
Abstract
Electrochemical urea oxidation offers a sustainable avenue for H2 production and wastewater denitrification within the water-energy nexus; however, its wide application is limited by detrimental cyanate or nitrite production instead of innocuous N2. Herein we demonstrate that atomically isolated asymmetric Ni–O–Ti sites on Ti foam anode achieve a N2 selectivity of 99%, surpassing the connected symmetric Ni–O–Ni counterparts in documented Ni-based electrocatalysts with N2 selectivity below 55%, and also deliver a H2 evolution rate of 22.0 mL h–1 when coupled to a Pt counter cathode under 213 mA cm–2 at 1.40 VRHE. These asymmetric sites, featuring oxygenophilic Ti adjacent to Ni, favor interaction with the carbonyl over amino groups in urea, thus preventing premature resonant C⎓N bond breakage before intramolecular N–N coupling towards N2 evolution. A prototype device powered by a commercial Si photovoltaic cell is further developed for solar-powered on-site urine processing and decentralized H2 production.
Similar content being viewed by others


Introduction
Urea is a milestone molecule in modern chemistry because of its substantial contribution to agricultural, industrial, and medical prosperity and is also a crucial N-containing biomolecule generated through mammalian protein catabolism1,2. However, the overuse of urea in agriculture, in conjunction with its runoff and overwhelming load of urea exceeding 58 megatons annually in municipal treatment facilities (Supplementary Notes 1), poses a latent threat to both ecosystem stability and human health3. Uncontrolled discharge of urea leads to eutrophication, intensifying the blooms of algae and other aquatic plants. The hydrolysis of urea yields ammonia, which, if released into the atmosphere, accelerates acid rain occurrence4. Generally, urea is treated through adsorption, biodegradation, reverse osmosis, and chemical oxidation methods, which have high operational complexity and consume large amounts of energy5. Notably, urea stands out as a decent hydrogen carrier (H2) with a gravimetric hydrogen content of 6.71 wt.%, fulfilling the present target as a reliable alternative to fossil fuels6. In the pursuit of transforming waste into valuable resources and achieving carbon neutrality, it is highly important to develop new strategies for urea-containing wastewater treatment and simultaneous H2 production7,8.
The electrochemical urea oxidation reaction (UOR) to innocuous N2 offers a sustainable solution for urea-containing wastewater denitrification, taking advantage of its facile working conditions and high processing capacity9,10. More importantly, compared with the oxygen evolution reaction (OER), the UOR offers a thermodynamically favorable pathway for supplying electrons for the coupled cathodic hydrogen evolution reaction (HER) (Fig. 1a)11. The development of UOR has been restricted by its dependence on expensive rare metals such as ruthenium, platinum, or iridium12. Recently, several earth-abundant and cost-effective transition metals, particularly nickel (Ni) and its oxides (NiO, NiOOH) with urease-like catalytic centers, have exhibited attractive UOR performances13,14,15,16. However, these documented Ni-based electrocatalysts frequently convert urea to deleterious cyanate (NCO–) and nitrite/nitrate (NOx–) species with N2 selectivities below 55%17,18, which might lower the stability of the integrated electrochemical system by poisoning the UOR anode and increase the environmental risk19. Structurally, urea contains two electron-donating amino (NH2) groups and an electron-withdrawing carbonyl (C = O) group that collectively engage in resonance to yield two C⎓N bonds with ~ 30% double bond character20,21. For the prevalent Ni-based catalysts under oxidative conditions, their connected symmetric Ni–O–Ni sites bind to urea in a symmetric bidentate-binuclear configuration via electron-donating NH2 ends22,23, blocking the resonance between the NH2 and C = O groups and thereby weakening the C⎓N bond, as evidenced by its transition from a quasi double bond to a single bond20. Theoretically, N2 formation proceeds through the pivotal intramolecular N–N bond coupling reaction during stepwise dehydrogenation of urea24. However, C⎓N weakening usually leads to its cleavage into cyanate (NCO–) and *NHx (where x = 1, 2), followed by their subsequent overoxidation to produce secondary NOx– (Fig. 1b)18. Therefore, the cleavage of the C⎓N bond in urea should be avoided for the selective conversion of urea to N2 during the electrochemical UOR, which might be achieved by donating electrons to the terminal O moiety of the C = O group to increase the electron accumulation on the resonant C⎓N bond (Fig. 1c).

a Scenario of the UOR towards clean water and energy production. b The UOR pathways for N2/CO2 or NOx–/NCO– production on connected symmetric Ni–O–Ni sites. c Illustration of strengthening the resonant C⎓N bond of urea by donating electrons to the terminal O moiety of the C = O group. d The bond formation energies of O with Ti, Ni, and other common transition metals at 298.15 K, defined as the standard enthalpy change for the formation of a diatomic species between O and the transition metal.
The C⎓N bond electron density of urea can be increased by pairing the catalytic Ni center with an auxiliary metal site that exhibits pronounced oxygenophilic affinity, which would facilitate electron donation to the terminal O moiety of the C = O group25,26,27. A careful assessment of the periodic table underscores the prominence of titanium (Ti) as an optimal choice owing to its bond formation energy of O (667 kJ mol–1), which is significantly greater than that of Ni (366 kJ mol–1), and other common transition metals (Fig. 1d)28,29. In this study, we endeavored to engineer atomically isolated Ni–O–Ti sites in an asymmetric configuration for electrochemical UOR into N2 by affixing single Ni atoms onto a commercially available Ti foam to preclude the formation of connected symmetric Ni–O–Ni sites and allow adjacent basal Ti atoms for strong C = O→Ti interactions26. Theoretical calculations in combination with in situ Raman spectroscopy, Fourier transform infrared spectroscopy, and on-line mass spectrometry were employed to reveal the formation of asymmetric Ni–O–Ti sites, electronically strengthened C⎓N bonds in urea and kinetically facilitated intramolecular N–N bond coupling, as well as their contributions to the N2 selectivity of the UOR. Furthermore, we conceptualized and engineered a prototype UOR–HER device by coupling a Ti foam monolithic electrode, which features atomically isolated asymmetric Ni–O–Ti sites, to a Pt cathode for efficient cathodic H2 evolution. Powered by a commercial Si photovoltaic cell, this device can harness solar energy for on-site urine processing and decentralized fuel production.
