
Abstract
Diabetic retinopathy (DR) is a common complication of diabetes mellitus, characterized by progressive neurodegeneration and vision impairment. The Ca2+/calmodulin-dependent protein kinase II alpha (CaMK2A) and cAMP response element-binding protein (CREB) signaling pathway has been implicated in various neurological disorders. However, its role in DR pathogenesis remains elusive. We established a DR mouse model by streptozotocin administration and performed histological, biochemical, and molecular analyses to investigate the involvement of CaMK2A/CREB signaling and its interplay with mitophagy. Additionally, we employed in vitro high-glucose (HG) treatment in primary mouse retinal ganglion cells to dissect the underlying mechanisms. Pharmacological and genetic modulations were utilized to target CaMK2A/CREB pathway and mitophagy. In the DR model, we observed retinal degeneration, increased apoptosis, and reduced neurotransmitter production, accompanied by enhanced mitophagy and activation of the CaMK2A/CREB pathway. HG induction in retinal ganglion cells recapitulated these findings, and autophagy inhibition partially rescued cell death but failed to suppress CaMK2A/CREB activation, suggesting mitophagy as a downstream consequence. CaMK2A knockdown or CREB phosphorylation inhibition attenuated HG-induced mitophagy, apoptosis, and neurotransmitter depletion, while CREB activation exacerbated these effects. CaMK2A silencing mitigated DR progression, oxidative stress, inflammation, and neuronal loss, akin to dopamine/carbidopa administration in DR mouse model. Our findings reveal the involvement of CaMK2A/CREB signaling activation and enhanced mitophagy in DR, suggesting these pathways may be therapeutically relevant targets for DR management.
Similar content being viewed by others


Introduction
Diabetic retinopathy (DR) is a microvascular complication of diabetes mellitus and a leading cause of vision loss and blindness worldwide. The pathogenesis of DR involves progressive neurodegeneration, characterized by the loss of retinal neurons, particularly dopaminergic neurons, which play a crucial role in regulating retinal function and vision processing1,2. The degeneration of dopaminergic neurons in DR is associated with several risk factors, including hyperglycemia, oxidative stress, inflammation, and mitochondrial dysfunction3,4. Current treatment strategies for DR primarily focus on managing hyperglycemia and targeting vascular abnormalities, but there is a need for neuroprotective therapies that can preserve retinal neuronal function5,6.
The Ca2+/calmodulin-dependent protein kinase II alpha (CaMK2A) and cAMP response element-binding protein (CREB) signaling pathway has been implicated in various neurological disorders, including Alzheimer’s disease, Parkinson’s disease, and Schizophrenia7,8. CaMK2A is a multifunctional serine/threonine kinase that plays a crucial role in regulating neuronal signaling, plasticity, and survival9. Upon activation, CaMK2A can phosphorylate and activate CREB, a transcription factor that regulates the expression of genes involved in neuronal survival, synaptic plasticity, and neurogenesis10,11. The dysregulation of CaMK2A/CREB signaling by calcium imbalance has been implicated in various neurodegenerative processes, such as oxidative stress, inflammation, and mitochondrial dysfunction12,13.
Mitochondria are essential organelles responsible for energy production, calcium homeostasis, and apoptosis regulation in retinal neurons14. Mitochondrial dysfunction and impaired mitochondrial quality control mechanisms, such as autophagy and mitophagy, have been implicated in the pathogenesis of DR15,16. Autophagy is a cellular process that facilitates the degradation and recycling of damaged or dysfunctional cellular components, including mitochondria (mitophagy)17. While moderate levels of autophagy and mitophagy are essential for maintaining cellular homeostasis, excessive or dysregulated mitophagy can lead to the excessive degradation of mitochondria, compromising energy production and promoting neuronal cell death18,19. Studies have shown that excessive mitophagy and autophagy can contribute to the degeneration of dopaminergic neurons in neurodegenerative diseases, such as Parkinson’s disease20,21.
The present study aimed to investigate the role of the CaMK2A/CREB signaling pathway in the pathogenesis of DR, with a particular focus on its interplay with mitophagy and neuronal degeneration. We hypothesized that dysregulated CaMK2A/CREB signaling contributes to excessive mitophagy and subsequent neuronal cell death in DR. Using a combination of in vivo and in vitro models, we explored the potential of targeting CaMK2A/CREB signaling as a therapeutic strategy for preserving retinal neuronal function and mitigating the progression of DR. Our findings offer valuable insights into the molecular mechanisms underlying DR pathogenesis and contribute to the development of novel neuroprotective therapies for this debilitating condition.
