
Abstract
Methanogenic hydrocarbon degradation can be carried out by archaea that couple alkane oxidation directly to methanogenesis, or by syntrophic associations of bacteria with methanogenic archaea. However, metagenomic analyses of methanogenic environments have revealed other archaea with potential for alkane degradation but apparent inability to form methane, suggesting the existence of other modes of syntrophic hydrocarbon degradation. Here, we provide experimental evidence supporting the existence of a third mode of methanogenic degradation of hydrocarbons, mediated by syntrophic cooperation between archaeal partners. We collected sediment samples from a hot spring sediment in Tengchong, China, and enriched Hadarchaeota under methanogenic conditions at 60 °C, using hexadecane as substrate. We named the enriched archaeon Candidatus Melinoarchaeum fermentans DL9YTT1. We used 13C-substrate incubations, metagenomic, metatranscriptomic and metabolomic analyses to show that Ca. Melinoarchaeum uses alkyl-coenzyme M reductases (ACRs) to activate hexadecane via alkyl-CoM formation. Ca. Melinoarchaeum likely degrades alkanes to carbon dioxide, hydrogen and acetate, which can be used as substrates by hydrogenotrophic and acetoclastic methanogens such as Methanothermobacter and Methanothrix.
Similar content being viewed by others


Introduction
Microbial transformation of hydrocarbons to methane is an environmentally significant phenomenon, which occurs in a wide variety of electron acceptor-limited habitats, including oil reservoirs, coal deposits, oil contaminated groundwater, and marinedeep sediments1,2,3,4,5,6. This process occurs in a series of steps and requires close syntrophic associations between fermentative bacteria and methanogenic archaea2,3,4,5,6. The well-known initial degradation of multi-carbon alkanes is carried out by syntrophic bacteria via addition to fumarate yielding alkyl-substituted succinates by alkylsuccinate synthases (ASS)1,2,3,4,5,6. The alkyl-succinates are further transferred to intermediates, such as acetate, hydrogen or carbon dioxide, which are then consumed by hydrogenotrophic and/or acetoclastic methanogenic archaea for methane production. These anaerobic alkane-degrading bacteria reported to be involved in methanogenic hydrocarbon degradation include Desulfatibacillum, Desulfatiferula, Desulfosporosinus, Smithellla, and their methanogenic partners including Methanobacterium, Methanococcus, Methanoculleus, Methanosarcina, Methanothermobacter, Methanothrix1,2,3,4,5,6.
Later, an alternative mode initially activating short- to long-chain alkanes into corresponding alkyl-CoMs by ACRs has been confirmed in cultured anaerobic alkane-oxidizing archaea, which belong to lineages within the phylum Halobacteriota7,8,9,10,11,12. The long-chain alkane-degrading archaea, Ca. Methanoliparia, encodes tetrahydromethanopterin S-methyltransferase (MTR) and an additional canonical methyl-coenzyme M (CoM) reductase (MCR), which allows to couple alkane degradation and methanogenesis in the single archaeal cell12,13. This is the other mechanism contributing to hydrocarbon degradation under methanogenic conditions. Other acr-containing archaea, such as Ca. Alkanophaga14, Ca. Argoarchaeum7, Ca. Cerberiarchaeon11, Ca. Ethanoperedens8 and Ca. Syntrophoarchaeum10 lack mcr and mtr genes, which oxidizes alkanes to carbon dioxide and transfer the reducing equivalents to partner bacteria that perform sulfate reduction. In addition, it has recently been revealed that Ca. Cerberiarchaeon from the Hadarchaeota phylum can thrive on long-chain alkanes (hexadecane) under sulfate-reducing conditions, transferring electrons produced during alkane degradation to sulfate-reducing bacteria11.
Furthermore, metagenomic studies found metagenome-assembled genomes (MAGs) with acr but not mcr genes distributed widely in methanogenic environments15,16,17,18,19. For example, several MAGs in the phylum Hadarchaeota (WYZ-LMO4-6, JZ-1 bin_103, and JZ-1 bin_103) were constructed from terrestrial hot springs18,19; two MAGs (BA1 and BA2) in the class Bathyarchaeia (phylum Thermoproteota) were constructed from a coalbed well12,17,19,20,21. However, their eco-physiological role on the process of hydrocarbon degradation has not been reported. Here we enriched an archaeon from the phylum Hadarchaeota, which grew with long-chain alkanes to generate methane in cooperation with methanogenic archaeon.