Results
Materials characterization
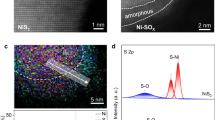
The atomically isolated asymmetric Ni–O–Ti sites were constructed by anchoring single Ni atoms onto a commercial monolithic Ti foam. The key to the synthesis of asymmetric Ni–O–Ti sites is the in situ generation of coordinated unsaturated Ti with dangling bonds on the amorphous TiOx surface layer of the Ti foam under a reductive 5.0% H2/Ar atmosphere (Supplementary Figs. 1, 2)30. These unsaturated Ti atoms could provide reliable immobilization sites for single Ni atoms via strong metal-support interactions. Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) revealed that the Ni atoms were atomically dispersed on the surface of the TiOx (Fig. 2a), which was highlighted by the corresponding line and 3D surface intensity profile aligned with the STEM simulation (Fig. 2b–d). Complementary STEM-mapping and X-ray diffraction (XRD) revealed the uniform dispersion of Ni atoms without discernible Ni clusters or nanoparticles (Fig. 2e and Supplementary Fig. 3). Inductively coupled plasma‒mass spectrometry (ICP-MS) analysis revealed a Ni loading of ~ 0.29 wt.% (Supplementary Table 1). For comparison, the connected symmetric Ni–O–Ni sites on the Ti foam anode were synthesized by increasing the Ni loading to 1.08 wt.% (Supplementary Fig. 4).

a Atomic-resolution HAADF-STEM image of asymmetric Ni–O–Ti sites with white circles marking Ni atoms. b Corresponding line intensity profile taken along the white dashed rectangle, and (c) 3D surface intensity profile in the yellow dashed square box. d 2D black and white STEM simulation images on the basis of the Z contrasts of atomic columns and corresponding structure diagram of asymmetric Ni–O–Ti sites. e STEM elemental mapping. f Ni K-edge X-ray absorption near-edge structure spectra of the atomically isolated asymmetric Ni–O–Ti sites and connected symmetric Ni–O–Ni sites on Ti foam. g FT-EXAFS spectra and (h) wavelet transformed (WT) k3-weighted χ(k)-function of Ni K-edge of the asymmetric Ni–O–Ti sites and symmetric Ni–O–Ni sites. Ni foil and NiO were used as the standard samples.
We then employed X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure spectroscopy (EXAFS) to decipher the chemical state and coordination environment of atomically isolated asymmetric Ni–O–Ti sites. The absorption edge energy of Ni in the asymmetric Ni–O–Ti sites was between those of standard Ni foil and NiO, suggesting the positively charged nature of the Ni atoms (Fig. 2f). In contrast, the absorption edge energy of connected symmetric Ni–O–Ni sites on Ti foam was similar to that on Ni foil, indicating of a more metallic state and consistent with the X-ray photoelectron spectroscopy (XPS) results (Supplementary Fig. 5). The Fourier-transformed EXAFS spectra of the asymmetric Ni–O–Ti sites displayed a dominant peak at 1.59 Å and a minor peak at 2.69 Å (Fig. 2g), which were attributed to Ni–O coordination in the first shell and Ni–Ti coordination in the second shell, respectively. The k-space of the minor peak located at 6.1 Å–1 differed from the 7.0 Å–1 k-space of the Ni–Ni coordination found in Ni foil, NiO, Ni(OH)2, or symmetric Ni–O–Ni sites according to the wavelet-transform EXAFS (Fig. 2h and Supplementary Fig. 6). Notably, the peak for metallic Ni–Ni scattering at 2.09 Å in the asymmetric Ni–O–Ti sites was absent, highlighting the atomically dispersed nature of the Ni atoms. Quantitative structural analysis indicated that each Ni atom in the asymmetric Ni–O–Ti sites was coordinated with approximately five adjacent Ti atoms in the second shell, which were interconnected by five O atoms (Supplementary Figs. 7, 8 and Table 2). As a comparison, the Ni atoms within the Ni–O–Ni sites on the Ti foam and conventional Ni-based materials were exclusively coordinated with Ni to form connected symmetric Ni–O–Ni sites.
Electrochemical performances
According to linear sweep voltammetry (LSV), the atomically isolated asymmetric Ni–O–Ti sites exhibited low potentials of 1.30 V and 1.33 V vs. reversible hydrogen electrode (VRHE) to achieve laboratory- and industrial-level current densities of 10 mA cm–2 and 100 mA cm–2, respectively, while the connected symmetric Ni–O–Ni counterparts required slightly higher potentials (Fig. 3a and Supplementary Figs. 9–12). For comparison, freestanding Ti foam was ineffective at catalyzing the UOR, while the Ni foam showed poor UOR activity, demanding considerably higher potentials of 1.38 and 1.54 VRHE to reach equivalent current densities of 10 mA cm–2 and 100 mA cm–2, respectively. Notably, the Ni–O–Ti sites had a substantially smaller Tafel slope (14.2 mV dec–1) than did the Ni–O–Ni sites (20.0 mV dec–1) and the Ni foam (20.8 mV dec–1), indicating a rapid electron transfer rate and superior reaction kinetics (Fig. 3b and Supplementary Fig. 13). Regarding the UOR potential, the asymmetric Ni–O–Ti sites outperformed most of the previously reported Ni-based electrocatalysts (Supplementary Fig. 14 and Table 3).