Materials and methods
Animals and DR model induction
Male C57BL/6J mice (8–10 weeks old, Beijing HFK Bioscience Co., Ltd. (Beijing, China.) were housed in a temperature-controlled environment (22 ± 2 °C) with a 12-hour light/dark cycle and free access to food and water. All animal experiments were conducted with the approval of Institutional Animal Care and Use Committee Ethical Review Committee for Animal Experiments of Kunming Medical University (Approval number: kmmu20241516). And all methods were performed in accordance with the relevant guidelines and regulations. The DR model was induced by intraperitoneal injection of streptozotocin (STZ, 60 mg/kg body weight, Sigma-Aldrich, St. Louis, MO, USA) dissolved in citrate buffer (pH 4.5) for five consecutive days. Age and sex-matched mice injected with citrate buffer served as the control group. Mice with blood glucose levels > 16.7 mmol/L for five consecutive days were considered to develop diabetes. For the dopamine/carbidopa group, diabetic mice received daily intraperitoneal injections of dopamine hydrochloride (10 mg/kg/day, Sigma-Aldrich) and carbidopa (2.5 mg/kg/day, Sigma-Aldrich) dissolved in saline for 4 weeks starting from the onset of diabetes. Carbidopa was co-administered with dopamine to inhibit peripheral dopamine decarboxylation and enhance its bioavailability in the retinal tissues. Lentiviral particles carrying CaMK2A shRNA (sh-CaMK2A) or control shRNA (sh-NC) were obtained from Cyagen Biotech (Guangzhou, China). For in vivo transduction, lentiviral particles (1 × 10^8 transducing units per week) were injected intravitreally into the eyes of DR mice. 4 weeks after the onset of diabetes and intervention, the mice were sacrificed by cervical dislocation and the retinal tissues were collected for histological analysis using Hematoxylin and Eosin Staining Kit (Beyotime, Beijing, China) and molecular examination.
Cell culture and treatments
Primary mouse retinal ganglion cells (RGCs) (PRI-MOU-00085, Shanghai Zhongqiao Xinzhou Biotechnology Co., Ltd., Shanghai, China) were maintained in specialized culture medium (PCM-M-85, Shanghai Zhongqiao Xinzhou Biotechnology Co., Ltd.) supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin-streptomycin (Invitrogen, Carlsbad, CA, USA). Cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2. The primary RGCs were isolated using a two-step immunopanning procedure and characterized by expression of RGC-specific markers (Brn3a, RBPMS, and Thy1.2) by the supplier. For high-glucose (HG) treatment, cells were exposed to 30 mM D-glucose (Sigma-Aldrich) for 48 h. The autophagy inhibitor 3-methyladenine (3-MA, 5 mM, Sigma-Aldrich) was used to inhibit autophagy. The CREB activator forskolin (10 µM, Sigma-Aldrich) and the CREB phosphorylation inhibitor 666 − 15 (10 µM, Sigma-Aldrich) were used to modulate CREB activity.
SiRNA transfection
CaMK2A siRNA (siCaMK2A, Santa Cruz Biotechnology, Dallas, TX, USA) and control siRNA (siControl, Santa Cruz Biotechnology) were transfected into RGCs using Lipofectamine RNAiMAX Reagent (Invitrogen) according to the manufacturer’s instructions. Cells were incubated with the siRNA complexes for 48 h before subsequent experiments.
Transmission electron microscopy (TEM)
Retinal tissues or RGCs were fixed in 2.5% glutaraldehyde, post-fixed in 1% osmium tetroxide, dehydrated, and embedded in Epon resin. Ultrathin Sect. (70 nm) were stained with uranyl acetate and lead citrate, and examined using a Hitachi H-7500 transmission electron microscope (Hitachi, Tokyo, Japan).
Western blotting
Retinal tissues (50 mg) or RGCs (1 × 10^6) were lysed in RIPA buffer (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with protease and phosphatase inhibitors (Roche, Basel, Switzerland). Proteins were separated by SDS-PAGE and transferred onto PVDF membranes (Millipore, Burlington, MA, USA). After blocking with 5% non-fat milk, membranes were incubated with primary antibodies against LC3BII (2775, 1:1000, Cell Signaling Technology), Parkin (ab77924, 1:1000, Abcam), PINK1 (ab23707, 1:1000, Abcam), Drp1 (8570, 1:1000, Cell Signaling Technology), Mfn2 (9482, 1:1000, Cell Signaling Technology), CaMK2A (3357, 1:1000, Cell Signaling Technology), CREB (9197, 1:1000, Cell Signaling Technology), phospho-CREB (9198, 1:1000, Cell Signaling Technology), and β-actin (A5441, 1:5000, Sigma-Aldrich) overnight at 4 °C for 16 h. Corresponding HRP-conjugated secondary antibodies (1:5000, anti-rabbit IgG (31460) and anti-mouse IgG (31430); Thermo Fisher Scientific) were used to detect the primary antibodies, and signals were detected using an enhanced chemiluminescence system (Thermo Fisher Scientific).
Enzyme-linked immunosorbent assay (ELISA)
Dopamine and 3,4-dihydroxyphenylacetic acid (DOPAC) levels in retinal tissues or cell culture supernatants were measured using commercially available ELISA kits from Abcam (Dopamine ELISA Kit, ab285238) and AFG Bioscience (DOPAC ELISA Kit, EK247749) according to the manufacturer’s instructions. The levels of interleukin-1β (IL-1β) (DY401), tumor necrosis factor-α (TNF-α) (DY410), and interleukin-6 (IL-6) (DY406) in retinal tissues were quantified using mouse-specific ELISA kits from R&D Systems (Minneapolis, MN, USA). 100 µL lysate from 50 mg tissues or 1 × 10^6 cells was used for measurement, and the relative concentration of each analyte was determined based on the linear regression of the standards included in each kit.
Reactive oxygen species (ROS) measurement
Oxidative stress was determined in tissue lysate by measuring ROS concentration (R0105, Jiancheng Bioengineering Institute, Nanjing, China). 100 µL lysate from 50 mg tissues was stained with 10 µM 2’,7’-Dichlorodihydrofluorescin diacetate for 45 min at 37 °C in the dark. The fluorescence intensity was assessed using a fluorescent spectrophotometer at an oscillation frequency wavelength of 485/530 nm.
