
Abstract
Previously, we demonstrated the potential of decellularized spinach leaves to act as prevascularized scaffolds for tissue engineering. In this follow-up study, we aim to further probe this technology’s efficacy through investigation of leaf scaffold biocompatibility. Decellularization protocols were modified to limit possible toxicity by lowering the time and concentration of detergents used. Decellularized leaves that were processed with this modified technique were found to have limited effects on cell viability in a leaching assay. To evaluate biocompatibility, decellularized leaves were implanted subcutaneously in Sprague Dawley rats. Leaves maintained a limited immunological response 4 weeks post-implantation. A separate group of decellularized leaves were pre-functionalized with an RGD-dopamine peptide and subsequently implanted. These functionalized leaf implants integrated into the host tissue within 1-week, with visible rat collagen deposition found within the leaf scaffold. These findings further demonstrate the suitability of leaves as scaffolding for tissue engineering and suggest exploration for clinical use.
Similar content being viewed by others

Introduction
There is a shortage of available organs for transplantation worldwide, with over 100,000 patients consistently on the organ waiting list in the United States alone1. While the number of patients on the waiting list has rapidly increased over the past 15 years, the number of available transplants has remained stagnant. As a result, researchers have actively pursued the advancement of organ and tissue engineering methodologies. This field, known as tissue engineering2, has shown great promise in potentially alleviating the organ shortage. As tissue engineering has progressed, there has been an emphasis on integrating cells, scaffolds, and scaffold biofunctionalizations into constructs that resemble native tissues.
Bioinspired analogs to human tissue structures have provided exciting opportunities for the development and improvement of engineered tissues. A prime example of applying biomimetic strategies in tissue engineering is evident in the careful design and choice of biomaterials intended for use as scaffolds. Human tissues are composed of cells embedded in a complex mixture of proteins, carbohydrates, and glycoproteins called the extracellular matrix (ECM)3,4. ECM was originally thought to be a mere supporting framework for the cells in a tissue or organ, but more recent research has revealed that the ECM has important roles in directing function at cellular, tissue, and organ levels5,6. Not only does the composition of the ECM affect the overall tissue function, but so does its hierarchical structure7. For instance, blood vessels that are integral to tissue viability must be considered when designing scaffolds for engineered tissues and organs8. In fact, one of the main hurdles currently affecting the clinical translation of tissue engineered grafts is the density and lack of properly sized vasculature, especially microvascular networks. A reliable, patent vascular supply is critical as it allows for blood and nutrient delivery to cells within the tissue. The delivery of oxygenated blood is limited by the diffusion limit of oxygen in tissue, which requires blood to be transported within 100–200 microns of cells within the tissue9. Without adequate blood distribution, engineered tissues will die when grown to clinically relevant sizes10, thereby limiting the ability to engineer larger tissue sections and whole organs. Overcoming this limitation will help enable the field to solve the organ shortage problem on a feasible scale.
Researchers have looked toward the native structure found in tissues and organs in the body and tried to harness those structures directly to create vascularized tissue. One such approach that has shown great promise is decellularization11. Decellularization is a technique where cells are washed away from a tissue or organ, leaving behind the ECM and the hierarchical structure of the tissue. This technique is commonly performed using different detergents, which work to disrupt the cell membrane thereby destroying the cells. These detergents can be perfused through the vasculature of the organ being decellularized which allows for retention of the hierarchical structure of the organ11,12. The ECM can then be studied13, used by itself as a therapeutic14, or engineered as a tissue scaffold15,16. Because the tissue or organ is devoid of cellular material, its usage of an implant should elicit minimal to no host immune rejection17.
However, the process of removing cells from tissues and organs has its own set of limitations, mostly due to the variability and availability of suitable tissues and organs for this procedure. The donor’s natural ECM is affected by factors like the donor’s health status18 and lifestyle habits19,20, making it challenging to establish a consistent and controllable baseline for human tissue quality. Furthermore, the requirement for donor tissue for this process presents a paradoxical obstacle to the initial goal of reducing donor dependence. While using animal tissue offers a potential extension, it remains inadequate to meet the current demand. Along with availability concerns, animal tissues offer high risks of infection from multiple pathogens and viral agents21. Animal sources also contribute to ecological concerns such as biodiversity loss, freshwater depletion, and deforestation22. These limitations have led our group to investigate the use of plant tissues, particularly spinach leaves, as prevascularized scaffolds for tissue engineering23. Despite the apparent differences between plant and animal tissue, there are many similarities in tissue architecture. The branching pattern of leaf vasculature closely resembles the branching pattern of the human cardiovascular system24, and these structures can withstand large fluid pressure gradients ranging to upwards of 1 MPa25. Harvesting plant tissues is both sustainable and highly controllable using techniques such as Good Agricultural Practices (GAPs) or hydroponic growth. We were successful in the decellularization and subsequent recellularization of plant tissues with functional human cells24. Furthermore, we were able to measure the diameter of the lumens within the vasculature of leaf scaffolds and found them suitable for the transport of red blood cells. These initial results and the potential of leaf scaffolds led us to expand upon our initial investigation.
Our previous plant decellularization method used a highly concentrated formulation of detergents, which had potential cytotoxic consequences. Decellularization can be performed with different detergents, acids and bases, hypotonic solutions, enzymes, and by physical means26. The most common methods include the use of detergents due to their effectiveness, ease of use, availability, and cost. Different detergents can be combined to maximize the process. For instance, sodium dodecyl sulfate (SDS) is an anionic detergent that is effective at removing nuclei from dense tissues27, whereas the nonionic surfactant, TritonX-100, is effective in dilapidation and removal of other detergents28. Because of these differences, SDS and TritonX-100 are often used serially when decellularizing tissue11,15. The timing of treatment and concentrations of the solution can affect the final decellularized tissue. Higher concentrations can reduce the time needed to decellularize, but there could be residual detergent leading to cytotoxic effects29,30. Likewise, lower detergent concentrations can lengthen decellularization but result in the tissue being exposed to cytotoxic agents for longer than necessary. Because of these concerns, we aimed to optimize a process to maximize the decellularization in the shortest amount of time with the minimum permissive concentration of detergents31,32,33. We have hypothesized that the removal of the waxy outer layer of the leaf, known as the cuticle, prior to decellularization would increase efficiency. The cuticle encases the leaf and works to protect it from invading pathogens and retention of water34. Furthermore, if these leaf scaffolds were employed in engineered tissues, their biocompatibility must be understood. Thereby, we aim to investigate the in vivo immunological response to decellularized leaf scaffolds. Furthermore, we previously functionalized the surfaces of decellularized leaves and stems with an arginine-glycine-aspartate-dopamine (RGD-dopamine) peptide35,36, allowing for large-scale cellular attachment and expansion over 50 days. Biofunctionalization of the leaves could offer potential immunomodulatory effects by attenuating the body’s response to the implanted leaf scaffold. Therefore, we investigated the biocompatibility and possible immunomodulatory effects of both non-functionalized and RGD-functionalized decellularized leaf scaffolds through subcutaneous implantation in a rat animal model.
Results
Removal of leaf cuticle allows for modification of decellularization process
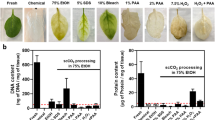
The modified decellularization protocol (Fig. 1B) decellularized the leaves in 5 days whereas the original process (Fig. 1A) required 9 days to complete. A tris-buffer wash was introduced post-decellularization to help change the pH of the leaf scaffold, which has previously been shown to be effective in releasing bound SDS from the tissue37.

The (A) original decellularization process was (B) modified to shorten the process and lower the concentration of detergents.
This process was verified through quantification measures of both DNA and protein. DNA analysis revealed that both the original and modified protocol produced fully decellularized leaves that had significantly (p < 0.05) lower amounts of DNA/mg of tissue compared to the non-decellularized control (Fig. 2A). Remaining protein content (Fig. 2B) was also quantified and exhibited a similar trend, in which both protocols resulted in significantly (p < 0.05) lower amounts of residual protein compared to the native spinach leaf. Interestingly, the modified decellularized processing protocol left leaves with more protein than the original, but this was not significantly different.