a LSV curves with 85% iR correction and (b) Tafel plots of the atomically isolated asymmetric Ni–O–Ti sites and connected symmetric Ni–O–Ni sites on Ti foam in 1.0 M KOH containing 0.33 M urea with a stirring rate of 500 rpm and 5 mV s–1. Freestanding Ti foam and Ni foam were used for comparison. The solution resistance is 2.1 ± 0.1 Ω. c The UOR selectivity of asymmetric Ni–O–Ti sites, symmetric Ni–O–Ni sites, and Ni foam as a function of the applied potential. Error bars represent the standard deviations derived from three distinct samples. d On-line MS for asymmetric Ni–O–Ti sites in 1.0 M KOH with 0.1 M CO(14NH2)2 and 0.1 M CO(15NH2)2. e The current density changes of asymmetric Ni–O–Ti sites after the introduction of 0.1 M NCO– and 0.1 M NO2– in 1.0 M KOH containing 0.33 M urea electrolyte. f Chronoamperometry measurements with the initial current density of 100 mA cm–2 in 1.0 M KOH containing 1.0 M urea, and the solution resistance is 2.0 ± 0.1 Ω.
Regardless of their comparable UOR potentials in catalyzing the UOR, asymmetric Ni–O–Ti sites and symmetric Ni–O–Ni sites exhibited significant differences in their selectivity for N2 evolution. Systematic analyses using ion chromatography and on-line mass spectrometry within a potential range of 1.40 to 1.70 VRHE (Supplementary Figs. 15–18) revealed that the asymmetric Ni–O–Ti sites primarily generated innocuous N2 (1), achieving a N2 selectivity exceeding 99% (Fig. 3c, Supplementary Figs. 19–21 and Supplementary Movie 1)17,18,31,32. Concurrently, a notable H2 evolution rate of 22.0 mL h–1 was delivered at the coupled Pt cathode under a current density of 213 mA cm–2 at 1.40 VRHE (2–3). Symmetric Ni–O–Ni sites, however, exhibited a poor N2 selectivity of 61% under 1.40 VRHE, which decreased progressively to 56%, 14%, and 9% as the potential increased to 1.50, 1.60, and 1.70 VRHE, respectively (Supplementary Figs. 22, 23 and Table 4), accompanied by a substantial production of NCO– and overoxidized NOx– (4). Moreover, conventional Ni foam displayed a disappointingly low N2 selectivity ranging from 10% to 30%, illustrating that Ni–O–Ni sites disfavored N2 production (Supplementary Figs. 24, 25). Furthermore, the indispensable contribution of Ti within the asymmetric Ni–O–Ti sites to the high N2 selectivity of the UOR was confirmed by the poor N2 selectivity (below 10%) of single Ni atoms dispersed on N-doped graphene (Ni1@NC) (Supplementary Fig. 26).
We subsequently utilized online mass spectrometry to analyze the mass-to-charge ratio of the produced N2 during UOR by feeding CO(14NH2)2 and CO(15NH2)2 in a 1:1 ratio. The observed mass spectral distribution of 14N2:15N2:14N15N at a 1:1:0 ratio offered compelling evidence of a dominant intramolecular N–N bond coupling reaction mediated by the asymmetric Ni–O–Ti sites (Fig. 3d)24. Control experiments indicated that the introduction of external 0.1 M NCO– and 0.1 M NO2– in the electrolyte led to the poisoning of asymmetric Ni–O–Ti sites, as evidenced by decreased current densities (Fig. 3e). To this end, the prevention of secondary NCO– and NOx– generation over the asymmetric Ni–O–Ti sites ensured the stability of the electrochemical system over 10,000 cycles and an extended period of 10 days with a well-maintained N2 selectivity of 99% during the laboratory test (Fig. 3f and Supplementary Figs. 27–30). A minor reduction in the current density was attributed to the depletion of urea as the UOR proceeded, which could be easily solved by refreshing the electrolyte. In contrast, the Ni–O–Ni sites and Ni foam exhibited more pronounced decreases in current density, retaining only 64.1% and 16.4% of their initial values, respectively.
Reaction mechanism of the UOR
In situ, Raman spectroscopy was utilized to clarify the role of atomically isolated asymmetric Ni–O–Ti sites in the selective oxidation of urea to N2 (Supplementary Fig. 31)33. The UOR on Ni-based electrocatalysts is known to follow a sequential electrochemical-chemical mechanism. That is, Ni first undergoes oxidation into high-valent Ni3+–O species. Subsequently, Ni3+–O reacts with the adsorbed urea to produce CO2, N2, and H2O, simultaneously regenerating the catalysts. This process was confirmed by the appearance of Ni3+–O on the asymmetric Ni–O–Ti sites at potentials ≥ 1.30 VRHE (Supplementary Fig. 32)34,35. A slightly greater potential was required for the formation of Ni3+–O species on the symmetric Ni–O–Ni sites and on the Ni foam (1.40 VRHE), which is consistent with the LSV curves of these materials. Upon the introduction of urea with a prototypical C⎓N stretching band at 1004 cm–1 (Fig. 4a)12, the Ni3+–O Raman peak rapidly diminished and completely vanished by 1.50 VRHE along with the emergence of adsorbed CO2 species (CO32–) at 1065 cm–1, highlighting Ni3+–O as the key species for the UOR (Supplementary Fig. 33)36. Interestingly, neither NCO– nor NOx– was observed on the asymmetric Ni–O–Ti sites (Supplementary Fig. 34), indicating that the C⎓N bond was well stabilized prior to intramolecular N–N coupling. In contrast, both the Ni–O–Ni sites and the Ni foam exhibited pronounced accumulations of NCO– (1221 and 1313 cm–1) and NO2– (1331 cm–1) species during the UOR, reflecting their poor selectivity for N2 evolution (Fig. 4b and Supplementary Fig. 35).