Results and discussion
Methanogenesis incubation with long-chain alkanes
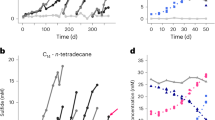
We collected sediments from Tengchong hot spring (Supplementary Fig. 1), which releases heated (65.7 °C), sulfate-depleted fluids containing various alkanes (Supplementary Data 1). We initially incubated these sediments in sulfate-free artificial freshwater medium (containing NaHCO3) with tetradecane as substrate at 60 °C, and observed a consecutive methane accumulation (data not shown). Then the culture was supplemented with hexadecane and sulfate-free artificial freshwater medium repeatedly during ~2 years of incubation (details see Materials and Methods, Fig. 1a). Continuous production of methane and carbon dioxide was observed during the incubation with hexadecane (Supplementary Fig. 2, Supplementary Data 2), while the non-alkane groups showed much lower methane and carbon dioxide production (Supplementary Fig. 3). when addition with 1,2-13C-labeled hexadecane during incubation at day 750, similar increasing of methane and carbon dioxide was observed, with 195.9–272.7 µmol methane and 62.8-76.6 µmol carbon dioxide production at day 810 (Fig. 1b, c). The ratio of methane to carbon dioxide produced was approximately 3:1, which was close to the theoretical value for methanogenic hexadecane degradation as depicted here:

Scheme of incubation experiments (a), concentration of methane (b) and carbon dioxide (c), carbon isotope of methane (d) and carbon dioxide (e), and concentration of 13C-labled methane (f) and carbon dioxide (g) in the cultures. r1 and r2 represent two replicates. The samples were incubated at 60 °C in Dec. 2021. Alkane groups: two bottles of slurry samples with the addition of 1% (volume/volume) tetradecane and fresh medium (with NaHCO3) during the initial cultivation; after 228 days culturing, 1% (volume/volume) tetradecane and 20% (v/v) fresh medium (with NaHCO3) were supplemented; after 386 days culturing, 0.1% (v/v) hexadecane and 20% (v/v) fresh medium (with NaHCO3) were supplemented, and the headspace in bottles was replaced with nitrogen; at day 614, after mixing the two samples thoroughly, divide the mixture into two equal parts, 0.1% (v/v) hexadecane and 100% (v/v) fresh medium (without NaHCO3) were supplemented, the headspace in the bottles was replaced with nitrogen; after 676 days culturing, 0.1% (v/v) hexadecane and 10% (v/v) fresh medium (without NaHCO3) were supplemented, and the headspace in the bottles was replaced with nitrogen; after 750 days culturing, 0.1% (v/v) 1,2-13C-labeled hexadecane and 10% (v/v) fresh medium (without NaHCO3) were supplemented, and the headspace in the bottles was replaced with nitrogen.
The methane production rate was ~0.04–0.05 µmol methane per ml of slurry per day. The δ13C values of methane and carbon dioxide also increased substantially (Fig. 1d, e). The amount of 13C-labeled methane produced were 3.9–6.2 µmol, and the amount of 13C-labeled carbon dioxide produced was 1.3–1.8 µmol (Fig. 1f, g). The ratio of 13C-labeled methane to 13C-labeled carbon dioxide produced was also approximately 3:1.
