51), Xiameng Hualin (J105), Xiameng Wenqing (J106) and Xiameng Xiaoxuan (J108). In contrast, 19 SSRs were identified in C. petelotii (Cpet). Moreover, Kagirohi (J94) and Songzi (J72) contained 17, and 15 SSRs, respectively. The distribution of SSRs was featured by significant heterogeneity, with the highest frequency observed in the LSC region, while the lowest detected in the SSC region. Tetranucleotide repeats was the most prevalent while pentanucleotide repeats were absent. AT/TA repeats were more frequently observed across the genomes. Three SSR types were exclusively identified in C. petelotii (Cept): ATT (12 bp), CTTTTT (18 bp), and AAAAAG (18 bp) (Table 1).
Dispersed repeat analysis was conducted across the nine Camellia accessions and three reference genomes: Camellia reticulata, Camellia oleifera, and Camellia sinensis cv. Wuyi Narcissus. A total of 600 dispersed repeats were identified, encompassing all four types, namely forward, reverse, palindromic and complement. Among them, 270 were palindromic sequences, accounting for 45% of all detected dispersed repeats (Fig. 3A). Length-based categorization revealed five distinct groups: 10–20 bp, 21–30 bp, 31–40 bp, 41–50 bp, and > 50 bp. The 10–20 bp category was the most prevalent, followed by 21–30 bp, with > 50 bp sequences being the least frequent. C. petelotii exhibited a significantly higher number of 10–20 bp repeats compared with other accessions. In contrast, C. reticulata displayed a markedly lower number of 41–50 bp repeat sequences, but a significantly higher number of > 50 bp sequences than the other Camellia accessions (Fig. 3B).

Dispersed repeat analysis. (A) Number of different types of dispersed repeats; (B) Number of dispersed repeats of different lengths.
Collinearity analysis
Collinearity analysis was conducted using the MCScanX tool within the Tbtools software on the nine Camellia genomes and three reference genomes. The results revealed a high degree of similarity among the cp genomes, with no large-scale inversions or rearrangements observed in their genetic structure (Fig. 4). Such a collinear relationship among genome structures and gene sequences demonstrate substantial homologous similarity across these cp genomes.

Collinearity analysis of cp genomes.
Expansion and contraction of the IR region
To investigate structural variations of Camellia cp genomes, a comparative analysis was conducted to evaluate the boundaries between the IR, LSC, and SSC regions of the nine Camellia accessions, combining the three reference genomes. The results revealed that the cp genomes were generally conserved, although some variations in the genome boundaries were observed (Fig. 5). Notably, C. petelotii exhibited a longer IR region and a shorter SSC region compared with other accessions, suggesting a potential expansion of the IR region during its evolutionary history. Except for the reference species C. reticulata and C. oleifera, the rps19 gene in the remaining 10 species spanned both the LSC and IRb regions, with an extension of 46 bp into the IRb. The rpl2 gene was consistently located within the IR region across all accessions. The ndhF gene in Massee lane, C. petelotii, and C. reticulata spanned both the IRb and SSC regions, whereas in the other nine Camellia accessions, the ndhF gene was confined to the SSC region. The ycf1 gene in all the 12 genomes spanned both the SSC and IRa regions, with the portion located within the IRa region showing little variation (ranging from 967 to 1,113 bp). With the exception of C. reticulata, the trnH gene in the remaining 11 accessions was located in the LSC region, with a uniform expansion of 1 bp towards the trnH gene from the IR region.

Comparative analysis of the expansion and contraction of IR regions in the cp genomes across the nine Camellia accessions, incorporating three reference genomes. JLB: junction between LSC and IRb; JSB: junction between IRb and SSC; JSA: junction between SSC and IRa; JLA: junction between IRa and LSC.