In situ Raman spectra of (a) atomically isolated asymmetric Ni–O–Ti sites and (b) connected symmetric Ni–O–Ni sites at different potentials during the UOR operations. The distinct Raman peaks at 476 and 556 cm–1 are attributed to Eg bending vibration and the A1g stretching modes of Ni3+–O, respectively. c The FTIR spectra of urea solution and adsorbed urea on atomically isolated asymmetric Ni–O–Ti sites and connected symmetric Ni–O–Ni sites. In situ FTIR spectra during the UOR on (d) atomically isolated asymmetric Ni–O–Ti sites and (e) connected symmetric Ni–O–Ni sites at different potentials.
To clarify the specific reaction pathways governed by the asymmetric Ni–O–Ti sites for the UOR, an in situ FTIR spectroscopy study was conducted37. Urea features ν(C = O) at 1670 cm–1, δ(NH2) at 1630 and 1600 cm–1, and νa(C⎓N) at 1466 cm–1 (Fig. 4c and Supplementary Fig. 36)38. As mentioned above, the C⎓N bond in urea possesses 30% hybridized double bonding characteristics due to the resonance between the NH2 and C = O groups. Upon interaction with symmetric Ni–O–Ni sites via NH2 groups, the C⎓N bond was weakened due to increased electron demand from donor N atoms, disrupting the resonance between the NH2 and C = O groups. This interaction is demonstrated by a redshift of νa(C⎓N) to 1445 cm–1 and a blueshift of ν(C = O) to 1694 cm–1, indicative of the weakening of the C⎓N bond and the strengthening of the C = O bond, respectively. These spectral shifts, in conjunction with the DFT simulations, corroborate the dual NH2 adsorption configuration of urea at symmetric Ni–O–Ni sites (Supplementary Fig. 37). In contrast, a blueshift of νa(C⎓N) to 1476 cm–1 was observed for urea adsorbed on the Ni–O–Ti sites, suggesting a strengthening of the C⎓N bond (Supplementary Fig. 38). Benefiting from the high oxygenophilicity of Ti, urea might bind to Ni–O–Ti sites in a new adsorption configuration via C = O→Ti interactions. With an increase in the potential to 1.30 VRHE, the IR peak assigned to urea gradually diminished, while a distinct peak attributed to the stretching vibration of the C–O single bond emerged at 1250 cm–1 (Fig. 4d)9,39. We believe that this peak is derived from the adsorption of the C = O group on Ni3+–O in the Ni–O–C = O→Ti configuration40. The peak at ~ 1380 cm–1 arose from CO32– in the electrolyte and gradually accumulated with increasing potential along with the physically adsorbed CO2 (2343 cm–1)41,42,43. The absence of NCO– or NOx– vibrational peaks in the spectra of asymmetric Ni–O–Ti sites across all potentials further confirms their high N2 selectivity. In contrast, symmetric Ni–O–Ni sites displayed an increasing NCO– peak at 2168 cm–1, associated with robust C⎓N bond cleavage of urea (Fig. 4e)44.
Theoretical investigation of the UOR
Building on the experimental results and discussions, comprehensive density functional theory (DFT) calculations were conducted to clarify the UOR pathways governed by asymmetric Ni–O–Ti sites (Supplementary Fig. 39 and Supplementary Data 1). As previously demonstrated, the Ni–O–Ti sites favored interactions with the C = O group of urea with higher adsorption energy (– 0.71 eV) than did interactions with the NH2 groups (– 0.22 eV). The electrochemically generated Ni3+–O then bound to the C moiety of the C = O group (Supplementary Fig. 40), while the oxygenophilic Ti atom was associated with the O moiety, forming an asymmetric bidentate binuclear intermediate (Fig. 5a). Significantly, this urea adsorption configuration on Ni–O–Ti sites manifested an electron accumulation on the two C⎓N bonds (+ 0.02 e and + 0.03 e), indicating a strengthened resonance between the NH2 and C = O groups (Fig. 5b). This was in stark contrast to the Ni–O–Ni sites where urea adsorption through its NH2 groups with a larger adsorption energy of – 1.50 eV led to a decrease in the electron density on the C⎓N bonds (– 0.40 e and – 0.49 e, Fig. 5c). The strengthened resonance within the urea molecule further shortened the C⎓N bond length from 1.38 Å to 1.35 Å and increased the bond strength from 4.94 eV to 5.40 eV, according to crystal orbital Hamilton population (COHP) analysis (Fig. 5d)45,46,47,48. In contrast, the resonant C⎓N bond was elongated to a single C–N bond of 1.47 Å with a destabilized bond strength of 4.22 eV49. Thus, the asymmetric Ni–O–Ti sites might prevent premature breakage of the primary C⎓N bond prior to intramolecular N–N coupling.

a The adsorption energies of urea on asymmetric Ni–O–Ti sites and symmetric Ni–O–Ni sites in different configurations. Charge density difference of urea adsorbed on (b) asymmetric Ni–O–Ti sites and (c) symmetric Ni–O–Ni sites. The yellow and blue iso-surfaces with an iso-value of 0.004 a.u. represent spatial charge accumulation and depletion, respectively. d The COHP analysis of C–N bond in the free and adsorbed urea on asymmetric Ni–O–Ti and symmetric Ni–O–Ni sites. e The UOR process diagram and Gibbs free energy change on asymmetric Ni–O–Ti and symmetric Ni–O–Ni sites.