Apoptosis detection
For retinal tissue analysis, the retinas were dissected, minced, and incubated with Accutase (Sigma-Aldrich) at 37 °C for 30 min with gentle shaking to dissociate into single-cell suspensions. The cell suspensions were filtered through a 40-µm cell strainer (Falcon, Durham, NC, USA) and centrifuged at 300 x g for 5 min. Cells from retinal tissues or cultured RGCs (1 × 10^6) in 1 mL staining buffer were then mixed with 5 µL Annexin V-FITC and 1 µL propidium iodide (PI) from an Annexin V-FITC Apoptosis Detection Kit (C1062M, Beyotime). After 15-minute staining, flow cytometry analysis was performed using a BD FACSAria III flow cytometer (BD Biosciences), and data were analyzed using FlowJo software (FlowJo LLC, Ashland, OR, USA).
Flow cytometry analysis of retinal cell populations
For microglial cell analysis, retinal tissues were dissociated into single-cell suspensions as described above. The cells were incubated with an Fc blocker (anti-mouse CD16/CD32, 553142, BD Biosciences) for 15 min at room temperature, followed by staining with anti-mouse Iba1-FITC (019-19741, Wako, Richmond, VA, USA) and anti-mouse CD68-APC (137801, BioLegend, San Diego, CA, USA) antibodies for 30 min at 4 °C. For retinal ganglion cell analysis, the dissociated retinal cells were stained with anti-mouse Brn-3a-FITC (sc-8429, Santa Cruz Biotechnology, Dallas, TX, USA) and anti-mouse ISL1-APC (ab109517, Abcam) antibodies. For retinal bipolar cell analysis, the cells were stained with anti-mouse PKCα-FITC (sc-8393, Santa Cruz Biotechnology) and anti-mouse CD15-APC (130-119-669, Miltenyi Biotec, Bergisch Gladbach, Germany) antibodies. After staining, the cells were washed, resuspended in PBS, and analyzed by flow cytometry using a BD FACSAria III flow cytometer (BD Biosciences).
EdU incorporation assay
Cell proliferation was assessed using the Click-iT EdU Alexa Fluor 594 Imaging Kit (Invitrogen) according to the manufacturer’s instructions. Cells were incubated with 10 µM EdU for 2 h, fixed, permeabilized, and stained with the Click-iT reaction cocktail for 1 h. After washing, EdU-positive cells were quantified using a fluorescence microscope (Nikon, Tokyo, Japan).
CREB transcription activity assay
The transcriptional activity of CREB was measured using the CREB Transcription Factor Assay Kit (ab207197, Abcam, Cambridge, MA, USA) following the manufacturer’s protocol. Briefly, nuclear extracts were prepared from retinal tissues or RGCs (1 × 10^6 cells) using the Nuclear Extraction Kit (ab113474, Abcam). The nuclear extracts were incubated with an immobilized oligonucleotide containing the CREB response element, allowing CREB binding. After washing, a CREB primary antibody was added, followed by the incubation with an HRP-conjugated secondary antibody. The colorimetric signal was quantified using a microplate reader (BioTek Instruments, Winooski, VT, USA) at 450 nm.
Statistical analysis
Data are presented as mean ± standard deviation (SD). Statistical analyses were performed using GraphPad Prism 8.0 (GraphPad Software, San Diego, CA, USA). One-way or two-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test was used for multiple comparisons. A p-value < 0.05 was considered statistically significant.
Results
Diabetic retinopathy (DR) is associated with enhanced mitophagy and the activation of CaMK2A/CREB pathway
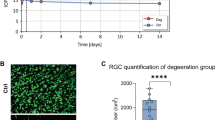
To study the potential involvement of CaMKII/CREB signaling in DR, we established a DR mouse model by STZ administration. After the confirmation of the onset of diabetes (blood glucose > 16.7mmol/L) for 4 weeks, the mice were sacrificed for retinal tissue collection. H&E staining revealed degeneration of retinal cells in different layers in the DR model group (Fig. 1A). Quantification of retinal thickness indicated the occurrence of retinopathy in the model group (Fig. 1B). Furthermore, apoptosis analysis demonstrated an increase of cell death in retinal tissues of the model group (Fig. 1C). This was accompanied by an reduced production of neurotransmitters (Dopamine and DOPAC) in DR tissues, as revealed by ELISA quantification (Fig. 1D). The above data confirmed the successful induction of DR model.

Morphological and histological changes in the diabetic retinopathy (DR) model. (A) H&E staining showing degeneration of retinal cells in the DR model group. (B) Quantification of retinal thickness indicating retinopathy in the model group. (C) Apoptosis analysis demonstrating increased cell death in retinal tissues of the model group. (D) ELISA quantification revealing reduced neurotransmitter production (Dopamine and DOPAC) in DR tissues. (E) Transmission electron microscopy (TEM) analysis showing engulfment of mitochondria by autophagosome membranes in DR tissues. (F) Protein expression levels of mitophagy markers (LCBII, Parkin, PINK1) and Drp1 in DR tissues. (G) Protein levels of CaMK2A and phosphorylated CREB in DR tissues. N=6 animals in each group. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Transmission electron microscopy (TEM) analysis showed that mitochondria tended to be engulfed by autophagosome membranes in DR tissues (Fig. 1E). Consistently, proteins related to mitophagy (LCBII, Parkin, PINK1) showed an increased expression in DR tissues. Besides, Drp1 (dynamin-related protein 1, a key mediator of mitochondrial fission) was also upregulated in DR tissues (Fig. 1F). The protein levels of Mfn (mitofusin 2, a key regulator of mitochondrial fusion) were comparable between the control and DR model group. We further examined the protein levels of CaMK2A and CREB, which revealed increased protein level of CaMK2A and enhanced phosphorylation of CREB in DR tissues (Fig. 1G). These data prompted us to further dissect the role of CaMK2A/CREB pathway in DR pathogenesis and its interplay with mitophagy.