A DNA analysis showed that both the original and modified decellularization protocols yielded similar levels of DNA/mg of tissue. Both were significantly lower than the DNA content of a native spinach leaf (n = 4, both p < 0.0001). B Protein levels were also similarly decreased because of the two processes, with both being significantly lowered when compared to native leaves (n = 4, both p < 0.0001). Qualitative measures of (C) native leaves, D leaves decellularized via the original method and E leaves decellularized via the modified method. F Relative Transparency was quantified for native leaves and that leaves that underwent both decellularization processes (n = 5, p = 1.4 × 10–6 for Modified Decell, p = 4.5 × 10–9 for Original Decell). $ denotes significance between Native Leaf and Modified Decellularization, # denotes significance between Native Leaf and Original Decellularization.
Qualitative measures demonstrated that prior to lyophilization, both protocols produced leaves with similar transparency (Fig. 2C–E). This was verified by quantifying the transparency of the different leaves (Fig. 2F). Optical intensities in multiple areas of leaves were measured and compared to control background imagery38. For this method, a value of 1 denotes a completely transparent item and a value of 0 denotes a completely opaque item. Leaves (n = 5) that were processed by both the modified (0.903, p = 0.0000014) and original (0.930, p = 0.0000000045) were significantly more transparent compared to native tissue (0.245) with no significant difference between the two decellularized groups.
The modified process included a lyophilization step (Fig. 3A). Lyophilization allows for long-term storage at room temperature and limits possible tissue degradation over time. One of the major advantages that the leaf scaffold possess is its innate vascular network. Because of the extra processing performed on the leaf scaffolds as a part of lyophilization, it was important to investigate the properties of the leaf vasculature after rehydration from lyophilization. Lyophilized leaves can be easily rehydrated and retain the same properties as leaves prior to lyophilization (Fig. 3B). Red dye perfusion through the rehydrated leaves showed that the vasculature within the leaf remains patent after the processing (Fig. 3C). This patency is similar to what was previously reported from our original decellularization investigations.

A, B Decellularized leaves after lyophilization can be rehydrated to reform its original properties. C Red dye perfusion through a rehydrated lyophilized leaf shows that the vasculature remains patent after processing and lyophilization.
Modified decellularization process yields limited cytotoxic effects
Optimization of the decellularization process aimed to not only shorten the amount of time necessary to perform the process but also to limit the amount of SDS used. Lower amounts of SDS at shorter times should lessen any cytotoxic effects29. To measure cytotoxicity, fibroblasts were cultured in standard polystyrene tissue culture wells. Equal amounts of leaves were diced and added into the medium in which the fibroblasts were being cultured in. The leaves were left floating in the medium to measure whether there would be cytotoxic agents that would leach out from the leaf (Fig. 4A).

A Fibroblasts were cultured in the bottom of a culture well. Pieces of the different treated decellularized leaves were introduced into the medium of the well plate and cytotoxicity was measured 24 h later. B There was no visible cytotoxicity seen in absence of leaves. C When leaves that had been decellularized with the original 10x SDS decellularization process, there was widespread cell death. D There was no apparent toxicity of leaves from the modified 1x SDS process. These cells appeared to be normally growing and had a phenotype akin to the no leaf condition. Fibroblasts maintained normal phenotype when cultured with (E) no leaf or with F leaves that underwent the modified decellularization process. Scale bars: (B–D)- 200 μm, (E, F)- 20 μm
A live-dead assay was run after 24 h of fibroblasts being cultured with the leaves floating in their media. When no leaf was introduced into the medium (Fig. 4B), normal phenotypic fibroblasts were seen with limited to no cell death found. In cultures that were introduced to leaves that had undergone decellularization with the original 10% SDS process (Fig. 4C), there were large areas where cells had detached from the culture plate indicating cell death occurred prior to the 24-h time point. Cells that remained attached to the culture plate were round and did not show the normal spindle-like phenotype of fibroblasts. Moreover, most remaining cells were positively stained red by the ethidium homodimer for the dead marker in their nuclei. Fibroblasts that were subjected to the modified decellularized 1% SDS leaves (Fig. 4D) had limited to no cytotoxicity. There were some small gaps of non-adherent areas indicating some cell death, but this was not widespread. Cells had the normal phenotype and were nearly the same as those that had no leaf in their medium, as verified through staining of F-actin (Fig. 4E, F). Furthermore, there was very little positive red dead stain found in the adherent cells.
Possible leaf cytotoxicity was further investigated by measuring the metabolic activity of fibroblasts using an MTT assay. Metabolic activity was measured at 6 h, 24 h, 3 days, and 7 days after introducing the leaf pieces into the medium. Metabolic activities were normalized to wells that contained no leaf pieces at each time point (Fig. 5A). At all four time points, fibroblasts subjected to the 10% SDS decellularized leaves had significantly lower levels of metabolic activity than the controls where fibroblasts were cultured without leaves. In contrast, fibroblasts cultured with the 1% SDS leaves showed no significant difference in metabolic activity compared to wells with no leaves, indicating no significant cytotoxic effects from these modified decellularized leaves.

A Fibroblast metabolic activity was measured using an MTT assay for fibroblasts grown in the culture shown in Fig. 4A. The activity was measured over 4 time points and was normalized to activity measured in conditions where no leaf was introduced into the medium. Leaves treated with the original 10x SDS process elicited significant cytotoxicity and fibroblasts showed limited and significantly lowered (Denoted by *, all time points p < 0.0001, n = 4) metabolic activity. There was no difference between the no leaf and the modified 1x SDS condition. B The percentage of viable wells was measured for these assays. Only 21% of the wells that had the original 10x SDS leaves introduced into them were usable, indicating large scale cell death. C Non-normalized metabolic activity for the different conditions was compared over time. The fibroblasts that were subjected to no leaves or to the modified 1x SDS followed very similar growth patterns. Fibroblasts that were treated with the original 10x SDS leaves had lowered metabolic activities that remained relatively the same over the time-course of the study. D Residual SDS was measured in both the original and modified processes. There were significantly greater amounts of residual SDS found in the original 10x SDS treated leaves and there were negligible amounts seen in the modified 1x SDS condition (Denoted by $, p < 0.0001, n = 4).
Furthermore, an interesting trend was seen where all the culture wells that had been introduced to no leaves or the 1% SDS leaves were usable for MTT assay at all the time points. Whereas only 21% of the plated wells that contained pieces of the 10% SDS leaves were viable for the MTT assay (Fig. 5B). This trend was likely due to large scale cell death and detachment from the culture vessels.
Comparing the fibroblast non-normalized metabolic activity over time also gave insight into the change in the metabolic activity over time of the fibroblasts. The fibroblasts that were subjected to no leaves have very similar temporal metabolic patterns when compared to the fibroblasts supplemented with the 1% SDS leaves. There was a sharp decrease in metabolic activity in both conditions going from the 6 to 24-h time point which is probably an artifact of the seeding of the cells. Going from 24-h to 3 days and subsequently to 7 days, the metabolic activity changed to a very similar degree. The slopes of the growth curves between the 1% SDS leaves and when there was no leaf introduced were similar. Whereas the fibroblasts that were subjected to the 10% SDS leaves had lowered metabolic activities starting at 6 h and these metabolic activity levels remained relatively unchanged throughout the study, indicating limited to no change in the metabolic activity of these fibroblasts (Fig. 5C). To fully elucidate difference between the two conditions, residual SDS was measured (Fig. 5D). 10% SDS leaves had statistically significantly greater amounts of SDS over the leaves processed in the modified 1% SDS process. These results validate the hypothesis that although residual SDS caused cytotoxicity, it could be limited by modifying the decellularization protocol.
Decellularized leaves illicit little immunological response
Leaves subjected to the modified decellularization process were implanted subcutaneously for 1, 2, and 4 weeks in female Sprague Dawley rats (n = 5–6 with 3 biological replicates). There was no systemic health issues observed in any of the rats during the time-course of the study. Animals were ethically sacrificed at the respective end time point and the abdomen was excised around the regions where the implants were made. Excised samples were cut to include all layers of skin, fat, and muscle layer to grossly study the impact that the host system had on the leaf implant. Excised tissue was prepared for histological analysis and embedded in paraffin.
Hematoxylin and Eosin (H&E) staining of excised tissue sections showed limited gross systemic immunological and inflammatory response to the leaf implant at all three time points (1, 2, and 4 weeks). At 1 week (Fig. 6A, D), there was a hematoma that had begun to form and encapsulate around the leaf scaffold and some cellular infiltration into the leaf implant was observed, which can be considered a limited initial inflammatory response suggesting the leaf could be viewed as an inert material. At 2 weeks (Fig. 6B, E), the encapsulation grew larger while also the cellular infiltration was greater with some remodeling seen. At week 4 (Fig. 6C, F), there was a higher level of cell infiltration into the implant while also the encapsulation was seen to have subsided at this time. There were cellular components found within the phloem and xylem (identified with black arrows) cross-sections of the leaf implant.