Analyses of nucleotide polymorphism (Pi) and selective pressure
To delve deeper into the variations in cp genome sequences across the nine Camellia accessions, nucleotide Pi analysis was conducted (Fig. 6). The results showed high sequence similarity, with Pi values ranging from 0 to 0.01. Five sites with high nucleotide diversity were identified: rpsbK, psbM, ndhJ, ndhF and ndhD. Among them, rpsbK, psbM and ndhJ were situated in the LSC region, while ndhF and ndhD were located in the SSC region.

Nucleotide polymorphism (Pi) analysis of cp genome sequences in the nine Camellia accessions. Five sites with high nucleotide diversity were identified: rpsbK, psbM, ndhJ, ndhF and ndhD.
The evolutionary rates of protein-coding genes in the cp genomes of the nine Camellia accessions were analyzed based on the ratio of non-synonymous substitution rate (dN) to synonymous substitution rate (dS). Ribosomal genes (rpoC1, rpoC2, and rpoB), photosynthesis-related genes (psbC, psaA, atpF, and psbA), and the fatty acid synthesis gene accD, were selected for analysis. Using the pyphy software, ka-Ks values for these genes within the cp genomes were visually represented. We found that the accD gene displayed a notably higher number of sites with dN/dS > 0. This underscores the exceptionally high evolutionary rate of the accD gene and further suggests that the accD-encoded protein may be under positive selective pressure. Moreover, the results revealed a significant increase in the number of sites with dN/dS < 0 for the CDSs of the remaining genes, indicating of purifying selection (Fig. 7).

Selective pressure analysis of eight protein-coding genes in the cp genomes of the nine Camellia accessions.
Phylogenetic analysis
To clarify the phylogenetic relationship of the Camellia genus, a phylogenetic tree was constructed using the cp genomes of 82 accessions from the genus, with Zea mays, Hordeum vulgare , Triticum aestivum , Brachypodium distachyon , Ficus microcarpa, four varieties of A. thaliana, as well as four species of the Diospyros genus included as the outgroup. Seven distinct clusters were identified. Four C. japonica varieties, namely Songzi, Sanyuecha, L.T.Dees, and Kagirohi, clustered with C. Oleifera within the Sect. Oleifera branch, In contrast, Massee Lane was classified under the Sect. Camellia branch. For Xiameng Hualin, Xiameng Wenqing, Xiameng Xiaoxuan, and Camellia petelotii, they grouped together in a clade closed to Camellia tamdaoensis within the Sect. Chrysantha branch. Additionally, a strong phylogenetic affinity was observed between Sect. Paracamellia and Sect. Oleifera. Moreover, Camellia Yunnanensis and Camellia liberistyloides within the Sect. Stereocarpus branch were found to be clustered with Sect. Camellia and Sect. Chrysantha, respectively (Fig. 8).

Phylogenetic tree encompassing 82 accessions from the Camellia genus, along with 13 outgroup accessions. A: Songzi; B: Sanyuecha; C: L.T.Dees; D: Kagirohi; E: Massee Lane;F: Xiameng Wenqing; G: Xiameng Hualin; H: Xiameng Xiaoxuan; I: Camellia petelotii.
Materials and methods
Sample collection, DNA extraction and sequencing
Plants of the nine Camellia accessions were cultivated at the International Flower Technology Innovation Center, Kunming, China. Fresh, healthy leaf samples were collected and immediately frozen in liquid nitrogen on February 12th, 2022. The plant materials used in this study are common species and not classified as endangered. Sampling was conducted in strict accordance with institutional, national, and international regulations. No specific permits were required for the collection of these materials. Species identification was performed by Wenzhong Huang, Huang-Wenzhong Camellia Planting Co., Ltd., Yunnan Province. All plant materials are registered in the Database of International Camellia Register (https://camellia.iflora.cn/). Leaf samples can be obtained upon reasonable request from the International Flower Technology Innovation Center, where the plants are actively growing.