High-glucose (HG) induces mitophagy in retinal ganglion cells and promotes the activation of CaMK2A/CREB pathway
Next, we applied high glucose (HG) in retinal ganglion cells (RGCs) since it was reported that HG can induces autophagy in neurons22,23. An autophagy inhibitor (3-MA) was applied together with HG induction as a potential rescue group. TEM showed that HG-induced mitophagy was suppressed by 3-MA (Fig. 2A). Furthermore, HG induced the upregulation of mitophagy markers (LCBII, Parkin, PINK1) and Drp1 protein. These effects were attenuated by 3-MA (Fig. 2B). EdU incorporation assay showed that HG induction inhibited cell proliferation and autophagy inhibition partially rescued this effect (Fig. 2C). Autophagy inhibition by 3-MA also attenuated HG-induced cell death in RGCs (Fig. 2D). However, we found that autophagy inhibition failed to suppress the increased expression of CaMK2A and CREB phosphorylation upon HG induction (Fig. 2E). Further, we conducted a CREB transcription activity assay, which also showed that autophagy inhibitor did not reduce CREB-dependent DNA binding under HG condition (Fig. 2F). These data suggest that the observed mitophagy may be a downstream consequence of CaMK2A/CREB pathway. Nevertheless, the rescue of cell death by 2-MA also promoted the production of dopamine and DOPAC in RGCs after HG induction (Fig. 2G).

High-glucose (HG) induction of mitophagy and its modulation by autophagy inhibition in RGCs. (A) TEM analysis demonstrating HG-induced mitophagy and its suppression by 3-MA, an autophagy inhibitor. (B) Protein expression levels of mitophagy markers (LCBII, Parkin, PINK1), Mfn2, and Drp1 in HG-induced RGCs with or without 3-MA treatment. (C) EdU incorporation assay showing HG-induced inhibition of cell proliferation and its partial rescue by autophagy inhibition. (D) Cell death analysis indicating the attenuation of HG-induced cell death by 3-MA. (E) Protein expression levels of CaMK2A and phosphorylated CREB in HG-induced RGCs with or without 3-MA treatment. (F) CREB transcription activity assay demonstrating no reduction in CREB-dependent DNA binding under HG condition in the presence of autophagy inhibitor. (G) Dopamine and DOPAC production in RGCs after HG induction with or without 3-MA treatment. N=3 independent experiments. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
CaMK2A knockdown reduces HG-induced CREB phosphorylation and mitohagy in RGCs
We next applied siRNA targeting CaMK2A to suppress the CaMK2A expression under HG condition. We also included a group with forskolin treatment (a chemical activator for cAMP production and CREB phosphorylation). We found that CaMK2A silencing reduced CREB phosphorylation, which could be re-activted by forskolin (Fig. 3A). Forskolin also increased the transcription activity of CREB after CaMK2A silencing (Fig. 3B). These data indicate CaMK2A is a positive regulator of CREB phosphorylation. Further, CaMK2A silencing suppressed the upregulation of mitophagy markers (LCBII, Parkin, PINK1) and Drp1 protein caused by HG induction. These effects was reversed by forskolin (Fig. 3C). TEM analysis showed that CaMK2A knockdown inhibited HG-induced mitophagy, and re-activation of CREB by forskolin promoted mitophagy (Fig. 3D). EdU assay demonstrated that CaMK2A silencing partially rescued cell division under HG condition, and forskolin treatment abrogated this effect (Fig. 3E). Forskolin treatment also abolished the rescue effect of CaMK2A silencing on apoptosis (Fig. 3F) and dopamine/DOPAC production (Fig. 3G) in HG-induced RGCs. These findings indicate that CaMK2A-mediated CREB activation contributes to HG-induced mitophagy and neuronal cell death.