Ca. Melinoarchaeum spp. archaea dominate in the alkane cultures
Genomic DNA extracted from the original sample and incubation experiments were subjected to the metagenomic analysis. In alkane incubations, Hadarchaeota stood out as the most dominant microbial group, with relative abundances of 42.8% and 56.5%; Thermoproteota (12.3–10.2%), and Methanobacteriota (7.6–5.1%) were also dominant; Chloroflexota (3.2–2.7%), Bipolaricaulota (1.7–1.4%), Patescibacteria (1.2–0.9%), Halobacteriota (1.2–0.8%), Asgardarchaeota (1.4–0.5%) and Nitrospirota (0.4–0.6%) showed lower abundance (Fig. 2a and Supplementary Data 3). A total of 68 high-quality MAGs (completeness > 80%, contamination < 5%) were obtained, belonging to the phyla Hadarchaeota, Halobacteriota, Methanobacteriota, Thermoplasmatota, Thermoproteota, Acidobacteriota, Bipolaricaulota, Caldisericota, Chloroflexota, Cyanobacteria, Desulfobacterota, Elusimicrobiota, Firmicutes, Nitrospirota, Verrucomicrobiota and WOR-3. The MAGs TC202112_7_001 and TC202112_8_001 of the Hadarchaeota group exhibited the highest relative abundances at 41.6% and 55.5%, respectively (Fig. 2b). Both were classified within the Hadarchaeia class, Hadarchaeales order, WYZ-LMO6 family, and WYZ-LMO genus based on the Genome Taxonomy Database (GTDB) taxonomy22 (Supplementary Data 3). Both MAGs carried the acr genes (Supplementary Data 4). According to the metabolic discussion below we named the dominant organism represented by these two MAGs as Ca. Melinoarchaeum fermentans DL9YTT1. In contrast, the abundance of Hadarchaeota MAGs was less than 0.6% in groups without the addition of alkanes, and we did not detect any Hadarchaeota MAGs in the metagenomic sequences of the original slurry (Supplementary Fig. 4).

a taxonomies of the MAGs in phylum level; Other, including the unclassified MAGs and MAGs with relative abundance <0.5%; Unmapped, including the relative abundance of reads which did not map to MAGs. b the relative abundance of Ca. Melinoarchaeum fermentans DL9YTT1, Methanothermobacter and Methanothrix. The samples for metagenomic and metatranscriptomic sequencing were collected after culturing for 791 days. c the abundance of Ca. Melinoarchaeum determined by Q-PCR of their 16S rRNA genes; the error bars are obtained from triplicate Q-PCR reaction. r1 and r2 represent two replicates.
Quantitative PCR (Q-PCR) revealed that following alkane incubations, the abundance of Ca. Melinoarchaeum spp. increased by 26–136 folds to 1.7–9.1 × 107 copies/ml compared to the sample collected on day 0 (Fig. 2c). These indicates that the growth of this archaeon depends on the availability of long-chain alkanes. However, when hexadecane was added under sulfate conditions, the growth of Ca.Melinoarchaeum spp. was not stimulated (Supplementary Fig. 5), indicating that their growth cannot be coupled with sulfate reduction. Fluorescence in situ hybridization with specific probes showed that Ca. Melinoarchaeum spp. grows in mono-species filaments, and does not exhibit tight association with other microbial cells (Supplementary Fig. 6).
The genome of Ca. Melinoarchaeum fermentans DL9YTT1 is highly similar to the previously published MAG JZ-2_bin_19918, which was also binned from Tengchong hot spring sediment metagenome with average nucleotide identity (ANI) of 90.0%. We combined the MAGs obtained in this study with data from public databases, resulting in a total of 48 MAGs of the phylum Hadarchaeota (Fig. 3a). These MAGs group into one class, two orders and six families based on GTDB taxonomy (Supplementary Data 5). Together with 12 other MAGs, Ca. Melinoarchaeum spp. forms the genus Ca. Melinoarchaeum. The genus Ca. Melinoarchaeaum and their sister genus Ca. Cerberiarchaeum form the family Ca. Cerberiarchaeaceae11.

a phylogenomic affiliation of the Hadarchaeota MAGs based on 37 conserved protein sequences and using 233 representative archaeal genomes. b phylogenomic affiliation of the AcrAs and McrAs based on the alignments with 461 aligned positions. The tree branches were classified into four distinct groups: the ACR clade; TACK clade; Class I&II clade and Class III clade, the last three are canonical McrA clades. ACR clade contains the AcrAs of Ca. Melinoarchaeum spp.; Class I&II clade contains the McrAs of Methanothermobacter and Methanothrix. Alignments were generated using MAFFT43 and then filtered with trimAl44, and the trees were built by the IQ-Tree45 method with the model LG + C60 + F + G with 1000 bootstrap replicates. Bootstrap values > 90% shown in gray dots.