The elemental steps of the UOR were then explored through broader theoretical calculations (Fig. 5e and Supplementary Figs. 41–43). Intriguingly, the urea adsorbed on the asymmetric Ni–O–Ti sites spontaneously underwent intramolecular N–N coupling during the initial dehydrogenation step, forming an *OCNH–NH2 intermediate while releasing 0.41 eV of energy. This pathway was more thermodynamically favorable than the alternative route, which involves the breaking of the C⎓N bond to form *ON + *OCNH2 intermediates, a reaction confronted with a large potential-independent energy barrier of + 1.03 eV. Subsequent stepwise dehydrogenation and desorption steps led to the proactive production of N2 with a relatively minor energy barrier of + 0.13 eV, consistent with the high UOR selectivity toward N2 on the asymmetric Ni–O–Ti sites. In contrast, at the symmetric Ni–O–Ni sites, urea necessitated a higher energy input (+ 0.34 eV) for the first dehydrogenation step to generate *NHCONH2. In the subsequent dehydrogenation stage, the generated *NHCONH was more inclined to undergo C⎓N bond cleavage into *HNO + *HNCO (Supplementary Fig. 44), a route that is more thermodynamically favorable by 0.23 eV over the formation of the *HN–NHCO intermediate via intramolecular N–N coupling, thus accounting for the excessive and uncontrolled oxidation of urea into NO2– and NCO– on conventional Ni-based catalysts during the UOR.
Solar-powered UOR and H2 evolution
To showcase the practical application of these asymmetric Ni–O–Ti sites, we designed a prototype UOR–HER device powered by a commercial monocrystalline Si solar panel (3.0 V, 49 cm2)50. The projected operation current of the designed system was estimated to be 183 mA according to the intersection of the J–V curve of the solar cell and the polarization curve of the electrolysis device (Fig. 6a). Compared with the OER, the potential required to reach a current density of 100 mA cm–2 on asymmetric Ni–O–Ti sites for the UOR was reduced by 0.28 V, resulting in an energy savings of ~ 17% for H2 production (Fig. 6b, Supplementary Fig. 45 and Supplementary Notes 2). Solar-powered UOR–HER electrolysis was then conducted on a clear summer day in Shanghai, China, using actual urine collected from public facilities and commercial Pt foil as the cathode electrode, delivering H2 and N2 production rates of approximately 3:1, with 75.7 and 25.2 mL h–1, respectively, and a solar-to-H2 conversion efficiency of 9.6% (Fig. 6c–e and Supplementary Fig. 46). The Faradaic efficiency of urea-to-N2 conversion and H2 production in this solar-powered reactor exceeded 99%. After 6 h of the electrochemical reaction, 96% of the N total in the urine was removed (Fig. 6f). In addition, the concentrations of NO3−, NO2−, and NH4+ were maintained at low levels throughout the other 4 cycles, revealing the high efficiency and stability of asymmetric Ni–O–Ti sites for practical UOR applications.

a J–V curve of the solar panel and the UOR polarization curves with the Ti foam containing atomically isolated asymmetric Ni–O–Ti sites as the anode and Pt foil as the cathode with a scan rate of 5 mV s–1. b LSV curves with 85% iR correction of the asymmetric Ni–O–Ti sites in 1.0 M KOH + 0.33 M urea and 1.0 M KOH for driving the UOR and OER, respectively. The on-line MS of H2 and N2/O2 collected from cathode and anode chambers, respectively, (c) in 1.0 M KOH + actual urine (urea: ~ 7 g L–1). d The digital image of the solar-powered reactor. e The collected H2 and N2 from actual urine. f Concentrations of N-total, NO3−, NO2− and NH3 during photo-electrochemical urine oxidation process.
Discussion
We have demonstrated the fabrication of atomically isolated asymmetric Ni–O–Ti sites on a Ti foam anode with an N2 selectivity of 99% for the UOR, surpassing the connected symmetric Ni–O–Ni counterparts in documented Ni-based electrocatalysts with N2 selectivity below 55% and delivering a H2 evolution rate of 22.0 mL h–1 when coupled to a Pt counter cathode under 213 mA cm–2 (1.40 VRHE). The oxygenophilic Ti enables urea adsorption on the asymmetric Ni–O–Ti sites via C = O, reinforcing the resonance between the NH2 and C = O groups to avoid premature breakage of the C⎓N bond and favor selective intramolecular N–N coupling to N2. Moreover, the prevention of poisoning by secondary NCO– and NOx– ensures the stability of the electrochemical system over an extended duration of 10 days. We have also conceptualized and engineered a prototype solar-powered UOR–HER device for on-site urine processing, which offers an eco-friendly solution to process urine in underdeveloped areas with limited infrastructure, reducing nitrogen pollution while supporting decentralized clean energy production.
Methods
Chemicals and materials
The Ti foam electrode (0.68 mm thick, 99.99%) utilized in the study was procured from Kunshan Guangjiayuan New Material Co., Ltd., China. Ni foam (1.0 mm thick, Tanqi New Materials Technology Department, Shanghai) and Pt foil (0.1 mm thick, 99.99%, GaossUnion) were used as received. The chemical reagents, nickel(II) chloride hexahydrate (NiCl2·6H2O, AR), potassium hydroxide (KOH, GR, 95%), potassium nitrate (KNO3, AR, ≥ 99.5%), potassium nitrite (KNO2, AR, 97%), potassium cyanate (KOCN, AR, 97%), heavy-oxygen water (H218O, 97atom% 18O), urea (CO(14NH2)2, 99.5%) and 15N-labeled urea (CO(15NH2)2, AR, 99atom%) and ethanol were sourced from Sinopharm Chemical Regent Company. All chemicals were used without further purification. Ultrapure water (R = 18.25 mΩ) has been applied in all the experiments.
Electrodes
The atomically isolated asymmetric Ni–O–Ti sites on the Ti foam were prepared using the droplet coating and annealing method. Typically, 50.0 μL, 100.0 μL and 200.0 μL homogeneous NiCl2·6H2O ethanol solution with a Ni mass concentration of 3.0 mg mL–1 was uniformly deposited onto 1.0 cm × 0.5 cm Ti foam, respectively. The deposition process was expedited by infrared lamp illumination, which accelerated solvent evaporation and prevented solvent aggregation arising from surface tension. Subsequently, the Ni-deposited electrodes were treated in a reductive atmosphere (5% H2/Ar) at 300 oC for 3 hours with a heating rate of 10 oC min–1 to yield the atomically isolated asymmetric Ni–O–Ti sites. The counterpart connected symmetric Ni–O–Ni sites were synthesized by increasing the volume of the homogeneous NiCl2·6H2O solution to 800.0 μL (Supplementary Table 1). The Ni1@NC was synthesized by high-temperature calcination (Supplementary Notes 3).