A, D 1 wk after implantation, leaves were seen to have a limited immunological response. There was apparent hematoma formed surrounding the leaves. B, E After 2 weeks of implantation, there was some cellular infiltration seen and the formation of some encapsulation around the leaf implant. C, F 4 wk after implantation, the leaves were infiltrated with more cells and some remodeling occurred. Panels (D– F) are magnified images of the insets of (A–C), respectively. Scale bars: (A–C)- 0.5 mm, (D–F)- 100 μm.
Masson’s Trichrome staining which stains collagen blue, muscle fibers red, and nuclei black (Fig. 7) showed the remodeling of the leaf by the body, as indicated by the deposition of collagen into the leaf scaffold. There was some collagenous encapsulation seen surrounding the different implants. At 1 week (Fig. 7A, D), there was limited collagen deposition seen within both the leaf scaffold and the surrounding tissue that was the beginning of the encapsulation. The encapsulation was remodeled into collagen by week 2 (Fig. 7B, E). There were also small, isolated areas of collagen deposition at the 2-week excision. By week 4 (Fig. 7C, F), the surrounding collagen capsule had shrunk in size and there were greater amounts of collagen deposition within the main sections of the leaf implant.

A, D 1 wk after implantation showing limited collagen deposition. B, E After 2 wk, there was some collagen deposited into the leaf scaffold indicating remodeling along with a thin collagen capsule being formed around the outside of the leaf. C, F After 4 weeks, there was more collagen deposition within the leaf and the collagen capsule began to shrink. Scale bars: A–C- 0.5 mm, D–F- 100 μm.
RGD-functionalized leaves are fully integrated into the body at 1 week
A separate set of decellularized leaves were functionalized with an RGD-Dopamine peptide prior to subcutaneous implantation to stimulate increased integration into the host tissue. Previous investigation into the effects of biofunctionalization of decellularized plants showed increased cellular attachment, alignment, and proliferation on plant tissues that were functionalized as opposed to non-functionalized35. RGD functionalized leaves were implanted subcutaneously and analyzed through histological staining in the same manner as the non-functionalized leaves. Implanted functionalized leaves are seen to be integrated to a greater degree than non-functionalized leaves after just one week (Fig. 8). H&E staining shows a limited negative immunological response. Leaf phloem and xylem (black arrows) are found being surrounded and assimilated by the native host’s tissue and there was a limited encapsulation seen after 1 week of implantation (Fig. 8A, D). After 2 weeks of implantation (Fig. 8B, E), there is a substantial amount of cellular infiltration from the host and tissue is seen being deposited into the leaf scaffold from the host. By 4 weeks of implantation (Fig. 8C, F), there was a large amount of tissue remodeling seen and the leaf was seen to be fully integrated into the host tissue with no negative response. The host tissue has grown to the edges of the phloem and xylem, whose structures remain present and appear to be left open and intact.

A, D H&E staining showed large scale integration of the functionalized leaf scaffold into the host tissue after one week of implantation. Black arrows indicate leaf vasculature ___location. B, E RGD-functionalized leaf integration improves after 2 weeks of implantation where there are large amounts of host tissue being deposited within the leaf scaffold. C, F Native host tissue deposition almost entirely overtakes the leaf scaffold and surrounds the leaf vasculature within the leaf scaffold after 4 weeks of implantation. Panels (D–F) are magnified images of the insets of (A–C), respectively. Scale bars: A–C- 0.5 mm, D–F- 100 μm.
Masson’s trichrome staining (Fig. 9) was performed to visualize any collagen encapsulation and deposition associated with the RGD-functionalized leaf implants. After 1 week of implantation (Fig. 9A, D), there was visible collagen deposition seen within the leaf scaffold with a very thin collagen layer encapsulating the leaf scaffold. The collagen encapsulation grew slightly and there was a visible greater amount of collagen being deposited into the scaffold within 2 weeks of implantation (Fig. 9B, E). There was substantial collagen deposited within the leaf scaffold 4 weeks (Fig. 9C, F) after implantation and the collagen encapsulation seemed to not grow any larger between weeks 2 and 4.

A, D Staining showed collagen deposition within the leaf scaffold after just one week of implantation. There was also a small collagenous encapsulation that had formed. There was an increase in the amount of collagen being deposited within the leaf scaffold after both two (B, E) and four (C, F) weeks of implantation. Panels (D–F) are magnified images of the insets of (A–C), respectively. Scale bars: (A–C)- 0.5 mm, (D–F)- 100 μm.
Comparison of non-functionalized and RGD-functionalized leaf scaffold integration into the host tissue
Cell infiltration into the leaf implant and border zone along with the thickness of the collagen encapsulation and the amount of collagen deposited within the leaf scaffold were quantified to compare the host response to the leaf scaffolds that did and did not undergo RGD functionalization prior to implantation (Fig. 10). At all three time points (1, 2, 4 weeks) there was a significantly greater amount of host cells found within the leaf scaffold when the scaffold was pre-functionalized with the RGD peptide (p = 0.00064 at 1 week, p = 0.010 at 2 weeks, and p = 0.0099 at 4 weeks). The number of cells increased over time in both RGD-functionalized and non-functionalized implants. Cells counted in the immediate border zone outside of the implant were found to be significantly greater in the non-functionalized implants when compared to the RGD-functionalized implants at all time points (p = 0.0066 at 1 week, p = 0.034 at 2 weeks, and p = 0.022 at 4 weeks), indicating the possibility of encapsulation in the non-functionalization implants whereas the functionalized implants promoted cell infiltration.

A The number of cells that infiltrated into the implanted scaffold was quantified and normalized to the area of the implant. There was significantly greater number of cells in the RGD-Functionalized Implant when compared to the Non-Functionalized (denoted by * at all time points. 1 week-p = 0.00064, 2 week-p = 0.01, 4 week-p = 0.0099, n = 5). B The number of cells that were found in the immediate border zone surrounding the implant were measured and normalized to the area of the implanted scaffold. There was significantly greater number of cells in the Non-Functionalized implant border zone compared to the RGD-Functionalized implant border zone (denoted by * at all time points. 1 week-p = 0.0066, 2 week-p = 0.034, 4 week-p = 0.022, n = 5). C The percentage of the total area of the implant that was overtaken by collagen deposition was measured for both non- functionalized and RGD-functionalized leaf scaffolds over the time-course of the study. There was a statistically significant greater amount of collagen deposition in all time points when comparing RGD-Functionalized Implants and Non-Functionalized Implants (Denoted by *, at all time points p < 0.0001, n = 5). There was a significantly greater amount of collagen deposition in the RGD-Functionalized Implant after 4 weeks when compared to 1 week (Denoted by $, p = 0.028, n = 5). D The thickness of the collagenous encapsulation surrounding the leaf implant. There was a statistically significant greater size of collagen encapsulation between Non-Functionalized Implants and RGD-Functionalized Implant at weeks 1 and 2 but not week 4 (Denoted by *, at both 1 week and 2 week, p < 0.0001, n = 5). At 2 weeks, the Non-Functionalized Implant had the greatest amount of encapsulation compared to the other two time points for Non-Functionalized Implants (Denoted by $, p < 0.0001, n = 5). Within RGD-Functionalized Implant time points, week 1 had significantly less amount of encapsulation compared to both week 2 and week 4 time points (Denoted by &, p = 0.037 between 2 week and 1 week, p < 0.0001 between 4 week and 1 week, n = 5).
Encapsulation thickness was measured in 7 different random regions across each sample and averaged to account for the irregularity seen within the collagen capsule. Non-functionalized leaf implant encapsulation increased from 1 week to 2 weeks when the capsule was at its thickest before subsiding in size by 4 weeks of implantation. The 2-week encapsulation thickness for the non-functionalized leaf implants was significantly thicker (p < 0.0001) than at both the 1-week and 4-week time points. RGD-functionalized leaf implant encapsulation increased over the course of the study from 1-week to 2-week to 4-week implant time points; 4-week (p < 0.0001) and 2-week (p = 0.037) encapsulation thickness were both found to be significantly thicker than the encapsulation seen at 1-week. Encapsulation around the non-functionalized leaf implants was found to be significantly thicker (p < 0.0001) than the RGD-functionalized leaf implants at 1 and 2 weeks but no significant difference was measured at 4 weeks.