Ca. Melinoarchaeum spp. activate alkanes with highly transcribed ACRs
In the entire metagenome, the ACR genes only present in two MAGs of Ca. Melinoarchaeum spp. (Supplementary Data 3). Both MAGs contain three copies of the acrA gene. Two of these acrA genes form a continuous operon with the acrB and acrG genes, while the third does not form an operon (Supplementary Data 4). Phylogenetic analysis of the AcrA-subunits showed that Hadarchaeota form a distinct cluster next to those of Ca. Bathyarchaeia and Ca. Methanoliparia (Fig. 3b). We also analyzed the gene expression level of Ca. Melinoarchaeum spp. during alkanes degradation. Ca. Melinoarchaeum spp. exhibited the highest transcriptional activity among all MAGs, with a relative abundance of 36.4 and 43.4% (Fig. 2b, Supplementary Data 3). The acr genes of Ca. Melinoarchaeum spp. were highly expressed (with fragments per kilobase of transcript per million mapped reads, FPKM = 179–852) (Fig. 4a, Supplementary Data 4).

a genome-inferred model and corresponding gene expression patterns for hexadecane degradation in Ca. Melinoarchaeum fermentans sp. The steps involving substrate activation (red arrows), β-oxidation (blue arrows), Wood-Ljungdahl pathway (yellow arrows) and energy conservation (black arrows). The non-confirmed steps for transform hexadecyl-CoM to hexadecanoate are indicated in dot line. b genome-inferred model and corresponding gene expression patterns for hydrogenotrophic methanogenesis of genus Methanothermobacter; c genome-inferred model and corresponding gene expression patterns for acetoclastic methanogenesis of Methanothrix. The names of genes not found in the genome were shown in gray font. Fdred, reduced ferredoxins; Fdox, oxidized ferredoxins. Squares indicate different gene transcription levels. List of related genes and gene transcription levels was showed in Supplementary Data 4.
We analyzed the hexadecane cultures for the formation of products of the ACR-based alkane activation using Q-Exactive Plus Orbitrap mass spectrometry (Fig. 5). We found peaks of hexadecyl-CoM (m/z = 365.21884) and its fragmentations products, including hexadecyl-thiol (m/z = 257.23082), ethenesulfonate (m/z = 106.98079) and sulfonate (m/z = 80.96517) (Fig. 5a, b). Moreover, cultures supplied with 1,2-13C-hexadecane contained a mass peak of 1,2-13C-hexadecyl-CoM (m/z = 367.22548) and the fragment of 1,2-13C-hexadecyl-thiol (m/z = 259.23739), their mass is shifted by 2 units compared to the unlabeled group (Fig. 5c, d). The retention times of the extracted metabolites of hexadecyl-CoM, as determined by HPLC-MS/MS, closely matched those of synthesized authentic standards (Supplementary Fig. 7). In contrast, the respective alkylsuccinates, the activation products formed by ASS were not detected in alkane cultures. This is in agreement with the lack of ass genes in Ca. Melinoarchaeum spp. and the entire assemblies within the whole metagenome of alkane cultures23,24,25. These findings excluded the activation of alkanes through fumarate addition pathway in our cultures.

a MS analysis of extracts of culture with unlabeled hexadecane showed a peak at m/z = 365.21884 (red), which matches the authentic hexadecyl-CoM standard (blue). b Fragmentation of the isolated m/z range of 365.0–365.4 yields hexadecyl-thiol (C16H33S−, m/z = 257.23082), ethenesulfonate (C2H3SO3−, m/z = 106.98079) and sulfonate (HSO3−, m/z = 80.96517). These peaks are also produced by hexadecyl-CoM standards. c MS analysis of extracts of culture with 1,2-13C-labeled hexadecane yielded a peak at m/z = 367.22548 (red). d Fragmentation of the isolated m/z range of 367.0–367.4 yielded 1,2-13C-labeled hexadecyl-thiol (C16H33S−, m/z = 259.23739), unlabeled ethenesulfonate (C2H3SO3−, m/z = 106.98084) and sulfonate (HSO3−, m/z = 80.96515). The mass errors for all mass peaks are <5 ppm.