Characterization
XRD patterns were collected using a Rigaku Miniflex-600 model with Cu Kα radiation (λ = 0.15406 nm), operated at 40 kV and 15 mA. HAADF-STEM images were acquired utilizing a JEOL JEM-ARM200F TEM/STEM, equipped with a spherical aberration corrector and operated at 200 kV. ICP-MS analyses were performed on a Thermo Scientific iCAP Q. XPS assessments were performed with a PHI 5000 Verasa scanning X-ray microprobe from ULAC-PHI, Inc., using Al Kα radiation. The binding energy calibration was standardized to the C 1 s peak at 284.6 eV. STEM mapping was obtained on an FEI Talos F200X. The X-ray absorption fine structure spectra of Ni K-edge were collected at the Singapore Synchrotron Light Source Center (Supplementary Notes 4). In situ Raman spectra were acquired using a Renishaw inVia Qontor instrument, employing a 532 nm semiconductor laser as the illumination source. The FT-IR spectra were recorded on a Nicolet iS50FT-IR spectrometer (Thermo, USA), scanning from 4000 to 800 cm–1 at room temperature. A chronoamperometry test was performed for 600 s to collect FT-IR and Raman spectra. Spin-polarized DFT calculations were conducted using the Vienna Ab initio Simulation Package (Supplementary Notes 5 and Supplementary Data 1)51,52.
Electrochemical measurements
Electrochemical data were collected in a conventional three-electrode system operated by a CHI 760E electrochemical analyzer (Shanghai Chenhua, China) at ambient conditions (25 ± 2 °C). The UOR electrodes, Pt foil, and Hg/HgO electrodes were used as working electrodes, counter electrodes, and reference electrodes, respectively. All of the electrodes were assembled within a H-type cell. The working and counter electrode was spaced 2.0 ± 0.5 cm and separated by a Nafion 117 membrane (4.5 × 4.5 cm2) with a thickness of 183 μm. The membrane underwent an initial treatment in 5 wt% H2O2 at 80 oC for an hour, before a half-hour soak in deionized water. Subsequently, it was boiled in 5 wt% H2SO4 at the same temperature for an hour. Finally, it was soaked in deionized water for half-hour. The immersed area of the working electrode was 0.25 cm2 (0.5 × 0.5 cm2) in 20.0 mL electrolyte. If not specified otherwise, all potentials reported in this work were referenced to an RHE (Supplementary Notes 6). Before each measurement, the electrolyte was purged with Ar (99.99%) for 30 min. LSV curves were recorded three times at a scan rate of 5 mV s−1 in a 1.0 M KOH that contains 0.33 M urea with a pH value of 13.7 ± 0.1. The electrolyte in the cathodic compartment was stirred at a rate of 500 rpm during electrolysis. Chronopotentiometry tests for the UOR selectivity were conducted at certain applied potentials within the UOR window for 30 min. The durability tests were carried out using the chronoamperometry measurements under 1.33 VRHE with the initial current density of 100 mA cm–2 in 1.0 M KOH that contains 1.0 M urea. The electrochemical impedance spectroscopy (EIS) measurements were carried out in the frequency range from 100 kHz to 0.1 Hz. The charge transfer resistance (Rct) obtained by fitting the EIS data was used for the iR compensation. The Faradaic efficiency (FE) was calculated on the basis of the following Eq. (5):
where n is the number of electrons transferred for each product molecule. Specifically, the values of n for NCO–, H2, O2, N2, and NO2– are 0, 2, 4, 6 and 6, respectively. F is Faraday’s constant (96,485 C mol–1), N is the mole number of products, and Q is the number of charges passing through the electrode.
Determination of urea and N-total
Urea was quantified by high-performance liquid chromatography (HPLC, Thermo Scientific™ UltiMate™ 3000) method using a mobile phase of acetonitrile-water (40:60, V/V) with a flow rate of 1.0 mL min–1. The HPLC column was Agilent ZORBAX SB-C18 (4.6 × 150 mm). The detection wavelength was 195 nm. The injection volume was 10 μL. The N-total concentration was determined by using UV spectrophotometry according to the Chinese Standard HJ636–2012. Briefly, the nitrogen of nitrogen compounds was converted into nitrate first by digestion using alkaline potassium persulfate for 30 min at 120 oC. The absorbance was measured at wavelengths of 220 nm and 275 nm after acidification with concentrated HCl. The calibrated absorbance was determined by taking the absorbance at 220 nm and subtracting double the absorbance at 275 nm, showing a clear proportional relationship to the total nitrogen content.
Solar-powered H2 production
A commercial monocrystalline Si solar panel was used for solar-driven H2 production from urine. The anode of the solar reactor was fabricated by atomically isolated Ni–O–Ti sites on 2.0 × 2.0 cm2 commercial Ti foam. Commercial Pt foil (1.0 × 2.0 cm2) was used as a cathode, separated by a Nafion 117 membrane. The solar-to-H2 efficiency (SE) was calculated according to the following Eq. (6):
where jUOR is the UOR current (183 mA). E is the stable cell potential (2.6 V). FE is the Faradaic efficiency of HER (99%). The Asolarpanel is the solar-panel area (49.0 cm2). The Pin is the optical power (100 mW cm−2). The urine electrolysis measurement under natural sunlight was conducted on a clear summer day at the ___location of 31°1′29.69″ N 121°25′48.02″ E Shanghai, China.
Data availability
The datasets analyzed and generated during the current study are included in the paper and its Supplementary Information. The data that support the findings of this study are available from the corresponding author upon request. Source data are provided with this paper.
References
Chen, C. et al. Coupling N2 and CO2 in H2O to synthesize urea under ambient conditions. Nat. Chem. 12, 717–724 (2020).