Collagen deposition within the leaf scaffold was measured to investigate active remodeling of the leaf scaffold being performed by the host tissue. Deposition was measured and compared as a total percentage of the leaf scaffold area within which it was occurring. There was an increase in the percentage of collagen being deposited over time within both non-functionalized and RGD-functionalized leaf scaffold implants. RGD-functionalized leaf implants had significantly greater (p < 0.0001) collagen deposition percentages when compared to non-functionalized leaf scaffolds at all three time points. The percentage of collagen deposition of RGD-functionalized leaves at 4 weeks was significantly greater (p = 0.028) that the deposition seen at 1 week within RGD-functionalized leaves.
Discussion
With the expressed need for reliable, available, and affordable organ transplants in the United States, there is a pressing need to advance tissue engineering technology. Tissue engineering’s potential to address the imbalance between supply and demand in organ transplantation positions this crossroads of health science and engineering as a necessity39. Despite considerable progress in the past two decades, the ability to generate clinically available tissue is still quite limited. A major limiting factor is the lack of properly sized and viable vasculature within engineered tissues9. Oxygen has a diffusion limit of 100–200 um40, thus the need for a microvascular network is imperative for engineered tissue survival.
There are a variety of different techniques being explored to produce such a vascularized tissue. 3D Bioprinting41,42,43 has been used in a large variety of vascularized tissue applications such as myocardium44,45 and liver46. The use of naturally derived ECM bioinks47 allow for conservation of ECM signaling while using the spatial control of bioprinting. Decellularization11,26,48 offers an alternative approach to the creation of vascularized tissue or even whole organs. Microvasculature remains conserved while keeping the cellular binding motifs within the ECM in decellularized tissues. Both approaches offer the potential for building vascularized tissue and need to be further developed to create a clinically-relevant solution.
We previously sought to create a prevascularized tissue engineering scaffold by exploiting the native vascular structures in spinach leaves23. Spinach leaves are attractive due to their availability, cost, flat planar structure, and accessible vascular network. To harness the leaf structure, we successfully modified perfusion-based decellularization techniques for the leaf. Leaf scaffolds had an intact vascular network that supported fluid and microparticle transport while also supporting recellularization with human cells. Because of this initial success, we aimed to further investigate the efficacy of using decellularized spinach leaves as a prevascularized scaffolds for tissue engineering.
One critical consideration when working with decellularized tissues is understanding whether the tissue would result in cell death upon recellularization or implantation. The decellularization process requires exposure to harsh chemical conditions to remove cell contents, making investigations into resultant cell viability a necessity. One commonly employed decellularization practice is the perfusion of detergent solutions throughout the tissue7,16,26. These solutions work to lyse the cells by destroying their cellular and nuclear membranes. Residual detergents, especially sodium dodecyl sulfate (SDS), have been shown to be cytotoxic29,30,32. SDS can also cause hierarchical structural changes to tissue that result in similar cytotoxicity37. Because of this, a decellularization process should aim to limit the amount of detergent used. Upon review of the original plant decellularization process23, it was found that the original process had a detergent concentration, and duration of decellularization greater than other methods7,26,49. Despite successful human cell adherence and survival on the decellularized plant tissues, potential cytotoxic effects due to these highly concentrated detergents remained worrisome, especially regarding future clinical applications. Therefore, we aimed to limit the cytotoxicity of decellularized leaf scaffolds by modifying the decellularization process.
Plant tissue was previously decellularized using a serial perfusion of 10x SDS for 5 days followed by 2 days of perfusing a 0.1% TritonX-100 in 10% bleach solution. This process was originally modified by previously used protocols to decellularize heart tissues3,11,13, which used 1% SDS followed by a 0.1% TritonX-100 solution. The SDS solution used for the plant decellularization was set at a higher concentration than the previously used protocols to break up the tougher plant cell-wall structures. Furthermore, bleach was incorporated to remove chloroplasts and sterilize the tissue50. The use of 10% SDS at the time seemed necessary but since working with plant tissue and understanding its phytotomy it became apparent that this concentration could be lowered through prior chemical treatment of the leaf.
The leaf has an outer waxy coating known as the cuticle51 which protects the epidermis of the leaf. The cuticle also acts to limit fluid loss by increasing the vascular resistance of the leaf. Previously, we removed the cuticle before recellularization via treatment with hexanes23. The cuticle’s hydrophobic properties make it a non-ideal surface for the attachment of human cells. Removal of the cuticle prior to decellularization should allow for increased fluid flow by reducing the vascular resistance. Considering this, we hypothesized that the removal of the cuticle prior to decellularization would lower the vascular resistance in the leaf, allowing for the decellularization process to be accomplished with lower concentrations of SDS in shorter amounts of time. By lowering the decellularization time and the SDS concentration, the likelihood that the leaf scaffold would elicit cytotoxic effects should be diminished.
Additional changes to the decellularization process were made to limit possible cytotoxicity. Residual SDS binds to proteins via lysine groups within decellularized tissue, however this binding can be disrupted by altering tissue pH37. A basic tris-buffer wash was added to change the pH of the tissue to disrupt any residual SDS bound within the tissues. Finally, spinach leaves were lyophilized for longer-term storage as opposed to the original storage method, which kept leaves in sterile deionized water at 4 °C. Lyophilization also helps to remove any residual bleach left behind on tissue. Bleach consists of a sodium hypochlorite and a small amount of sodium hydroxide. Lyophilization removes gaseous chlorine while leaving traces of sodium chloride on the surface, which is easily washed away when a leaf is rehydrated prior to further experimentation52.
The modified decellularization process was similar in outcome to the original process. Both methods produced leaves with significantly lower levels of DNA and protein compared to native spinach leaves. Residual DNA levels in decellularized samples were low enough to be considered fully decellularized by proposed standards26. Both processes resulted in leaves that had similar quantified transparencies, which were both significantly more transparent compared to native non-decellularized leaves. The modified decellularized leaves were stored lyophilized which should increase their shelf-life and improve any potential storage degradation. Lyophilized leaves were able to be rehydrated, and the rehydrated leaves still had patent vasculature, demonstrating the efficacy of this approach.
The cytotoxic effects of decellularized leaf scaffolds were tested through a modified indirect contact cell viability assay53, as described by ISO standard 10993-5. Equal weights of leaves that underwent either the modified and original decellularization process were cut up and suspended in the media of a cell culture well containing cultured human dermal fibroblasts. The viability of the fibroblasts was measured over time and through both a live/dead assay54 and a MTT assay55. Aligning with our hypothesis, the leaves that underwent the original process caused higher levels of cell death and limited the proliferation of the cultured fibroblasts. The leaves that underwent the modified process had limited cytotoxic affects and allowed for proliferation that matched the controls where no leaves were introduced into the cell culture medium. Interestingly, there was a decrease in viability in all three cases after 24 h of culture before the viability being rescued prior to day 3 in the conditions where leaves that underwent the modified decellularization process or no leaves were inserted into the culture wells. This initial decrease over all the conditions is likely an artifact from fibroblast seeding and initial inclusion of foreign material. The fact that there is further cell death in the original decellularization condition was significant. The toxicity observed was hypothesized to be the resultant of residual SDS in the decellularized leaves, which was verified through measurement of residual SDS in both leaf types. There was a significant difference in the residual SDS found in leaves processed with both decellularization techniques. The modified protocol had significantly less amounts of residual SDS compared to the original protocol. We have thereby demonstrated that the leaf decellularization protocol can be modified to limit cytotoxicity. The modified decellularization protocol was used throughout the rest of this study and will be used for studies going forward.
It is imperative to investigate a body’s gross biologic and immunological response to a new biomaterial. The properties of the biomaterial can elicit different levels of response from the host. For example, surface porosity56, roughness57, or binding of cellular adhesion peptide motifs such as RGD58 can modulate the host response from unfavorable to favorable, depending on the biomaterial’s intended application. At the same time, if the body determines the implant is unsuitable, it will begin to encapsulate the material to wall it off from the rest of the body59. Encapsulation can lead to fibrosis and ultimately, rejection of a material60.