Methanogenic alkane degradation in cooperation with multiple archaea
The degradation of the alkyl-CoMs generated by the ACRs involves their conversion into fatty acids. The pathways and enzymes involved in this reaction are yet unknown9,10,20,26 (Fig. 4a). The Ca. Melinoarchaeum spp. encodes long-chain acyl-CoA synthetase (ACSL/FadD) and a complete β-oxidation pathway. This allows the activation of long-chain fatty acids as CoA-bound acyl units, and splitting of those compounds into acetyl-CoA units. The acetyl-CoA decarbonylase/synthase (ACS/CODH) complex could work in the cleaving of acetyl-CoAs into carbon dioxide and methyl-H4MPT, then the H4MPT-bound methyl groups are fully oxidized to carbon dioxide with the downstream part of the Wood-Ljungdahl (WL) pathway.
Ca. Melinoarchaeum fermentans sp. and all other ACR-containing Hadarchaeota do not encode MTR and canonical MCR, the crucial elements of methanogenesis (Supplementary Data 5). In contrast, Ca. Melinoarchaeum spp. might use the reduced ferredoxin, F420H2 and NADH produced from β-oxidation and WL pathway, to form hydrogen (Fig. 4a). Enzymes for this process are ferredoxin hydrogenase (Mvh), the beta subunit of F420-reducing NiFe-hydrogenase (FrhB) and sulphohydrogenase (HydAD). The acetyl-CoA synthetase (ACSS) might be used to convert acetyl-CoAs, the product of β-oxidation, into acetate. Ca. Melinoarchaeum spp. encodes related genes and all of them were highly expressed (FPKM > 100) (Fig. 4a, Supplementary Data 4).
Based on the metabolic pathway predication and gene expression in our cultures, two groups of methanogens capable of acetate fermentation and carbon dioxide reduction were most likely associated with the alkane degradation process (Fig. 4b, c and Supplementary Data 3). The MAGs of the genus Methanothermobacter (TC202112_7_002 and TC202112_8_003) in the phylum Methanobacteriota and the genus Methanothrix (TC202112_7_009 and TC202112_8_014) in phylum Halobacteriota were detected in our long-chain alkane cultures, with relative abundance of 4.8–6.9% and 0.6–0.9% respectively. The Methanothermobacter and Methanothrix MAGs contained the complete pathway of hydrogenotrophic methanogenesis and acetoclastic methanogenesis respectively. The mcr genes of Methanothermobacter exhibited high expression levels in alkane cultures, with average FPKM values of 1254–1466; the expression of mcr genes in Methanothrix was relatively lower, with average FPKM values of 39–47 (Fig. 4b, c and Supplementary Data 4). Therefore, we speculate that the degradation proceeds in a syntrophic interactions of Ca. Melinoarchaeum spp. and methanogens. Ca. Melinoarchaeum spp. might degrade alkanes to hydrogen, acetate and carbon dioxide. Then, the hydrogenotrophic methanogens (Methanothermobacter) might use hydrogen and carbon dioxide for methane production; and acetoclastic methanogens (Methanothrix) might use the acetate for methane and carbon dioxide production. These steps might degrade the model compound hexadecane according to:
At standard conditions, the breakdown of alkanes in to hydrogen, acetate, and carbon dioxide is highly endergonic, hence very low intermediate concentrations would need to be maintained to allow growth on these organisms27. The hexadecane cultures contained low concentration of acetate (8.8–15.2 μM) and hydrogen (8.8–25.6 pa) (Supplementary Data 6). Such low of intermediate concentrations were also described for the syntrophic associations of alkane-degrading bacteria and methanogenic archaea6,28.
Most previously cultured ACR-containing archaea were considered to transfer electrons from alkane oxidation to sulfate reducers7,8,10,14. In contrast, our cultures do not contain sulfate, but produces methane as electron sink. The interaction between alkane degrading archaea and methanogens likely differs from that suggested for consortia of alkane degrading archaea and their sulfate-reducing partners7,8,10,14. Both Ca. Melinoarchaeum spp. and the methanogens (Methanothermobacter and Methanothrix) do not encode type IV pilin (PilA) and multi-heme c-type cytochromes (MHCs). These proteins were suggested to form the filamentous nanowires that mediate direct interspecies electron transfer10,14. The lack of enzymes to establish direct interspecies electron transfer agrees with the members of Ca. Melinoarchaeum spp. appear as single cells rather than form consortia with other cells (Supplementary Fig. 6).