Effects of CaMK2A knockdown on HG-induced CREB phosphorylation and mitophagy in RGCs. (A) Protein expression levels of CaMK2A, phosphorylated CREB, and total CREB in HG-induced RGCs with or without CaMK2A siRNA and forskolin treatment. (B) CREB transcription activity assay showing the influence of CaMK2A silencing and forskolin treatment on CREB-dependent DNA binding. (C) Protein expression levels of mitophagy markers (LCBII, Parkin, PINK1), Mfn2, and Drp1 in HG-induced RGCs with or without CaMK2A knockdown and forskolin treatment. (D) TEM analysis demonstrating the effect of CaMK2A knockdown and forskolin treatment on HG-induced mitophagy. (E) EdU incorporation assay indicating the rescue of cell division by CaMK2A silencing under HG condition and its abrogation by forskolin treatment. (F) Apoptosis analysis showing the protective effect of CaMK2A knockdown and its reversal by forskolin treatment. (G) Dopamine and DOPAC production in RGCs after HG induction with or without CaMK2A knockdown and forskolin treatment. N=3 independent experiments. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Inhibiting CREB phosphorylation shows rescuing effect on HG-induced mitophagy and cell death
To further show the critical role of CREB activation in HG-induced neuronal defect, we applied a CREB activator (forskolin) and a CREB phosphorylation inhibitor (666 − 15). Under HG induction condition, forskolin further increased CREB phosphorylation and 666 − 15 showed the opposite effect. The protein levels of CaMK2A was not affected by these chemicals (Fig. 4A). Consistently, CREB transcription activity was enhanced by forskolin and reduced by 666 − 15, respectively (Fig. 4B). Immunoblotting analysis showed that forskolin treatment enhanced the upregulation of mitophagy markers (LCBII, Parkin, PINK1) and Drp1 protein caused by HG induction, while 666 − 15 treatment suppressed their upregulation (Fig. 4C). Meanwhile, we observed augmented mitophagy in HG + forskolin condition, and 666 − 15 attenuated HG-induced mitophagy (Fig. 4D). EdU assay demonstrated that forskolin treatment further arrested cell division under HG condition, and 666 − 15 showed a rescue effect (Fig. 4E). Similar results were observed regarding the apoptosis induction, with forskolin showing aggravating effect and 666 − 15 showing protective effect (Fig. 4F). Moreover, the reduced cell survival by forskolin treatment and enhanced survival by 666 − 15 were associated with the reduced and increased production of dopamine and DOPAC, respectivey (Fig. 4G). Therefore, CREB-dependent transcription mediates HG-induced mitophagy and neuronal cell death.

Modulation of HG-induced mitophagy and cell death by CREB activation and phosphorylation. (A) Protein expression levels of CaMK2A, phosphorylated CREB, and total CREB in HG-induced RGCs treated with forskolin (a CREB activator) or 666-15 (a CREB phosphorylation inhibitor). (B) CREB transcription activity assay demonstrating enhanced CREB-dependent DNA binding by forskolin and reduced binding by 666-15. (C) Protein expression levels of mitophagy markers (LCBII, Parkin, PINK1), Mfn2, and Drp1 in HG-induced RGCs treated with forskolin or 666-15. (D) TEM analysis showing the modulation of HG-induced mitophagy by forskolin and 666-15. (E) EdU incorporation assay indicating the further arrest of cell division by forskolin under HG condition and its rescue effect by 666-15. (F) Apoptosis analysis demonstrating the aggravating effect of forskolin and the protective effect of 666-15 on HG-induced cell death. (G) Dopamine and DOPAC production in RGCs after HG induction with forskolin or 666-15 treatment. N=3 independent experiments. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Targeting CaMK2A suppresses mitophagy and DR progression
To further corroborate the role of CaMK2A in DR pathogenesis, we applied lentivirus carrying control shRNA (sh-NC) or CaMK2A shRNA in the DR model after the onset of diabetes. The administration of dopamine/carbidopa was used as a positive control group to show neuron protection. We observed that similar to the dopamine/carbidopa rescue group, silencing CaMK2A attenuated DR pathogenesis and increased retinal thickness (Fig. 5A). Flow cytometry analysis of retinal cell death also proved the protective effect of CaMK2A silencing in the DR model (Fig. 5B). As expected, CaMK2A knockdown increased the levels of dopamine and DOPAC in the retinal tissues (Fig. 5C), and attenuated the phosphorylation of CREB (Fig. 5D). Of note, dopamine administration also attenuated CaMK2A/CREB pathway in DR tissues, suggesting a feedback regulation between dopamine and CaMK2A/CREB pathway. We also analyzed the features of mitophagy by immunoblotting and TEM. We found that both CaMK2A knockdown or dopamine administration suppressed mitophagy in DR tissues by TEM analysis (Fig. 5E), and prevented the upregulation of mitophagy markers (LCBII, Parkin, PINK1) and Drp1 (Fig. 5F). These data demonstrated the neuroprotective effect of CaMK2A silencing during DR progression.

Effects of CaMK2A knockdown on DR pathogenesis and mitophagy in retinal tissues. (A) Evaluation of DR pathogenesis and retinal thickness in the DR model with CaMK2A knockdown or dopamine/carbidopa administration. (B) Flow cytometry analysis of retinal cell death in the DR model with CaMK2A knockdown or dopamine/carbidopa administration. (C) Dopamine and DOPAC levels in retinal tissues of the DR model with CaMK2A knockdown or dopamine/carbidopa administration. (D) CaMK2A, phosphorylated CREB, and total CREB levels in retinal tissues of the DR model with CaMK2A knockdown or dopamine/carbidopa administration. (E) TEM analysis showing the effect of CaMK2A knockdown or dopamine/carbidopa administration on mitophagy in retinal tissues of the DR model. (F) Protein expression levels of mitophagy markers (LCBII, Parkin, PINK1), Mfn2, and Drp1 in retinal tissues of the DR model with CaMK2A knockdown or dopamine/carbidopa administration. N=6 animals in each group. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Targeting CaMK2A attenuates oxidative stress, inflammation, and neuronal loss in retinal tissues of DR model
We further assessed the oxidative stress, inflammatory cytokines and different neuronal cells in the retinal tissues. ROS measurement showed the reduction of oxidative stress in DR tissues after CaMK2A knockdown or dopamine/carbidopa treatment (Fig. 6A). CaMK2A knockdown or dopamine/carbidopa administration also attenuated the production of inflammatory cytokines (IL-1β, TNF-α and IL-6) in DR tissues (Fig. 6B). Concomitantly, flow cytometry analysis showed that the activation of Iba1 + CD68 + microglial cells was suppressed in retinal tissues after CaMK2A knockdown or dopamine/carbidopa treatment (Fig. 6C). The detection of Brn-3a + ISL1 + retinal ganglion cells (Fig. 6D) and PKCα + CD15 + retinal bipolar cells (Fig. 6E) further revealed the protective effect of CaMK2A knockdown or dopamine/carbidopa treatment against neuronal cell loss in the DR model. Together, our data suggest that the activation of CaMK2A/CREB pathway triggers mitophagy in retinal neurons, leading to neuronal degeneration. CaMK2A silencing not only protect against neuronal degeneration, but also mitigates oxidative stress and inflammation in DR progression.