The host response to implantation of cellulose, the major extracellular component of the leaf scaffold, has been extensively studied and found to be highly biocompatible61,62. Cellulose, a major structural component of the leaf, is a widely studied biomaterial with several biomedical applications. It has been used in a wide variety of regenerative medicine approaches, such as cartilage tissue engineering63,64, bone tissue engineering65,66, and wound healing61,67. These studies used cellulose derived from bacteria to control fiber geometries and chemical properties but are not from plant derived sources. More recently, there has been some work on plant-based cellulose. Modulevsky et al., generated cellulose-based scaffolds from the decellularized hypanthium tissue of an apple slice68, which was found to be biocompatible and promote angiogenesis when implanted in mice69. The success of the plant derived cellulose from the apple tissue provides some insight into a possible reaction to our decellularized leaf scaffolds, but it is not a perfect analog. The apple hypanthium tissue is comprised of just the plant cells with mainly cellulosic cell walls and is void of the more complex structures, such as the vascular system, seen in our leaf scaffolds. These complex structures in the leaf scaffolds are mainly composed of various plant-derived biopolymers such as hemicellulose, lignin and pectin70. These plant-based biopolymers differ greatly from what is seen in the mammalian extracellular matrix, which is a complex collection of proteins, glycosaminoglycans, and polysaccharides3. Because of these differences to what has been studied and what is in mammalian tissue, it became imperative to investigate the immunological response to the decellularized leaf scaffold.
When a biomaterial is implanted into the body, there is an injury that occurs to the region of the implant. This injury elicits an inflammatory and wound healing response71 which range from normal to abnormal depending upon the material that is being implanted. In short, neutrophils in the blood recognize the material and start to absorb proteins onto its surface. The proteins cause activation of macrophages and in turn macrophages fuse together to form foreign body giant cells. These cells work to encapsulate the foreign body isolating it from the rest of the body. Immunological response to any foreign body is inevitable but can be modulated to allow for proper integration into the body. Immunosuppressive drugs72 can effectively subdue the immune system to allow for material integration but there are many complications73 that can occur due to this therapy such as infection74. Newer techniques have looked to altering the surface of the material such that it is seen as more favorable by the body75. Establishing an immunomodulatory biomaterial59 has been a promising approach as it allows for a finely-tuned immunological response that is favorable given the intended application76. The establishment of a novel biomaterial requires analysis of its ability to activate the innate host immune system, and its suitability for immunomodulatory alteration to achieve a favorable response.
The body has a complex response to a foreign body implantation, which cannot be simulated in vitro. Therefore, to fully elucidate the biocompatibility of the decellularized leaf scaffold, decellularized leaf scaffolds were implanted subcutaneously in the abdomen of Sprague Dawley rats. The host response to the implanted scaffolds was analyzed through histological analysis at three distinct time points: 1-, 2-, and 4-weeks post-implantation. There was, at most, a weak immunological and inflammatory response to the decellularized leaf. Although a thin collagen capsule formed along the outer border of the leaf, it decreased in size by the fourth week after implantation. Despite formation of the thin capsule, there was still appreciable cellular infiltration observed, along with visible collagen deposition, onto the leaf scaffold–all positive indications that decellularized leaves are biocompatible. Capsule formation is not an ideal response depending on the application of an implant, but if these leaves were to be used as scaffolding for a tissue-engineered graft, the presence of patient-specific host cells could modulate the host response and limit capsule formation77.
Further alteration of the leaf should improve its ability to integrate into host tissue and a separate set of leaves were functionalized with an RGD-dopamine peptide prior to implantation. Previously, we generated an RGD-dopamine peptide to functionalize decellularized plant structures35. The RGD peptide sequence is the major cellular binding motif found within the amino acid structure of fibronectin78, and dopamine plays a role in generating adhesive coatings on the outside of inert materials, therefore these two molecules were conjugated for the best outcome79,80. This peptide was designed to permit dopamine to bind to the leaf surface while leaving the RGD ___domain accessible as a cellular binding site. Functionalization increased cellular infiltration and promoted better integration into host tissue. After one week, these functionalized leaves were well integrated into the host, with the leaf almost totally overtaken by native tissue. The leaf’s vasculature was still visible, and collagen deposition and positive remodeling were observed. This positive response continued over the next two timepoints, where large amounts of collagen were deposited within the implant despite some degree of implant encapsulation.
Collagen accumulation is seen as advantageous as it is an indicator that the host’s cells are infiltrating and functional within the plant tissue. Furthermore, by including the pre-functionalization, this increase of cellular infiltration and subsequent collagen deposition showcase the potential in the leaf scaffold for future therapeutic development. Depending on the intended result, cell’s infiltrating into a leaf scaffold could be useful. For example, if the leaf scaffold is intended to act in a regenerative medicine approach, allowing for good cell ingrowth into the leaf would only increase the regenerative potential of the scaffold. Similarly, cellulose is non-degradable within the human body. This can be advantageous as the strength and form of the scaffold should be retained over time which would allow for better control over the implanted tissue, especially in more dynamic parts of the body such as the heart. However, in the applications where there is a need for the scaffold to degrade away, there could be steps such as a pre-digestion of the cellulose prior to implantation with cellulase. Ultimately, these scaffolds can be fine-tuned towards whichever specific application they are intended for, making them a powerful and sustainable scaffold material. While we investigated implants lasting up to 4 weeks, if these scaffolds were to be used in a permanent manner, longer term implants would need to be considered to fully understand long term risks of using a cellulosic material. Because of the widespread use of bacterial-based cellulose, we hypothesize that any long-term risks would be mitigated but because of the complexity of using plant-based cellulose, it would be important to investigate such claims. Lastly, cell ingrowth would also help form vascular networks within the leaf and connecting to the host tissue. The current scaffolds are being used as prevascularized tissue but are not being attached to the host’s vascular network. Future studies will look to integrate the vascular networks together to form fully function cross-kingdom vascular networks.
These functionalized leaves demonstrate that the leaf platform can be modified for intended use. Considering that leaves can be easily modified to express different human proteins or therapeutic biomolecules81,82,83, the development of plants with an inherent immunomodulatory effect is feasible. This could be achieved by engineering a plant’s structural proteins to suit the desired application of the implant while maintaining patent leaf vasculature, the xylem and phloem.
Methods
Leaf decellularization process and its modification
Spinach leaves were acquired from a local market and were used prior to expiration dates listed on the packaging or within 5 days of purchase, whichever was more recent. Their petioles were cannulated as described previously23. One set of leaves was decellularized according to our previously reported procedure (Fig. 1A). In short, leaves were perfused through their petioles with 10% SDS for 5 days, followed by perfusion with 0.1% TritonX-100 in 10% bleach solution for 2 days, and finished with perfusion of sterile deionized water for 2 days. After decellularization, leaves were stored in sterile deionized water at 4 °C until needed.
A second set of leaves were decellularized with our modified decellularization procedure (Fig. 1B). Prior to the perfusion of different detergent solutions, the leaves were treated to remove their cuticles through three one-minute serial washes with hexanes (98%, Mixed Isomers, Alfa Aesar, Haverhill, MA) and 1x PBS. 1% SDS in deionized water was perfused through the leaf for 1 day, followed by another 1 day perfusion of 0.1% TritonX-100 in 10% bleach and followed by 1 day perfusion of deionized water. After decellularization was complete, leaves were washed in 10 mM, pH 9.0 tris-HCl buffer (Sigma Aldrich, St. Louis, MO) for 12 h. The leaves were then frozen overnight prior to being lyophilized for 24 h, then stored at room temperature. Prior to any investigation, lyophilized leaves were rehydrated in the same tris buffer solution for at least 3 h before being washed twice with 1x PBS. Leaves were further sterilized with UV light exposure for 20 mins.
DNA and protein quantification
Leaves were processed using either the original decellularization process or the newer modified protocol. DNA and protein were extracted and measured in both groups as previously described23 to determine if the modified protocol decellularizes the tissue with the same efficacy of the original protocol. In short, leaves were flash frozen in liquid nitrogen and ground in a chilled mortar and pestle then passed through a 25-gauge needle and sonicated with 5 pulses x3 to further fragment them using a probe sonicator. A CyQuant Direct Cell Assay (Thermo Fisher, Waltham, MA) was used to detect DNA in samples. Protein was quantified using a Bradford Assay kit (Thermo Fisher).
Leaf transparency quantification
Transparency of native leaves and leaves that were processed through either the modified or original decellularization method were quantified. Integrated optical densities were measured using ImageJ Software in 5 different locations within the different leaf samples and corresponding backgrounds. Each measure was done in conjunction with a background control with the same region of interest area size. The measured intensity of the area behind the leaf was divided by the corresponding intensity of the background without the leaf in front of it to obtain the relative transparency value. A value of 1 denotes full transparency and a value of 0 denotes full opacity.