Lee, J. S. et al. Urea cycle dysregulation generates clinically relevant genomic and biochemical signatures. Cell 174, 1559–1570 (2018).
Rollinson, A. N., Jones, J., Dupont, V. & Twigg, M. V. Urea as a hydrogen carrier: a perspective on its potential for safe, sustainable and long-term energy supply. Energy Environ. Sci. 4, 1216–1224 (2011).
Dong, H., Laguna, C. M., Liu, M. J., Guo, J. & Tarpeh, W. A. Electrified ion exchange enabled by water dissociation in bipolar membranes for nitrogen recovery from source-separated urine. Environ. Sci. Technol. 56, 16134–16143 (2022).
Urbańczyk, E., Sowa, M. & Simka, W. Urea removal from aqueous solutions–a review. J. Appl. Electrochem. 46, 1011–1029 (2016).
Li, J. et al. A review of hetero-structured Ni-based active catalysts for urea electrolysis. J. Mater. Chem. A 10, 9308–9326 (2022).
Xu, X. et al. Fluorinated Ni-O-C heterogeneous catalyst for efficient urea-assisted hydrogen production. Adv. Funct. Mater. 33, 2303986 (2023).
Zhang, X., Feizpoor, S., Humayun, M. & Wang, C. Urea oxidation reaction electrocatalysts: Correlation of structure, activity, and selectivity. Chem. Catal. 4, 100840 (2024).
Geng, S.-K. et al. Nickel ferrocyanide as a high-performance urea oxidation electrocatalyst. Nat. Energy 6, 904–912 (2021).
Zhao, H. et al. Facile, controllable, and ultrathin NiFe-LDH in situ grown on a Ni foam by ultrasonic self-etching for highly efficient urine conversion. Environ. Sci. Technol. 57, 2939–2948 (2023).
Boggs, B. K., King, R. L. & Botte, G. G. Urea electrolysis: direct hydrogen production from urine. Chem. Commun. 4859–4861 https://doi.org/10.1039/B905974A (2009).
Zheng, X. et al. Dual-atom support boosts nickel-catalyzed urea electrooxidation. Angew. Chem. Int. Ed. 62, e202217449 (2023).
Cai, M. et al. Formation and stabilization of NiOOH by introducing α-FeOOH in LDH: composite electrocatalyst for oxygen evolution and urea oxidation reactions. Adv. Mater. 35, 2209338 (2022).
Lu, X. F., Zhang, S. L., Sim, W. L., Gao, S. & Lou, X. W. Phosphorized CoNi2S4 yolk–shell spheres for highly efficient hydrogen production via water and urea electrolysis. Angew. Chem. Int. Ed. 60, 22885–22891 (2021).
Ma, Y., Ma, C., Wang, Y. & Wang, K. Advanced nickel-based catalysts for urea oxidation reaction: challenges and developments. Catalysts 12, 337 (2022).
Suárez, D., Díaz, N. & Merz, K. M. Ureases: quantum chemical calculations on cluster models. J. Am. Chem. Soc. 125, 15324–15337 (2003).
Li, J. et al. Deciphering and suppressing over-oxidized nitrogen in nickel-catalyzed urea electrolysis. Angew. Chem. Int. Ed. 60, 26656–26662 (2021).
Tatarchuk, S. W., Medvedev, J. J., Li, F., Tobolovskaya, Y. & Klinkova, A. Nickel-catalyzed urea electrolysis: from nitrite and cyanate as major products to nitrogen evolution. Angew. Chem. Int. Ed. 61, e202209839 (2022).
Hou, W., Yao, Y. C. & Zhang, L. Z. Advances in electrochemical reductive removal of oxyanions in water. Acta Chim. Sinica 81, 979–989 (2023).
Penland, R. B., Mizushima, S., Curran, C. & Quagliano, J. V. Infrared absorption spectra of inorganic coördination complexes. X. studies of some metal-urea complexes. J. Am. Chem. Soc. 79, 1575–1578 (1957).
Climent, V. et al. On the electrochemical and in-Situ fourier transform infrared spectroscopy characterization of urea adlayers at Pt(100) electrodes. Langmuir 13, 2380–2389 (1997).
Zhang, L. et al. A lattice-oxygen-involved reaction pathway to boost urea oxidation. Angew. Chem. Int. Ed. 131, 16976–16981 (2019).
Wang, P. et al. Directed urea-to-nitrite electrooxidation via tuning intermediate adsorption on Co, Ge Co-doped Ni sites. Adv. Funct. Mater. 33, 2300687 (2023).
Chen, W. et al. Unveiling the electrooxidation of urea: intramolecular coupling of the N−N bond. Angew. Chem. Int. Ed. 60, 7297–7307 (2021).
Zhang, K., Liu, C., Graham, N., Zhang, G. & Yu, W. Modulation of dual centers on cobalt-molybdenum oxides featuring synergistic effect of intermediate activation and radical mediator for electrocatalytic urea splitting. Nano Energy 87, 106217 (2021).
Overbury, S. H. et al. Complexity of intercalation in MXenes: destabilization of urea by two-dimensional titanium carbide. J. Am. Chem. Soc. 140, 10305–10314 (2018).
Wang, T. et al. Interfacial engineering of Ni3N/Mo2N heterojunctions for urea-assisted hydrogen evolution reaction. ACS Catal. 13, 4091–4100 (2023).
Kepp, K. P. A quantitative scale of oxophilicity and thiophilicity. Inorg. Chem. 55, 9461–9470 (2016).
Haynes, W. M. CRC Handbook of Chemistry and Physics. CRC Press: Boca Raton, FL, (2014).
Yao, Y. et al. Single atom Ru monolithic electrode for efficient chlorine evolution and nitrate reduction. Angew. Chem. Int. Ed. 61, e202208215 (2022).