Total genomic DNA was extracted from the samples using cetyltrimethylammonium bromide following a modified protocol13. The quality and concentration of the extracted DNA products were assessed using 1% agarose gel electrophoresis and spectrophotometry. After quality assessment, the DNA was fragmented by ultrasonic treatment, and 270 bp DNA fragments were selected for polymerase chain reaction (PCR) amplification. The PCR products were purified by removing the primer dimers, and the resulting fragments were used to construct the library. The prepared library was sequenced on the Illumina platform (Illumina, USA), generating 150 bp paired-end (PE150) reads.
Cp genome assembly and annotation
Fastp was used to filter out low-quality sequences from raw reads. For each accession, approximately 6 GB of clean data were successfully obtained. The clean data were assembled using the GetOrganelle software14. Leveraging the c p genome sequences of C. reticulate (KJ806278), C. oleifera (OP953554), and C. sinensis cv. Wuyi Narcissus (MT612435) as reference sequences from the National Center for Biotechnology Information (NCBI), we predicted and annotated the cp genome sequences of the nine Camellia accessions with Dual Organellar Genome Annotator (DOGMA)15 and CPGAVAS216. Following the prediction of CDS, rRNA, and tRNA of the cp genomes, we visualized the complete cp genomes of the nine Camellia accessions using Chloroplot17.
Analysis of codon preference and collinearity
Gffread was used to extract CDSs. The codonW software18 was utilized to carry out statistical analysis on the codon preference and to calculate RSCU. The results were visualized with stacked bar charts using the R software (version 4.3.1). For collinearity visualization of the cp genomes, MCScanX19 within the Tbtools software was used.
Analysis of cp genome features
The repeat sequences in the cpDNA of the nine Camellia accessions, including forward, reverse, palindromic and complement repeats were identified using REPtuter20. The parameters for repeat identification were set to a maximum of 50 repeats, with a minimum repeat length of eight nucleotides. SSRs were detected using MISA21 with the minimum repeat numbers set at 10, 5, 4, 3, 3, and 3 for mononucleotide, dinucleotide, trinucleotide, tetranucleotide, pentanucleotide, and hexanucleotide, respectively. The minimum distance between two SSRs was set to 100 bp. Based on the annotated GeneBank files, the local IRscope software22 was used to visualize the boundaries of the IR region and to observe the expansion and contraction of the IR boundary in the cp genome.
Pi and selective pressure analyses
Nucleotide polymorphisms of the complete cp genome sequence of the nine Camellia accessions were calculated using DnaSP software23. The parameters were set as follows: step size = 400 bp, and window length = 600 bp. The Ka/Ks values for each gene were computed using KaKs Calculator software24) and the MLWL method.
Phylogenetic analysis
Gblocks was applied to extract the conserved protein sequences from the cp genomes of 82 accessions of the Camellia genus, which were obtained from the NCBI database, with Zea mays, Hordeum vulgare, Triticum aestivum, Brachypodium distachyon, Ficus microcarpa, and four varieties of A. thaliana, as well as four species of Diospyros included as the outgroup. Orthofinder was then used to identify sequences of single-copy homologous genes. Sequence alignment was conducted by Muscle. Subsequently, an optimal model was predicted using ProtTest software (v.3.4). Ultimately, the RAxML software25 was used to construct a phylogenetic tree based on the JTT + I + G + F model (Bootstrap = 1000). The resulting evolutionary tree was visualized using MEGA (v 11.0.11).
Discussion
This study sequenced and assembled the complete cp genomes of nine Camellia accessions, including Massee Lane, L.T.Dees, Songzi, Kagirohi, Sanyuecha, Xiamen Hualin, Xiamen Wenqing, Xiamen Xiaoxuan, and Camellia petelotii. Furthermore, annotation of these genomes revealed that the cp genomes of the nine accessions exhibited a typical quadripartite structure26. The cp genome lengths of the nine Camellia accessions ranged from 156 to 157 kb, consistent with the reported lengths of cp genomes within the Camellia genus in previous studies1,27,28. After thorough comparison, we found that the primary factor contributing to the variation in the lengths of the cp genomes was in the differences in the LSC region, aligning with findings from a previous study1. The GC content of genomes serves as a crucial metric for assessing phylogenetic relationships and sequence stability across species29. In the cp genomes of the nine Camellia accessions, the average GC content was found to be highest in the two IR regions. This suggests that the IR region may be the most conserved segment of the cp genomes, likely due to the presence of rRNA genes in the region. In this study, the cp genomes of the nine Camellia accessions were annotated, identifying 87 protein-coding genes and 8 rRNA genes. No rearrangements or deletions were observed. Notably, in many angiosperm lineages, genes such as infA and rps16 are frequently lost from the cp genome11,30, but these genes were retained in the cp genomes of our study.