Fibroblast and mesenchymal stem cell culture
P5-P12 human dermal fibroblasts (CRL-2352, American Type Culture Collection, Manassas, VA) were cultured in fibroblast maintenance media (Iscove’s Modified Dulbelcco’s Medium supplemented with 10% fetal bovine serum, 2% Glutamax and 1% pen-strep), as previously described84. When fibroblasts were ready for use in the cytotoxicity assays, they were plated in 24-well plates at a seeding density of 25,000 cells/cm2 for 12 h prior to initiation of assays. Plating medium was removed and replaced with 1 mL of fibroblast maintenance media. Equal weights of either the original decellularized leaves or the modified decellularized leaves were cut up and placed in the media of different respective wells. The cells were cultured in these different conditions for up to 7 days for use in the different assays subsequently described.
Live/dead assay
Fibroblasts were incubated in the different material supplemented media for 24 h, after which a Live/Dead cell viability assay (L-3224, Thermo Fisher) was performed. Control wells for dead cells were treated with ethanol for 30 mins prior to staining. Cells were incubated in 8 μM ethidium homodimer and 4 μM calcein AM in DMEM for 30 mins. Cell nuclei were counterstained with Hoechst for 5 mins prior to fixation in 4% paraformaldehyde. Ethidium homodimer stains a dead nucleus red, calcein AM stains a live cell’s cytoplasm green, and the Hoechst counterstain stains the live cell nucleus blue. Phenotype of cells that were grown with either no leaf or with leaves that underwent the modified decellularization protocol was determined through staining of filamentous actin via a GFP conjugated phalloidin stain (A12379, Thermo Fisher). Fluorescent images were acquired on a DMIL inverted microscope (Leica Microsystems, Buffalo Grove, IL).
MTT metabolic activity assay
The metabolic activity of the fibroblasts cultured in different conditions was measured at 6 and 24 h, and 3 and 7 days after the medium was supplemented with the different types of decellularized leaves. The metabolic activity was measured using an MTT assay85. A stock 10 mg/mL solution of MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide, Thermo Fisher) in PBS was made. A 0.5 mL aliquot of the MTT stock solution was added to 4.5 mL of media and sterile filtered. This working solution was added to each well of cells at appropriate time points and allowed to incubate for 2 h at 37 °C. After incubation, the unreacted solution was aspirated to waste. The activated MTT was solubilized in dimethyl sulfoxide for 10 min and loaded into wells of a 96-well plate. Absorbance at 540 nm was read on a Molecular Devices SpectraMax250 plate reader with a premix for 5 secs. Metabolic activity was measured at each timepoint, and the activity of different conditions was compared to a control that lacked MTT supplementation into the media of the well.
SDS quantification
Decellularized leaves from either the original or the modified decellularization protocols were put into 1.5 mL centrifuge tubes that were then subjected to a liquid nitrogen bath and subsequently ground in a mortar and pestle. Fragments were suspended in 0.5 mL of diH2O and further processed by pulling through a 25-gauge syringe needle and by sonication with 5 pulses performed 3x to reduce leaf fragment size. Four samples from each leaf were taken and analyzed for SDS content by treatment with a Stains-All dye reagent (1-Ethyl-2-[3-(1-ethylnaphtho[1,2-d]thiazolin-2-ylidene)-2-methylpropenyl]naphtho[1,2-d]thiazoliumbromide,3,3′-Diethyl-9-methyl-4,5,4′,5′-dibenzothiacarbocyanine, E9379, Sigma Aldrich)86. Samples taken from the 10% SDS decellularized leaves were diluted 5x in diH2O. The samples were quantified using a standard curve of known SDS amounts ranging from 0 to 2000 g. Samples were read on a Molecular Devices SpectraMax250 plate reader at absorbance of 438 nm with a premix for 5 secs.
RGD functionalization of decellularized leaf
Spinach leaves that were decellularized by the modified decellularization process (1% SDS) were functionalized with a synthesized RGD-Dopamine peptide, as previously reported35. Decellularized leaves were washed in a 10 mM tris buffer solution (pH 9.0) for 30 mins. Leaves were then placed in a 1 mg/mL RGD-Dopamine solution in 10 mM tris buffer and were lightly agitated for 24 h. After 24 h, leaves were again rinsed in tris buffer to remove any unbound solution and then frozen overnight and lyophilized for 24 h. Functionalized leaves were stored lyophilized at 4 °C.
Subcutaneous implant surgery
Both non-functionalized and RGD-functionalized lyophilized leaves were cut into 1 cm x 1 cm square pieces and subsequently rehydrated in sterile 1x PBS for 20 min and placed under UV light for 20 mins to enhance sterilization. Leaves were washed once more in sterile PBS and placed in a 37 °C incubator until use in surgeries that same day.
All animal experiments were performed under the guidance of and approved by the Institutional Animal Care and Use Committee at Worcester Polytechnic Institute. Female Sprague Dawley rats weighing between 350 and 500 g were used in the study. Rats were anesethsized by injection of a combination of Ketamine (87 mg/kg) and Xylazine (5 mg/kg). Anesthesia was maintained by respiration of Isoflurane (1–2.5% mL) in oxygen through a nose-cone. A ~ 3-cm long incision was made in the rat abdominal skin along its midline. The abdominal skin was then bluntly dissected from the underlying tissue for 3-cm lateral to the end incision on both sides, creating 2 skin pockets. Two pieces of leaf were then implanted in one of the created pockets, with one leaf being placed on the cranial side of the pocket and another leaf being placed in the caudal side. The other created pocket was used as a control sham operation. Skin was then closed using 5–0 nylon suture. Animals were sacrificed ethically at the different reported time points (1 week, 2 weeks, and 4 weeks) post implantation and the tissues were then excised. Excised tissue was then fixed in 4% paraformaldehyde prior to histological processing.
Histological analysis
Fixed excised tissues were processed in alcohol, xylene, and paraffin in a Tissue-Tek Vip 6AI tissue processor (Sakura, Torrance, CA) in a 14 h processing cycle. After processing, tissues were embedded in paraffin blocks and 10 µm sections of the tissue were cut using a Leica RM 2235 microtome (Leica Microsystems, Buffalo Grove, Il). Sectioned tissues were stained using either a Hematoxylin and Eosin (H&E)87 or a Masson’s Trichrome stain88.
Cell infiltration and cell count in border zone measurements
Images of Hematoxylin and Eosin stained sections of implants were used to quantify the cells that had infiltrated into the implanted scaffold and into the immediate border zone of the implant. Cells were counted using ImageJ Software and were normalized to the size of the implanted material.
Collagen encapsulation and deposition measurements
Images of Masson’s Trichrome stained sections of implants were used to quantify collagen encapsulation thickness and deposition within leaf implants. All measurements were performed using ImageJ Software. Collagen encapsulation thickness was measured in 7 different random regions along the entirety of the implant to account for any observed irregularities56. Deposition area was measured and compared as a percentage of the total area of the leaf implant.
Statistical analysis
All results are presented as mean ± standard error of independent replicates. Statistical comparisons were made through either a t test, one-way ANOVA or a two-way ANOVA with a Tukey’s post hoc test depending upon the specific experimentation. The statistical tests were performed using SigmaPlot (Systat Software Inc., San Jose, CA). Statistical significance was determined to be p < 0.05.
Data availability
All data are available upon request to the corresponding author.
References
Schladt, D. P. & Israni, A. K. OPTN/SRTR 2021 Annual Data Report: Introduction. Am. J. Transplant. 23, S12–S20 (2023).
Langer, R. & Vacanti, J. P. Tissue engineering. Science 260, 920–926 (1993).
Gershlak, J. R. et al. Mesenchymal stem cells ability to generate traction stress in response to substrate stiffness is modulated by the changing extracellular matrix composition of the heart during development. Biochem. Biophys. Res. Commun. 439, 161–166 (2013).
Borg, T. K., Gay, R. E. & Johnson, L. D. Changes in the distribution of fibronectin and collagen during development of the neonatal rat heart. Collagen Relat. Res. 2, 211–218 (1982).
Bowers, S. L. K., Banerjee, I. & Baudino, T. A. The extracellular matrix: At the center of it all. J. Mol. Cell. Cardiol. 48, 474–482 (2010).