Wang, D., Vijapur, S. H., Wang, Y. & Botte, G. G. NiCo2O4 nanosheets grown on current collectors as binder-free electrodes for hydrogen production via urea electrolysis. Int. J. Hydrogen Energy 42, 3987–3993 (2017).
Shen, Z., Qi, Y., Ge, W., Jiang, H. & Li, C. Highly selective electrooxidation of urea to nitrogen on copper/nickel boride interface under alkaline condition. Ind. Eng. Chem. Res. 62, 8736–8743 (2023).
Chen, Z. & Wang, Y. Direct monitoring of reaction intermediates through in situ characterization to promote the selectivity of N-integrated electrocatalytic CO2 reduction. Fund. Res. https://doi.org/10.1016/j.fmre.2023.03.019 (2023).
Qin, H. et al. Synergistic engineering of doping and vacancy in Ni(OH)2 to boost urea electrooxidation. Adv. Funct. Mater. 33, 2209698 (2023).
Zhu, Y. et al. Multistep dissolution of lamellar crystals generates superthin amorphous Ni(OH)2 catalyst for UOR. Adv. Mater. 35, 2301549 (2023).
Ji, Z. et al. Pathway manipulation via Ni, Co, and V ternary synergism to realize high efficiency for urea electrocatalytic oxidation. ACS Catal. 12, 569–579 (2021).
Cao, S. et al. Construction of an OCP-ATR-FTIR spectroscopy device to in situ monitor the interfacial reaction of contaminants: competitive adsorption of Cr(VI) and oxalate on hematite. Environ. Sci. Technol. 57, 16532–16540 (2023).
Gao, Y. et al. Engineering spin states of isolated copper species in a metal–organic framework improves urea electrosynthesis. Nano Micro Lett. 15, 158 (2023).
Liu, Z. et al. Interfacial water tuning by intermolecular spacing for stable CO2 electroreduction to C2+ products. Angew. Chem. Int. Ed. 62, e202309319 (2023).
Han, W. K. et al. Activating lattice oxygen in layered lithium oxides through cation vacancies for enhanced urea electrolysis. Angew. Chem. Int. Ed. 61, e202206050 (2022).
Sun, H. et al. Atomic metal–support interaction enables reconstruction-free dual-site electrocatalyst. J. Am. Chem. Soc. 144, 1174–1186 (2022).
Qin, Y. et al. Homogeneous vacancies-enhanced orbital hybridization for selective and efficient CO2-to-CO electrocatalysis. Nano Lett. 23, 9227–9234 (2023).
Zemtsova, V. M., Oshchepkov, A. G. & Savinova, E. R. Unveiling the role of iron in the nickel-catalyzed urea oxidation reaction. ACS Catal. 13, 13466–13473 (2023).
Gao, X. et al. Boosting urea electrooxidation on oxyanion-engineered nickel sites via inhibited water oxidation. Nat. Commun. 14, 5842 (2023).
Maintz, S., Deringer, V. L., Tchougreeff, A. L. & Dronskowski, R. LOBSTER: A tool to extract chemical bonding from plane-wave based DFT. J. Comput. Chem. 37, 1030–1035 (2016).
Nelson, R. et al. LOBSTER: Local orbital projections, atomic charges, and chemical-bonding analysis from projector-augmented-wave-based density-functional theory. J. Comput. Chem. 41, 1931–1940 (2020).
Dronskowski, R. & Blochl, P. E. Crystal orbital Hamilton populations (COHP): energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J. Phys. Chem. 97, 8617–8624 (1993).
Deringer, V. L., Tchougreeff, A. L. & Dronskowski, R. Crystal orbital Hamilton population (COHP) analysis as projected from plane-wave basis sets. J. Phys. Chem. A 115, 5461–5466 (2011).
Zhao, L. et al. A universal approach to dual-metal-atom catalytic sites confined in carbon dots for various target reactions. Proc. Natl. Acad. Sci. USA 120, e2308828120 (2023).
Sun, H. et al. Highly efficient overall urea electrolysis via single-atomically active centers on layered double hydroxide. Sci. Bull. 67, 1763–1775 (2022).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (U22A20402, L.Z.), (22206124, H.L.), (22102100, Y.Y.), (22306119, Y.S.), the National Key Research and Development Program of China (2022YFA1505000, H.L.), (2021YFA1201701, Y.Y.), the Natural Science Foundation of Shanghai (23ZR1431000, H.L.), (22ZR1431700, Y.Y.), Shenzhen Science and Technology Program (JCYJ20220818095601002, Y.Y.), the China Postdoctoral Science Foundation (2022M722080, G.Z.), (2021M702117, J.D.), (2022M712049, 2023T160419, Y.S.), and the Shanghai Postdoctoral Excellence Program (2022381, G.Z.), (2021182, J.D.). The authors acknowledge the support from the Instrumental Analysis Center of Shanghai Jiao Tong University, the Instrumental Analysis Center of the School of Environmental Science and Engineering, Shiyanjia Lab (www.shiyanjia.com), and ceshihui (www.ceshihui.cn). The computations were run on the π 2.0 cluster supported by the Center for High-Performance Computing at Shanghai Jiao Tong University.
Author information
Authors and Affiliations
Contributions
L. Z. Z., H. L., and Y. C. Y. supervised the project. G. M. Z., H. L., and Y. C. Y. conceived and designed the experiments. G. M. Z. conducted material synthesis and characterizations, electrochemical experiments, and DFT calculations. L. F. H. contributed to the electrochemical experiments. L. Z. contributed to the material characterizations. J. D., Q. Z., X. Y. Z., Y. B. S., J. X. W., and W. H. commented on the paper. G. M. Z., H. L., and L. Z. Z. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xueqiang Qi and the other anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhan, G., Hu, L., Li, H. et al. Highly selective urea electrooxidation coupled with efficient hydrogen evolution. Nat Commun 15, 5918 (2024). https://doi.org/10.1038/s41467-024-50343-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50343-8