Repetitive sequences, which carry significant genetic information and are closely associated with sequence variation, tend to be more abundant in species with greater evolutionary complexity31,32. In comparison to the Gramineae (132 bp) and Leguminosae (287 bp) families7, the repeat sequences within the cp genomes of the nine Camellia accessions exhibited a comparatively shorter length (ranging from 10 to 50 bp), with very few sequences exceeding 50 bp. Collectively, these observations suggest that the evolution of cp genomes in the Camellia genus may be relatively slow and conservative, which is consistent with a previous report6. The contraction and expansion of the IR region significantly influences the length of cp genome33, and play crucial roles in cp evolution34. In this study, genes located near the IR boundary (rps19, rpl2, ndhF, ycf1, and trnH) were analyzed. The results revealed that in Massee Lane, C. petelotii, and C. reticulata, the ndhF gene spanned both the IRb and SSC regions. These three accessions exhibited position shifts of the ndhF gene, as well as length variation, while the other nine accessions showed minimal differences in the distance between the ndhF gene and the IR boundary. We propose that the IR/LSC boundary was more conserved than the IR/SSC boundary, which holds significant implications for the study of IR region boundaries, offering valuable insights for phylogenetic analysis and species identification.
SSR analysis identified five types of nucleotide repeats in the cp genomes of the nine Camellia accessions. Notably, no pentanucleotide repeats were detected, and hexanucleotide repeats were only found in C. petelotii. The remaining four repeat units were consistently observed across all nine accessions. This finding aligned with previous research, which indicates the rare occurrence of pentanucleotide and hexanucleotide repeats within cp genomes35. A/T nucleotide repeats are a common feature in most angiosperms36. In this study, the most prevalent SSRs were mononucleotide repeats, all consisting of A/T, with a notable A/T base preference also observed in di- to hexanucleotide repeats. Codon usage analysis further revealed that nearly all codons with an RSCU value > 1 terminated with A/T. This suggests that the A/T preference is a crucial evolutionary characteristics of the cp genome. The high proportion of A/T repeats may be one of the factors contributing to the elevated A/T content observed in cp genomes37. Accurate species identification in the Camellia genus based on morphological traits is often difficult. However, DNA barcoding techniques using cp genomes have become increasingly effective for species identification in recent years38. For example, the rpl16 and psbA-tmH sequences have been shown to reliably identify Camellia pubipetala39, while rbcL, matK and ycf1 have been used as DNA barcodes for other plant species40. In our study, Pi analysis identified five highly variable nucleotide sites: rpsbK, psbM, ndhJ, ndhF and ndhD. These regions possessed strong potential as DNA barcodes and molecular markers. Furthermore, previous research has demonstrated that the ndhF gene in the genus Rheum contains three positively selected amino acid sites, which are likely associated with environmental adaptation41.