Potential habitats of the members in Ca. Melinoarchaeum
Although culturable Ca. Melinoarchaeum and Ca. Cerberiarchaeon, both exhibiting activity in the degradation of long-chain alkanes, belong to the same family (Ca. Cerberiarchaeaceae) within the Hadarchaeota phylum, Ca. Cerberiarchaeon was enriched from marine hydrothermal vent sediments11, while here Ca. Melinoarchaeum was sourced from terrestrial hot springs. Additionally, they exhibit distinct metabolic pathways; Ca. Cerberiarchaeon interacts with sulfate-reducing bacteria under sulfate conditions, whereas Ca. Melinoarchaeum cooperate with methane-producing archaea under methane-producing conditions. All 13 MAGs of genus Ca. Melinoarchaeum derived from continental hot spring sediments, including Tengchong hot spring in China, Yellowstone National Park and little hot spring in United States18,19 (Supplementary Data 5). Furthermore, samples of Yellowstone National Park contain acrA genes and gene transcripts clustering with Ca. Melinoarchaeaum29. Thus, the members of genus Ca. Melinoarchaeum are widely distributed among hot spring environments, where they likely grow as alkane degraders. All MAGs in genus Ca. Melinoarchaeum have the genes for alkane degradation, which include the acr operon, a complete β-oxidation and WL pathway for further degradation of alkanes to carbon dioxide (Supplementary Fig. 8). Only the less complete MAG GEM56 (58.46%), lacks most of the WL pathway. All Ca. Melinoarchaeum MAGs have very similar electron cycling complexes, such as Mvh, FrhB and HydAD, but lack of respiratory pathways. This indicates incomplete degradation of alkanes into acetate and/or hydrogen would be a common trait for the cluster of Ca. Melinoarchaeum.
Hot springs are dynamic geobiologically active environments and often rich in alkanes, including short-, mid-, and long-chain alkanes and aromatic hydrocarbons15,30,31,32,33. These alkanes are considered as abiogenic and formed in the deep subsurface, from where they migrate with rising fluids to reach the surface15,30,31,32,33. In addition to hot spring environments, the 16S rRNA genes of the genus Ca. Melinoarchaeum were also detected in mud volcanoes and subsurface aquifers (Supplementary Data 7). These environments also communicate with the deep Earth. Thus, we hypothesize that the members of genus Ca. Melinoarchaeum, are more abundant in the deep subsurface, and are occasionally transported and emplaced within the surface hot springs. Future studies will reveal if Ca. Melinoarchaeum or related Hadarchaeota have a larger role in reservoir biochemistry.
Our study revealed that the archaeon Ca. Melinoarchaeum spp., belonging to the phylum Hadarchaeota, grows as an ACR-based alkane degrader together with methanogens, thereby establishing an archaea-archaea syntrophy for methanogenic hydrocarbon degradation (Fig. 6). This finding explains the frequent observation of MAGs of ACR-containing archaea without canonical methanogenesis pathways in methanogenic environments15,16,17,18,19. Our study expands our understanding of archaeal alkane degradation and significantly extends present knowledge of syntrophy in methanogenic alkane degradation.

Three metabolic modes have been identified for the execution of methanogenic hydrocarbon degradation: (I) a symbiotic relationship between syntrophic bacteria and methanogens, (II) a standalone archaeon, and (III) a archaea-archaea syntrophy.
Methods
Etymology
Melinoarchaeum fermentans. Me.li.no.ar.chae’um. N.L neut. n. Melinoe, the greek goddess of nightmares, a resident of Hades, the underworld. N.L. neut. n. archaeum, ancient one, archaeon; an archaeon named after Melinoe, fer.men’tans. L. part. adj. fermentans, fermenting.
Sample collection
Our samples were collected from a hot spring sediment in Tengchong, China (98.82350°N, 25.04233°W) in January 2020 (Supplementary Fig. 1). The water temperature of the sampling site was 65.7 °C and pH was 6.7. The sediment samples for culturing were collected and kept in anaerobic bottles with nitrogen as the head space at room temperature. Sediment samples for sequencing and upper water samples for chemical parameter measurement were stored in dry ice during transportation to the laboratory, then transferred into a −80 oC refrigerator.