Targeting CaMK2A attenuates oxidative stress, inflammation, and neuronal loss in retinal tissues of the DR model. (A) ROS measurement showing the reduction of oxidative stress in DR tissues after CaMK2A knockdown or dopamine/carbidopa treatment. (B) Quantification of inflammatory cytokines (IL-1β, TNF-α, and IL-6) in DR tissues with CaMK2A knockdown or dopamine/carbidopa administration. (C) Flow cytometry analysis of Iba1+CD68+ microglial cell activation in retinal tissues of the DR model with CaMK2A knockdown or dopamine/carbidopa treatment. (D) Detection of Brn-3a+ISL1+ retinal ganglion cells in the DR model with CaMK2A knockdown or dopamine/carbidopa administration. (E) Detection of PKCα+CD15+ retinal bipolar cells in the DR model with CaMK2A knockdown or dopamine/carbidopa administration. N=6 animals in each group. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Discussion
The present study provides evidence that dysregulated CaMK2A/CREB signaling could contribute to excessive mitophagy and subsequent neuronal degeneration in DR. Our findings demonstrate that hyperglycemia induces CaMK2A activation and CREB phosphorylation, leading to excessive mitophagy. Modulating the CaMK2A/CREB pathway or inhibiting autophagy can suppress high-glucose induced neuronal cell death in vitro and in vivo. These findings highlight the importance of maintaining a delicate balance between mitophagy and mitochondrial biogenesis in retinal neurons and identify the CaMK2A/CREB pathway as a potential therapeutic target for neuroprotection in DR.
Previous evidence has indicated that the hyperglycemic condition of diabetes results in the overstimulation of autophagy, causing harm to the target organs. For instance, in the case of diabetic retinopathy, neurons’ excess activation of autophagy results in heightened apoptosis of retinal nerve cells. Conversely, suppressing their autophagy levels can mitigate the toxicity caused by hyperglycemia and enhance retinal functionality24. Findings from animal trials have demonstrated that diabetes-related cognitive impairment is also associated with excessive autophagy activation, and mitigating this activity can enhance cognitive performance25. These findings are in agreement with our study, showing that hyperglycemia-induced autophagy can undermine neuronal function. Our data further indicate that excessive autophagy in DR condition may compromise retinal function by disrupting mitochondrial homeostasis in retinal neurons.
It is important to note that the role of autophagy and mitophagy in neuronal survival is complex and context-dependent. While moderate levels of mitophagy are essential for maintaining mitochondrial quality control and cellular homeostasis, excessive or dysregulated mitophagy can be detrimental to neuronal function and retinal cells19,20,26,27. Our findings suggest that the high glucose conditions in DR tip the balance towards excessive mitophagy, leading to the depletion of mitochondria in retinal neurons. This imbalance may be exacerbated by other DR-associated factors, such as oxidative stress and inflammation, which can further compromise mitochondrial function and promote neuronal cell death28,29.
CaMK2A mediated signaling has been implicated in various diabetic complications, including retinal vascular and neuronal abnormalities. Previous studies have shown that CaMK2A activity is increased in the retinas of diabetic mice, contributing to pericyte loss and vascular leakage30. Moreover, genetic ablation of CaMK2A or pharmacological inhibition of CaMK2A activity ameliorated diabetes-induced retinal vascular dysfunction and inflammation31. Our findings extend these observations by demonstrating that CaMK2A/CREB signaling also plays a critical role in regulating mitophagy and neuronal survival in DR. The dysregulation of this pathway leads to excessive mitophagy, which ultimately culminates in the degeneration of dopaminergic neurons, a key pathological feature of DR32,33.
CREB is a transcription factor that mediates the expression of genes involved in neuronal development, synaptic plasticity, and neuroprotection11,34. In the retina, CaMKII-mediated CREB signaling has been implicated in the regulation of retinal ganglion cell (RGC) survival35,36. Our findings demonstrated that high glucose conditions lead to aberrant activation of the CaMK2A/CREB pathway, indicating that excessive activation of this pathway can be detrimental in retinal cells. Several studies have highlighted the role of CREB in mediating autophagy in different cellular contexts. For instance, Seok et al.37 demonstrated that the transcription factor farnesoid X receptor (FXR) induces autophagy through a CREB-dependent mechanism. They found that FXR activation leads to the phosphorylation and activation of CREB, which subsequently upregulates the expression of autophagy-related genes, including Atg7 and Beclin1. In another study, Carloni et al.38 showed that rapamycin can activate autophagy in a neonatal hypoxia-ischemia model through the Akt/CREB signaling pathway. Furthermore, Wang et al.39 reported that thioperamide, a histamine H3 receptor antagonist, can promote CREB-mediated autophagy in an animal model of Alzheimer’s disease. In the context of our findings, silencing CaMK2A/CREB pathway attenuated DR condition by blunting mitophagy in retinal tissues, suggesting novel targets for ameliorating DR progression.