Phylogenetic analysis revealed that Songzi, Sanyuecha, L.T.Dees, Kagirohi were closely related to Sect. Camellia and Sect. Oleifera. Massee Lane was found to be clustered with the Sect. Camellia branch, while the other four Camellia accessions Xiameng Hualin, Xiameng Wenqing, Xiameng Xiaoxuan, Camellia petelotii were classified into the same branch, close to Camellia tamdaoensis within the Sect. Chrysantha branch. This analysis elucidates the phylogenetic positions of the nine Camellia accessions, and provides a theoretical foundation for investigating the genetic relationships among Camellia species. The taxonomic position of C. yunnanensis has long been a subject of debate. In the classification systems of Hongda Zhang42 and Tianlu Min43, C. yunnanensis has been placed in Sect. Stereocarpus and Sect. Heterogenea, respectively. Our results showed that C. yunnanensis was under Sect. Stereocarpus, which is consistent with the classification of Hongda Zhang. It clustered with Sect. Camellia, suggesting a potentially close phylogenetic relationships between these sections. Meanwhile, we also discovered that Sect. Theopsis clustered with two species within Sect. Eriandria, which is consistent with previous research44,45. Based on previous findings and the results of the current study, we propose that Sect. Eriandria and Sect. Theopsis form a monophyletic branch. A significant body of literature has suggested that Sect. Chrysantha may represent a paraphyletic or polyphyletic group, raising questions about its validity as a distinct taxonomic unit8,46. Li et al. found that species of Sect. Camellia are predominantly distributed across two branches1. Our findings indicate that Sect. Chrysantha and Sect. Camellia may not form a monophyletic group. Additionally, Wu et al. reported a close relationship between Sect. Camellia and Sect. Oleifera4. The combination of Sect. Paracamellia and Sect. Oleifera has long been a contentious issue in the classification of Camellia. The Hongda Zhang classification system treats these two groups as distinct entities42. Jiang et al. have found significant differences in flower traits between C. Oleifera and the C. Breviflora47. Shen et al. have also clearly distinguished C. Oleifera and C. Breviflora using FTIR technology48. Tianlu Min demonstrated that Sect. Oleifera should be merged with Sect. Paracamellia49. The results of this study showed that Sect. Oleifera and Sect. Paracamellia were closely related. Therefore, we supports the merge of Sect. Paracamellia and Sect. Oleifera, which is consistent with findings from multiple studies4,38,50.
Data availability
The datasets generated during and/or analysed during the current study are available from GenBase repository under the project PRJCA030917.
References
Li, L., Hu, Y., He, M., Zhang, B. & Hong, Y. Comparative chloroplast genomes: Insights into the evolution of the chloroplast genome of Camellia sinensis and the phylogeny of Camellia. BMC Genom. 22(1), 138 (2021).
Zhang, T., Qiu, F., Chen, L., Liu, R. & Wang, X. Identification and in vitro anti-inflammatory activity of different forms of phenolic compounds in Camellia oleifera oil. Food Chem. 344(49), 128660 (2020).
Zhou, L. et al. Optimization of the extraction of polysaccharides from the shells of Camellia oleifera and evaluation on the antioxidant potential in vitro and in vivo. J. Funct. Foods 86, 104678 (2021).
Wu, Q. et al. Comparative transcriptomic analysis unveils the deep phylogeny and secondary metabolite evolution of 116 Camellia plants. Plant J. Cell Mol. Biol. 111(2), 406–421 (2022).
Chunmei, C., Chunlei, M. A., Jianqiang, M. A., Shengchuan, L. & Liang, C. Sequencing of chloroplast genome of Camellia sinensis and genetic relationship for camellia plants based on chloroplast DNA sequences. J. Tea Sci., 34(4), 371–380 (2014).
Jun-Bo, Y. et al. Comparative chloroplast genomes of Camellia Species. PLoS ONE 8(8), e73053 (2013).
Huang, H., Shi, C., Liu, Y., Mao, S. Y. & Gao, L. Z. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Biol. 14(1), 151–151 (2014).
Wei, F., Jun-Bo, Y., Shi-Xiong, Y. & Li, D.-Z. Phylogeny of Camellia sects. Longipedicellata, chrysantha and longissima (theaceae) based on sequence data of four chloroplast DNA loci. Acta Botanica Yunnanica 32(1), 1–13 (2010).
Daniell, H., Lin, C. S., Yu, M. & Chang, W. J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 17(1), 1–29 (2016).
Xing, S. C. & Liu, C. J. Progress in chloroplast genome analysis. Prog. Biochem. Biophys. 35(1), 21–28 (2008).