Cultivation conditions
Before incubation, the sediments were washed twice with sulfate free artificial freshwater medium34, through centrifugation at 13,800 × g for 10 min in anaerobic chamber. Then, the sediment was mixed again with anaerobic artificial freshwater medium (with 5 mM NaHCO3). Then, 100 ml slurries were dispensed into 156 ml serum bottles, which were sealed with butyl rubber stoppers (Bellco Glass) and aluminum caps. The ratio of liquid to sediment in slurries was ~10:1. The following tetradecane were added with 1% (volume/volume, v/v) into two bottles. The samples were incubated at 60 °C in Dec. 2021 (Fig. 1a). After 228 days culturing, 1% (v/v) tetradecane and 20% (v/v) fresh medium (with NaHCO3) were supplemented. After 386 days culturing, 0.1% (v/v) hexadecane and 20% (v/v) fresh medium (with NaHCO3) were supplemented, and the headspace in bottles was replaced with nitrogen. At day 614, after mixing the two samples thoroughly, divide the mixture into two equal parts, 0.1% (v/v) hexadecane and 100% (v/v) fresh medium (without NaHCO3) were supplemented, the headspace in the bottles was replaced with nitrogen. Due to the addition of sodium bicarbonate in the culture medium, which would affect the detection of carbon dioxide produced during alkane degradation (Supplementary Figs. 2 and 3), we replaced the medium with one without sodium bicarbonate at this time. After 676 days culturing, 0.1% (v/v) hexadecane and 10% (v/v) fresh medium (without NaHCO3) were supplemented, and the headspace in the bottles was replaced with nitrogen. After 750 days culturing, 0.1% (v/v) 1,2-13C-labeled hexadecane and 10% (v/v) fresh medium (without NaHCO3) were supplemented, and the headspace in the bottles was replaced with nitrogen. Two bottles of slurry samples without the addition of alkanes were subjected to identical treatments at various time points. The samples for metagenomic and metatranscriptomic sequencing were collected after culturing for 791 days. Cultures were centrifuged at 13,800 × g for 10 min to separate the sediment and supernatant. Then samples stored at −80 °C for sediment DNA/RNA isolation and acetate measurements in supernatant. 1 ml of headspace gas was extracted and injected into a 10 ml serum bottle (filled with nitrogen) then for the detection of methane and carbon dioxide; 1 ml headspace gas was taken directly from the culture bottles for hydrogen detection.
To test whether Ca. Melinoarchaeum fermentans sp. can grow under sulfate conditions, the sediment samples were incubated at 60 °C in Sep. 2022 with three different treatment groups (Supplementary Fig. 5). Hexadecane+sulfate groups: two bottles of slurry samples with the addition of fresh medium (with 5 mM NaHCO3), 1% (volume/volume) hexadecane and 10 mM sodium sulfate; hexadecane groups: two bottles of slurry samples with the addition of sulfate free medium (with 5 mM NaHCO3) and 1% (volume/volume) hexadecane; sulfate groups: two bottles of slurry samples with the addition of fresh medium (with 5 mM NaHCO3) and 10 mM sodium sulfate. After 480 days culturing, 2 ml cultures were collected for DNA isolation and Q-PCR of Ca. Melinoarchaeum 16S rRNA genes.
Metagenomic and metatranscriptomic sequencing
Total DNA for metagenome sequencing was obtained using FastDNA® Spin Kit for Soil (MPBIO, USA). Libraries for metagenome sequencing were generated using TruSeq Nano DNA LT Library Prep Kit (Illuminia, USA), following the manufacturer’s recommendations with the addition of index codes. Total RNA for metatranscriptome was recovered from alkane cultures by using RNA PowerSoil® Total RNA Isolation Kit (Qiagen, Germany). Reverse transcription and mRNAseq libraries were generated using Illumina’s TruSeq Stranded mRNA LT Sample Prep Kit (Illuminia, USA). The metagenomic and metatranscriptomic libraries were sequenced on an Illumina Novaseq6000 platform (Illumina, USA), generating 150 bp paired-end reads at Personal Biotechnology Co., Ltd. (China).