Several limitations of our study should be acknowledged. First, while our findings demonstrate a strong association between CaMK2A/CREB pathway activation and enhanced mitophagy in DR, the precise molecular mechanisms linking these two processes remain to be fully elucidated. Future studies using molecular interaction approaches and pathway-specific interventions are needed to determine whether CaMK2A/CREB signaling directly regulates mitophagy or acts through intermediate pathways. Second, our study focused primarily on retinal ganglion cells, and the effects of CaMK2A/CREB pathway modulation on other retinal cell types require further investigation. Third, while our short-term intervention showed promising results, long-term safety and efficacy studies of CaMK2A pathway inhibition in DR treatment are needed before clinical translation can be considered.
Conclusion
In summary, our study unravels a novel mechanistic link between the dysregulated CaMK2A/CREB signaling pathway, excessive mitophagy, and dopaminergic neuronal degeneration in DR. We demonstrated that high glucose conditions in DR lead to aberrant activation of the CaMK2A/CREB pathway, promoting excessive mitophagy, disrupting mitochondrial homeostasis, and ultimately contributing to neuronal cell death. These findings suggest CaMK2A/CREB pathway as a potential therapeutic target for neuroprotective interventions in DR. Future studies should explore the cell-type-specific effects of CaMK2A/CREB signaling on mitophagy and neuronal survival, which could pave the way for more targeted therapeutic approaches in DR and other neurodegenerative diseases.
Data availability
Data is provided within the manuscript.
References
Kern, T. S. & Barber, A. J. Retinal ganglion cells in diabetes. J. Physiol. 586 (18), 4401–4408 (2008).
Gastinger, M. J., Kunselman, A. R., Conboy, E. E., Bronson, S. K. & Barber, A. J. Dendrite remodeling and other abnormalities in the retinal ganglion cells of Ins2 Akita diabetic mice. Investig. Ophthalmol. Vis. Sci. 49 (6), 2635–2642 (2008).
Wan, T. T., Li, X. F., Sun, Y. M., Li, Y. B. & Su, Y. Recent advances in Understanding the biochemical and molecular mechanism of diabetic retinopathy. Biomed. Pharmacother. 74, 145–147 (2015).
Cecilia, O. M. et al. Ricardo Raúl RR, Adolfo Daniel RC. Oxidative stress as the main target in diabetic retinopathy pathophysiology. J. Diabetes Res. 2019, 8562408 (2019).
Stitt, A. W. et al. The progress in Understanding and treatment of diabetic retinopathy. Prog. Retin. Eye Res. 51, 156–186 (2016).
Simó, R., Hernández, C. & European Consortium for the Early Treatment of Diabetic Retinopathy (EUROCONDOR). Neurodegeneration in the diabetic eye: new insights and therapeutic perspectives. Trends Endocrinol. Metab. 25 (1), 23–33 (2014).
Wang, H., Xu, J., Lazarovici, P., Quirion, R. & Zheng, W. cAMP response Element-Binding protein (CREB): A possible signaling molecule link in the pathophysiology of schizophrenia. Front. Mol. Neurosci. 11, 255 (2018).
Lisek, M., Tomczak, J., Boczek, T. & Zylinska, L. Calcium-Associated proteins in neuroregeneration. Biomolecules 14 (2), 183 (2024).
Wayman, G. A., Lee, Y. S., Tokumitsu, H., Silva, A. J. & Soderling, T. R. Calmodulin-kinases: modulators of neuronal development and plasticity. Neuron 59 (6), 914–931 (2008).
Altarejos, J. Y. & Montminy, M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat. Rev. Mol. Cell. Biol. 12 (3), 141–151 (2011).
Lonze, B. E. & Ginty, D. D. Function and regulation of CREB family transcription factors in the nervous system. Neuron 35 (4), 605–623 (2002).
Baev, A. Y. et al. Interaction of mitochondrial calcium and ROS in neurodegeneration. Cells 11 (4), 706 (2022).
Sama, D. M. & Norris, C. M. Calcium dysregulation and neuroinflammation: discrete and integrated mechanisms for age-related synaptic dysfunction. Ageing Res. Rev. 12 (4), 982–995 (2013).
Perkins, G. A., Ellisman, M. H. & Fox, D. A. Three-dimensional analysis of mouse rod and cone mitochondrial Cristae architecture: bioenergetic and functional implications. Mol. Vis. 9, 60–73 (2003).
Zhang, S. M., Fan, B., Li, Y. L., Zuo, Z. Y. & Li, G. Y. Oxidative Stress-Involved mitophagy of retinal pigment epithelium and retinal degenerative diseases. Cell. Mol. Neurobiol. 43 (7), 3265–3276 (2023).
Skeie, J. M. et al. Mitophagy: an emerging target in ocular pathology. Investig. Ophthalmol. Vis. Sci. 62 (3), 22 (2021).
Levine, B. & Kroemer, G. Biological functions of autophagy genes: a disease perspective. Cell 176 (1–2), 11–42 (2019).
Stavoe, A. K. H. & Holzbaur, E. L. F. Autophagy in neurons. Annu. Rev. Cell. Dev. Biol. 35, 477–500 (2019).