Ling, W., Wen-Pan, D. & Shi-Liang, Z. Structural mutations and reorganizations in chloroplast genomes of flowering plants. Acta Bot. Boreal. Occident. Sin. 32(6), 1282–1288 (2012).
Gu, L., Su, T., An, M. T. & Hu, G. X. The complete chloroplast genome of the vulnerable Oreocharis esquirolii (Gesneriaceae): Structural features, comparative and phylogenetic analysis. Plants 9(12), 1692 (2020).
Allen, G. C., Flores-Vergara, M., Krasynanski, S., Kumar, S. & Thompson, W. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nature Protoc. 1(5), 2320–2325 (2006).
Jin, J.-J. et al. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 1–31 (2020).
Wyman, S., Jansen, R. & Boore, J. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20(17), 3252–3255 (2004).
Linchun, S. et al. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucl. Acids Res. 47(W1), W1 (2019).
Zheng, S., Poczai, P., Hyvnen, J., Tang, J. & Amiryousefi, A. Chloroplot: An online program for the versatile plotting of organelle genomes. Front. Genet. 11, 576124 (2020).
Ingvarsson, P. K. Gene expression and protein length influence codon usage and rates of sequence evolution in Populus tremula. Mol. Biol. Evol. 24(3), 836 (2007).
Yupeng, W. et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucl. Acids Res. 40(7), e49–e49 (2012).
Kurtz, S. et al. REPuter: The Manifold Applications of Repeat Analysis on a Genomic Scale Vol. 22 (Oxford University Press, Oxford, 2001).
Sebastian, B., Thomas, T., Thomas, M., Uwe, S. & Martin, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 16, 2583 (2017).
Amiryousefi, A., Hyvonen, J. & Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34(17), 3030–3031 (2018).
Librado, P. & Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25(11), 1451–1452 (2009).
Zhang, Z. et al. KaKs_Calculator: Calculating Ka and Ks through model selection and model averaging. 基因组蛋白质组与生物信息学报: 英文版 4(4), 259–263 (2006).
Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics (Oxford, England), 1312–1313 (2014).
Wicke, S., Schneeweiss, G. M., Depamphilis, C. W., Müller, K. F. & Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Molecular Biology 76(3–5), 273–297 (2011).
Ping, L. et al. Comparative genomic analysis uncovers the chloroplast genome variation and phylogenetic relationships of Camellia Species. Biomolecules 12(10), 1474–1474 (2022).
Yu, X. Q. et al. Insights into the historical assembly of East Asian subtropical evergreen broadleaved forests revealed by the temporal history of the tea family. New Phytol. 215(3), 1235–1248 (2017).
Wu, F. et al. Genome-wide analysis of DNA G-quadruplex motifs across 37 species provides insights into G4 evolution. Commun. Biol. 4(1), 98 (2021).
Gang, Z. et al. Comparative analyses of chloroplast genomes from 13 Lagerstroemia (Lythraceae) species: Identification of highly divergent regions and inference of phylogenetic relationships. Plant Mol. Biol. 102(6), 659–676 (2020).
Shu-Fen, L. et al. Chromosome evolution in connection with repetitive sequences and epigenetics in plants. Genes 8(10), 290 (2017).
Goon-Bo, K. et al. Comparative chloroplast genome analysis of Artemisia (Asteraceae) in East Asia: Insights into evolutionary divergence and phylogenomic implications. BMC Genom. 21(1), 415 (2020).
Alexandre, M. & Normand, B. Recombination and the maintenance of plant organelle genome stability. New Phytol. 186(2), 299–317 (2010).
Wang, W., Chen, S. & Zhang, X. Whole-genome comparison reveals divergent IR borders and mutation hotspots in chloroplast genomes of herbaceous bamboos (Bambusoideae: Olyreae). Molecules 23(7), 1537–1537 (2018).
Meixiu, Y., Shujie, D., Qiuyi, G., Qin, X. & Yuqing, G. Comparative chloroplast genome analysis of four Polygonatum species insights into DNA barcoding, evolution, and phylogeny. Sci. Rep. 13(1), 16495–16495 (2023).