Badylak, S. F., Freytes, D. O. & Gilbert, T. W. Extracellular matrix as a biological scaffold material: Structure and function. Acta Biomaterialia 5, 1–13 (2009).
Song, J. J. & Ott, H. C. Organ engineering based on decellularized matrix scaffolds. Trends Mol. Med. 17, 424–432 (2011).
Rouwkema, J., Rivron, N. C. & Blitterswijk, C. A. van. Vascularization in tissue engineering. Trends Biotechnol. 26, 434–441 (2008).
Novosel, E. C., Kleinhans, C. & Kluger, P. J. Vascularization is the key challenge in tissue engineering. Adv. drug Deliv. Rev. 63, 300–311 (2011).
Griffith, C. K. et al. Diffusion limits of an in vitro thick prevascularized tissue. Tissue Eng. 11, 257–266 (2005).
Ott, H. C. et al. Perfusion-decellularized matrix: using nature’s platform to engineer a bioartificial heart. Nat. Med. 14, 213–221 (2008).
Guyette, J. P. et al. Perfusion decellularization of whole organs. Nat. Protoc. 9, 1451–1468 (2014).
Gershlak, J. R. & Black, L. D. Beta 1 integrin binding plays a role in the constant traction force generation in response to varying stiffness for cells grown on mature cardiac extracellular matrix. Exp. cell Res. 330, 311–324 (2015).
Singelyn, J. M. & Christman, K. L. Injectable materials for the treatment of myocardial infarction and heart failure: the promise of decellularized matrices. J. Cardiovascular Transl. Res. 3, 478–486 (2010).
Guyette, J. P. et al. Bioengineering human myocardium on native extracellular matrix. Circulation Res. 118, CIRCRESAHA.115.306874-72 (2015).
Badylak, S. F., Taylor, D. & Uygun, K. Whole-organ tissue engineering: decellularization and recellularization of three-dimensional matrix scaffolds. Annu. Rev. Biomed. Eng. 13, 27–53 (2011).
Keane, T. J., Londono, R., Turner, N. J. & Badylak, S. F. Consequences of ineffective decellularization of biologic scaffolds on the host response. Biomaterials 33, 1771–1781 (2012).
Sullivan, K. E., Quinn, K. P., Tang, K. M., Georgakoudi, I. & Black, L. D. 3rd Extracellular matrix remodeling following myocardial infarction influences the therapeutic potential of mesenchymal stem cells. Stem Cell Res. Ther. 5, 1–16 (2014).
Snedeker, J. G. & Gautieri, A. The role of collagen crosslinks in ageing and diabetes - the good, the bad, and the ugly. Muscles Ligaments Tendons J. 4, 303–308 (2014).
Kwak, H.-B. Aging, exercise, and extracellular matrix in the heart. J. Exerc. Rehab. 9, 338–347 (2013).
Fishman, J. A., Scobie, L. & Takeuchi, Y. Xenotransplantation-associated infectious risk: a WHO consultation. Xenotransplantation 19, 72–81 (2012).
Thyden, R. et al. An edible, decellularized plant derived cell carrier for lab grown meat. Appl. Sci. 12, 5155 (2022).
Gershlak, J. R. et al. Crossing kingdoms: Using decellularized plants as perfusable tissue engineering scaffolds. Biomaterials 125, 13–22 (2017).
McCulloh, K. A., Sperry, J. S. & Adler, F. R. Water transport in plants obeys Murray’s law. Nature 421, 939–942 (2003).
Burgess, S. S. O., Pittermann, J. & Dawson, T. E. Hydraulic efficiency and safety of branch xylem increases with height in Sequoia sempervirens (D. Don) crowns. Plant, cell Environ. 29, 229–239 (2006).
Crapo, P. M., Gilbert, T. W. & Badylak, S. F. An overview of tissue and whole organ decellularization processes. Biomaterials 32, 3233–3243 (2011).
Nakayama, K. H., Batchelder, C. A., Lee, C. I. & Tarantal, A. F. Renal tissue engineering with decellularized rhesus monkey kidneys: age-related differences. Tissue Eng. Part A 17, 2891–2901 (2011).
Feil, G. et al. Investigations of urothelial cells seeded on commercially available small intestine submucosa. Eur. Urol. 50, 1330–1337 (2006).
Friedrich, E. E. et al. Residual sodium dodecyl sulfate in decellularized muscle matrices leads to fibroblast activation in vitro and foreign body response in vivo. J. Tissue Eng. Regenerative Med. https://doi.org/10.1002/term.2604 (2017).
Cebotari, S. et al. Detergent decellularization of heart valves for tissue engineering: toxicological effects of residual detergents on human endothelial cells. Artif. Organs 34, 206–210 (2010).
Akhyari, P. et al. The quest for an optimized protocol for whole-heart decellularization: a comparison of three popular and a novel decellularization technique and their diverse effects on crucial extracellular matrix qualities. Tissue Eng. Part C. Methods 17, 915–926 (2011).
Caralt, M. et al. Optimization and critical evaluation of decellularization strategies to develop renal extracellular matrix scaffolds as biological templates for organ engineering and transplantation. Am. J. Transplant. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 15, 64–75 (2015).
Hudson, T. W., Liu, S. Y. & Schmidt, C. E. Engineering an improved acellular nerve graft via optimized chemical processing. Tissue Eng. 10, 1346–1358 (2004).
Yeats, T. H. & Rose, J. K. C. The Formation and Function of Plant Cuticles. Plant Physiol. 163, 5–20 (2013).
Fontana, G. et al. Biofunctionalized Plants as Diverse Biomaterials for Human Cell Culture. Adv. Healthc. Mater. 6, 1601225 (2017).
Adamski, M. et al. Two Methods for Decellularization of Plant Tissues for Tissue Engineering Applications. J. Vis. Exp. 135, 57586 (2018).
Gratzer, P. F., Harrison, R. D. & Woods, T. Matrix alteration and not residual sodium dodecyl sulfate cytotoxicity affects the cellular repopulation of a decellularized matrix. Tissue Eng. 12, 2975–2983 (2006).
Webber, A. C. Method for the measurement of transparency of sheet materials. J. Opt. Soc. Am. 47, 785 (1957).
Nerem, R. M. Tissue engineering: the hope, the hype, and the future. Tissue Eng. 12, 1143–1150 (2006).
Jain, R. K., Au, P., Tam, J., Duda, D. G. & Fukumura, D. Engineering vascularized tissue. Nat. Biotechnol. 23, 821–823 (2005).
Gershlak, J. R. & Ott, H. C. Bioprinting organs-progress toward a moonshot idea. Transplantation 104, 1310–1311 (2020).
Murphy, S. V., Coppi, P. D. & Atala, A. Opportunities and challenges of translational 3D bioprinting. Nat. Biomed. Eng. 4, 370–380 (2019).
Murphy, S. V. & Atala, A. 3D bioprinting of tissues and organs. Nat. Biotechnol. 32, 773–785 (2014).
Cui, H. et al. 3D printing of thick myocardial tissue constructs with anisotropic myofibers and perfusable vascular channels. Biomater. Adv. 153, 213579 (2023).
Noor, N. et al. 3D printing of personalized thick and perfusable cardiac patches and hearts. Adv. Sci. Weinh. Baden. -Wurtt. Ger. 6, 1900344 (2019).
Liu, X. et al. 3D liver tissue model with branched vascular networks by multimaterial bioprinting. Adv. Healthc. Mater. 10, e2101405 (2021).
Kim, J. J. & Cho, D.-W. Advanced strategies in 3D bioprinting for vascular tissue engineering and disease modelling using smart bioinks. Virtual Phys. Prototyp. 19, e2395470 (2024).
Gorbenko, N., Rinaldi, G., Sanchez, A. & Merna, N. Small-caliber vascular grafts engineered from decellularized leaves and cross-linked gelatin. Tissue Eng. Part A 29, 397–409 (2023).
Cheng, Y.-W., Shiwarski, D. J., Ball, R. L., Whitehead, K. A. & Feinberg, A. W. Engineering aligned skeletal muscle tissue using decellularized plant-derived scaffolds. ACS Biomater. Sci. Eng. 6, 3046–3054 (2020).
Heath, R. L. & Packer, L. Photoperoxidation in isolated chloroplasts: I. Kinetics and stoichiometry of fatty acid peroxidation. Arch. Biochem. Biophys.125, 189–198 (1968).