Kubli, D. A. & Gustafsson, Å. B. Mitochondria and mitophagy: the Yin and Yang of cell death control. Circ. Res. 111 (9), 1208–1221 (2012).
Wang, Y., Liu, N. & Lu, B. Mechanisms and roles of mitophagy in neurodegenerative diseases. CNS Neurosci. Ther. 25 (7), 859–875 (2019).
Martinez-Vicente, M. Neuronal mitophagy in neurodegenerative diseases. Front. Mol. Neurosci. 10, 64 (2017).
Pan, Y., Qiu, D., Chen, S., Han, X. & Li, R. High glucose inhibits neural differentiation by excessive autophagy via peroxisome proliferator-activated receptor gamma. Eur. J. Histochem. 67 (2), 3691 (2023).
Xiong, L. et al. The protective effects of melatonin in high glucose environment by alleviating autophagy and apoptosis on primary cortical neurons. Mol. Cell. Biochem. 478 (7), 1415–1425 (2023).
Wang, W. et al. Histone HIST1H1C/H1.2 regulates autophagy in the development of diabetic retinopathy. Autophagy 13 (5), 941–954 (2017).
Ma, L. Y. et al. Autophagy-lysosome dysfunction is involved in Aβ deposition in STZ-induced diabetic rats. Behav. Brain Res. 320, 484–493 (2017).
Intartaglia, D., Giamundo, G. & Conte, I. Autophagy in the retinal pigment epithelium: a new vision and future challenges. FEBS J. 289 (22), 7199–7212 (2022).
Szatmári-Tóth, M. et al. Clearance of autophagy-associated dying retinal pigment epithelial cells - a possible source for inflammation in age-related macular degeneration. Cell. Death Dis. 7 (9), e2367 (2016).
Duh, E. J., Sun, J. K. & Stitt, A. W. Diabetic retinopathy: current understanding, mechanisms, and treatment strategies. JCI Insight. 2 (14), e93751 (2017).
Kowluru, R. A. Cross talks between oxidative stress, inflammation and epigenetics in diabetic retinopathy. Cells 12 (2), 300 (2023).
Kim, Y. H. et al. CaMKII regulates pericyte loss in the retina of early diabetic mouse. Mol. Cells. 31 (3), 289–293 (2011).
Chen, J. et al. CaM kinase II-δ is required for diabetic hyperglycemia and retinopathy but not nephropathy. Diabetes 70 (2), 616–626 (2021).
Eggers, E. D. & Carreon, T. A. The effects of early diabetes on inner retinal neurons. Vis. Neurosci. 37, E006 (2020).
Potilinski, M. C., Lorenc, V., Perisset, S. & Gallo, J. E. Mechanisms behind retinal ganglion cell loss in diabetes and therapeutic approach. Int. J. Mol. Sci. 21 (7), 2351 (2020).
Sakamoto, K., Karelina, K. & Obrietan, K. CREB: a multifaceted regulator of neuronal plasticity and protection. J. Neurochem. 116 (1), 1–9 (2011).
Guo, X. et al. Preservation of vision after CaMKII-mediated protection of retinal ganglion cells. Cell 184 (16), 4299–4314e12 (2021).
Xia, X. et al. Ca2+/Calmodulin-Dependent Protein Kinase II Enhances Retinal Ganglion Cell Survival But Suppresses Axon Regeneration after Optic Nerve Injury. eNeuro. 11(3). (2024).
Seok, S. et al. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature 516 (7529), 108–111 (2014).
Carloni, S. et al. Activation of autophagy and Akt/CREB signaling play an equivalent role in the neuroprotective effect of Rapamycin in neonatal hypoxia-ischemia. Autophagy 6 (3), 366–377 (2010).
Wang, J. et al. Activation of CREB-mediated autophagy by thioperamide ameliorates β-amyloid pathology and cognition in Alzheimer’s disease. Aging Cell. 20 (3), e13333 (2021).
Acknowledgements
The following funds are gratefully acknowledged for supporting this study: Yunnan Science and Technology Talents and Platform Plan (No. 2019HB050), Yunnan Youth Talent Support Program (No. YNWR-QNBJ-2018-315), Yunnan Province Applied Basic Research Program Project(202101AY070001-275).
Funding
This work was supported by Yunnan Science and Technology Talents and Platform Plan (No. 2019HB050), Yunnan Youth Talent Support Program (No. YNWR-QNBJ-2018-315), Yunnan Province Applied Basic Research Program Project(202101AY070001-275).
Author information
Authors and Affiliations
Contributions
All authors were involved in the conceptualization and design of the study. Xiaochun Yang provided experimental ideas and financial support. Xiaochun Yang, Yuxin Zhang and Yinkun Zhou completed the first draft of the manuscript. Mingzhi Liu and Haiyan Zhao performed the material preparation, data collection and analysis. Yang Yang and Jianyun Su were mainly responsible for reviewing the final manuscript. All authors participated in the editorial revision of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The protocol of this study was approved by Ethical Review Committee for Animal Experiments of Kunming Medical University (Approval number: kmmu20241516). Each process of this study was conducted in compliance with the guidelines of the Declaration of Helsinki.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, X., Zhang, Y., Zhou, Y. et al. CaMK2A/CREB pathway activation is associated with enhanced mitophagy and neuronal apoptosis in diabetic retinopathy. Sci Rep 15, 12516 (2025). https://doi.org/10.1038/s41598-025-97371-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-97371-y