Dong, W., Xu, C., Wen, J. & Zhou, S. Evolutionary directions of single nucleotide substitutions and structural mutations in the chloroplast genomes of the family Calycanthaceae. BMC Evolutionary Biology 20, 1–12 (2020).
Xiaojun, N. et al. Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). Plos One 7(5), e36869 (2012).
Bei-Bei, W. et al. DNA barcodes for identification of closely related species in Camellia. Guangdong Agric. Sci. 44(1), 55–56 (2017).
Li, D. M. et al. Thirteen complete chloroplast genomes of the costaceae family: Insights into genome structure, selective pressure and phylogenetic relationships. BMC Genom. 25(1), 68–68 (2024).
Li, W., Zhang, C., Guo, X., Liu, Q. & Wang, K. Complete chloroplast genome of Camellia japonica genome structures, comparative and phylogenetic analysis. PLoS ONE 14(5), e0216645 (2019).
Zhao, B., Li, J., Yuan, R. & Mao, S. Adaptive evolution of the rbcL gene in the genus Rheum (Polygonaceae). Biotechnol. Biotechnol. Equip. 31(3), 493–498 (2017).
Chang, H. T. A taxonomy of the genus Camellia. J. Sun. Yatsen. Univ. 1, 68–69 (1981).
Ming, T. A revision of Camellia sect. Thea. Acta Botanica Yunnanica 14(2), 115–132 (1992).
Vijayan, K., Zhang, W. J. & Tsou, C. H. Molecular taxonomy of Camellia (Theaceae) inferred from nrITS sequences. Am. J. Bot. 96(7), 1348–1360 (2009).
Xiao, T. J. & Parks, C. R. Molecular analysis of the genus Camellia (2002).
Sha, T., Qian-Hui, Z., Yong-Fang, H., Wen-Hong, H. & Ke-Na, X. Pollen morphology of three species in sect. Chrysantha studied by scanning electron microscope. Guihaia (2016).
Jiang, W. et al. Floral morphology resolves the taxonomy of Camellia L. (Theaceae) sect. Oleifera and sect. Paracamellia. Bangladesh J. Plant Taxon. 19(2), 155–165 (2012).
Shen, J. B., Lu, H. F., Peng, Q. F., Zheng, J. F. & Tian, Y. M. FTIR spectra of Camellia sect. Oleifera, sect. Paracamellia, and sect. Camellia (Theaceae) with reference to their taxonomic significance. J. Syst. Evol. 46(2), 194–204 (2008).
Ming, T. L. The classification, differentiation and distribution of the genus Camellia sect Camellia. Acta Bot. Yunnanica 20(4–5), 127–148 (1998).
Feng-Ying, L. I., Yu-Guo, W. & Shao-Qing, T. Characters of leaf epidermis in section chrysantha series chrysantha (Theaceae, Camellia) and their systematic significance. J. Guangxi Normal Univ. (Nat. Sci.) 19, 75–79 (2001).
Funding
This work was supported by the National Natural Science Foundation of China (32260413), Science and Technology Talent and Platform Plan in Yunnan Province (YNWR-QNBJ-2019-292), International Science and Technology Cooperation Base (GHJD-2021024), Innovation Guidance and Technology Oriented Enterprise Cultivation Plan (202404BI090009), and China Agriculture Research System of MOF and MARA (CARS-23-G56).
Author information
Authors and Affiliations
Contributions
YFC: conception, analysis, writing-original draft. MT: conception, acquisition, writing-original draft. YJY: analysis, interpretation of data. ZMS: acquisition, analysis. PFZ: conception, writing-reviewing. JHW: writing-reviewing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cai, Y., Tian, M., Yang, Y. et al. Nine complete chloroplast genomes of the Camellia genus provide insights into evolutionary relationships and species differentiation. Sci Rep 15, 8783 (2025). https://doi.org/10.1038/s41598-025-87764-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-87764-4