Lee, B., Botany, J. P. A. The plant cuticle. I. Its structure, distribution, and function. JSTOR. https://doi.org/10.2307/43236936 (1924).
Guimarães, M. et al. Preparation of cellulose nanofibrils from bamboo pulp by mechanical defibrillation for their applications in biodegradable composites. J. Nanosci. Nanotechnol. 15, 6751–6768 (2015).
Wang, M. O. et al. Evaluation of the in vitro cytotoxicity of cross-linked biomaterials. Biomacromolecules 14, 1321–1329 (2013).
Proulx, M. K. et al. Fibrin microthreads support mesenchymal stem cell growth while maintaining differentiation potential. J. Biomed. Mater. Res. Part A 96, 301–312 (2011).
Bush, K. A., Downing, B. R., Walsh, S. E. & Pins, G. D. Conjugation of extracellular matrix proteins to basal lamina analogs enhances keratinocyte attachment. J. Biomed. Mater. Res. Part A 80, 444–452 (2007).
Sussman, E. M., Halpin, M. C., Muster, J., Moon, R. T. & Ratner, B. D. Porous implants modulate healing and induce shifts in local macrophage polarization in the foreign body reaction. Ann. Biomed. Eng. 42, 1508–1516 (2014).
Yim, E. K. F. & Leong, K. W. Significance of synthetic nanostructures in dictating cellular response. Nanomed.: Nanotechnol., Biol., Med. 1, 10–21 (2005).
Kao, W. J., Lee, D., Schense, J. C. & Hubbell, J. A. Fibronectin modulates macrophage adhesion and FBGC formation: The role of RGD, PHSRN, and PRRARV domains. J. Biomed. Mater. Res. 55, 79–88 (2001).
Franz, S., Rammelt, S., Scharnweber, D. & Simon, J. C. Immune responses to implants - a review of the implications for the design of immunomodulatory biomaterials. Biomaterials 32, 6692–6709 (2011).
Jones, K. S. Effects of biomaterial-induced inflammation on fibrosis and rejection. Semin. Immunol. 20, 130–136 (2008).
Czaja, W. K., Young, D. J., Kawecki, M. & Brown, R. M. The future prospects of microbial cellulose in biomedical applications. Biomacromolecules 8, 1–12 (2007).
Helenius, G. et al. In vivo biocompatibility of bacterial cellulose. J. Biomed. Mater. Res. Part A 76A, 431–438 (2006).
Svensson, A. et al. Bacterial cellulose as a potential scaffold for tissue engineering of cartilage. Biomaterials 26, 419–431 (2005).
Müller, F. A. et al. Cellulose-based scaffold materials for cartilage tissue engineering. Biomaterials 27, 3955–3963 (2006).
Liuyun, J., Yubao, L. & Chengdong, X. Preparation and biological properties of a novel composite scaffold of nano-hydroxyapatite/chitosan/carboxymethyl cellulose for bone tissue engineering. J. Biomed. Sci. 16, 65 (2009).
Zaborowska, M. et al. Microporous bacterial cellulose as a potential scaffold for bone regeneration. Acta Biomater.6, 2540–2547 (2010).
Czaja, W., Krystynowicz, A., Bielecki, S. & Brown, R. M. Microbial cellulose—the natural power to heal wounds. Biomaterials 27, 145–151 (2006).
Modulevsky, D. J., Lefebvre, C., Haase, K., Al-Rekabi, Z. & Pelling, A. E. Apple derived cellulose scaffolds for 3D mammalian cell culture. PLoS One 9, e97835 (2014).
Modulevsky, D. J., Cuerrier, C. M. & Pelling, A. E. Biocompatibility of subcutaneously implanted plant-derived cellulose biomaterials. PLoS One 11, e0157894 (2016).
Pettolino, F. A., Walsh, C., Fincher, G. B. & Bacic, A. Determining the polysaccharide composition of plant cell walls. Nat. Protoc. 7, 1590–1607 (2012).
Anderson, J. M., Rodriguez, A. & Chang, D. T. Foreign body reaction to biomaterials. Semin. Immunol. 20, 86–100 (2008).
Makinodan, T., Santos, G. W. & Quinn, R. P. Immunosuppressive drugs. Pharmacol. Rev. 22, 189–247 (1970).
Kang, I. & Park, S. H. Infectious complications in SLE after immunosuppressive therapies. Curr. Opin. Rheumatol. 15, 528–534 (2003).
Fishman, J. A. & Rubin, R. H. Infection in organ-transplant recipients. N. Engl. J. Med. 338, 1741–1751 (1998).
Morais, J. M., Papadimitrakopoulos, F. & Burgess, D. J. Biomaterials/tissue interactions: possible solutions to overcome foreign body response. AAPS J. 12, 188–196 (2010).
Vishwakarma, A. et al. Engineering immunomodulatory biomaterials to tune the inflammatory response. Trends Biotechnol. 34, 470–482 (2016).
Babensee, J. E., Anderson, J. M., McIntire, L. V. & Mikos, A. G. Host response to tissue engineered devices. Adv. drug Deliv. Rev. 33, 111–139 (1998).
Ruoslahti, E. & Pierschbacher, M. D. New perspectives in cell adhesion: RGD and integrins. Science 238, 491–497 (1987).
Lee, H., Dellatore, S. M., Miller, W. M. & Messersmith, P. B. Mussel-inspired surface chemistry for multifunctional coatings. Science 318, 426–430 (2007).
Ku, S. H., Ryu, J., Hong, S. K., Lee, H. & Park, C. B. General functionalization route for cell adhesion on non-wetting surfaces. Biomaterials 31, 2535–2541 (2010).
Xu, J., Dolan, M. C., Medrano, G., Cramer, C. L. & Weathers, P. J. Green factory: plants as bioproduction platforms for recombinant proteins. Biotechnol. Adv. 30, 1171–1184 (2012).
Dolan, M. C., Wu, D., Cramer, C. L. & Xu, J. Hydroxyproline-O-glycosylated peptide tags enhance recombinant protein yields in tobacco transient expression. Process Biochem. 49, 490–495 (2014).
Medrano, G., Dolan, M. C., Condori, J., Radin, D. N. & Cramer, C. L. Quality assessment of recombinant proteins produced in plants. Methods Mol. Biol. (Clifton, N. J.) 824, 535–564 (2012).
Page, R. L. et al. Induction of stem cell gene expression in adult human fibroblasts without transgenes. Cloning Stem Cells 11, 417–426 (2009).
Twentyman, P. R. & Luscombe, M. A study of some variables in a tetrazolium dye (MTT) based assay for cell growth and chemosensitivity. Br. J. Cancer 56, 279–285 (1987).
Rusconi, F., Valton, E., Nguyen, R. & Dufourc, E. Quantification of sodium dodecyl sulfate in microliter-volume biochemical samples by visible light spectroscopy. Anal. Biochem. 295, 31–37 (2001).
Badylak, S. F. et al. The use of extracellular matrix as an inductive scaffold for the partial replacement of functional myocardium. Cell Transpl. 15, S29–S40 (2006).
Hansen, K. J. et al. Functional effects of delivering human mesenchymal stem cell-seeded biological sutures to an infarcted heart. BioRes. Open Access 5, 249–260 (2016).
Acknowledgements
This work is supported in part by the National Heart, Lung and Blood Institute (R01HL115282 to G.G.) and National Science Foundation (DGE1144804 to J.G., T.D., G.G.). The authors declare that they have no competing interests. All the data needed to evaluate the conclusions made in this paper are present within the data presented in the paper. Additional data may be requested from the authors.
Author information
Authors and Affiliations
Contributions
J.G. performed and analyzed decellularization protocols, perfusion studies, cellular cytoxicity assays, animal implantation studies, and histology. C.B. acquired and analyzed cellular cytotoxicity assays and histology. G.F. provided and created the RGD-dopamine peptide. L.P. and R.T. provided histology assistance. L.S. and A.dS. performed decellularization protocols. P.W. provided plant biology supervision. T.D. provided tissue engineering and animal study supervision. W.M. provided tissue engineering supervision. J.G. and G.G. designed the study. J.G. collected and assembled the data. G.G. supervised the study. J.G., P.W., T.D., G.G. wrote the manuscript. All authors reviewed and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gershlak, J.R., Burgess, C.K., Fontana, G. et al. Biocompatibility of decellularized spinach leaves. npj Biomed. Innov. 2, 23 (2025). https://doi.org/10.1038/s44385-025-00028-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s44385-025-00028-8