Metagenomic assembly, binning, and annotation
Trimmomatic (v0.39) was used to remove possible adaptors and low-quality bases for each read35. The dereplicated, trimmed, and paired-end DNA reads were assembled using Megahit (v1.2.9)36. For assembled contigs longer than 1 kb, open reading frames (ORFs) were predicted and translated using prodigal (v2.6.3) with -p meta parameters37. Clean reads were mapped onto their assembled contigs using bowtie2 (v2.2.8) with -very sensitive mode38. Assembled contigs (longer than 1 kb) were binned into putative taxonomic groups based on abundance information using MaxBin (v2.2.4) with the run MaxBin.pl script39. The genome completeness and contamination were assessed based on lineage-specific marker sets with CheckM (v1.1.3)40. The retrieved MAGs were classified using the GTDB22 and annotated using eggnog-mapper-1.03 in the EggNOG database with an e-value of 10-10. The completeness of specific pathways and functions within the MAGs was assessed based on the canonical pathways available in KEGG Pathway Database (www.kegg.jp). For the ASS genes annotation, all predicted ORFs were also checked with Blastp41 against ASS reference sequences for amino acid identity (>30%) and coverage (>80%). The Ass related sequences (AssA, AssB, AssC and AssD; or/and BssA, BssB, BssC and BssD) were collected and downloaded from NCBI-nr and uniprot (https://www.uniprot.org/) databases. Relative abundances of MAGs were calculated by mapping the trimmed reads to the MAGs with CoverM (v0.6.1; https://github.com/wwood/CoverM).
Metatranscriptomic analysis
For metatranscriptomic analysis, sequence reads were filtered to remove possible adaptors and low-quality reads using fastp (v0.20.0). The rRNA reads were eliminated using bbduk (v39.06) with the SILVA SSU138 database (https://www.arb-silva.de/) as a reference. Then the filtered reads mapped to metagenome assembled contigs with bowtie2 (v2.2.8)38. Expected FPKM values were used to estimate the expression level of each gene using Cufflinks (v2.2.1) scripts (http://cole-trapnell-lab.github.io/cufflinks/). Relative abundances of MAGs in metatranscriptome were calculated by mapping the filtered reads to the MAGs with CoverM (v0.6.1; https://github.com/wwood/CoverM).
Phylogenetic analyses
These reference genomes and the MAGs from this study were used to construct a phylogenomic tree based on a concatenated alignment of 37 conserved marker genes19,42. Specifically, each of 37 marker protein sequences was aligned using MAFFT43 (v7.313) with parameters --ep 0 --genafpair --maxiterate 1000 and filtered with trimAl44 (v1.4.rev2) with parameter -automated1. Then, all 37 marker genes were concatenated into a single alignment and phylogenetic trees were built using both IQ-Tree45 (v1.6.6) with the model LG + C60 + F + G and a bootstrap value of 1000. For the phylogenetic analysis of functional marker proteins (AcrA and McrA), the respective protein sequences were retrieved from the MAGs, and additional reference sequences were obtained from the NCBI. Alignment, filtering and tree building were carried out with the same programs described above. iTOL (http://itol.embl.de/) was used to modify the phylogenetic trees, including merging branches into triangles and representing bootstrap values greater than 90% with dots.
Ca. Melinoarchaeum specific 16S rRNA primer design
The 16S rRNA gene was extracted form MAGs of Ca. Melinoarchaeum fermentans DL9YTT1, another six sequences (EU924239.1, FJ638503.1, KP784726.1, JQ245670.1, AB050223.1 and AB802277.1) which show high similarity (>98%) to Ca. Melinoarchaeum fermentans DL9YTT1 were download from NCBI database. Homolog sequences were individually aligned with ClustalX46, then two aligned regions were selected as primer sequences: Melino-395F: 3’-CGCTGGTCTCACGATCGGCG-5’; Melino-736R: 3’-TGCCCGCTACCCTCAGCCT-5’.
Q-PCR
DNA extraction for Q-PCR was performed using the PowerSoil® DNA Isolation Kit (Qiagen, Germany). The Ca. Melinoarchaeum spp. 16S rRNA genes were quantified by Q-PCR and were amplified using the primer Melino-395F/Melino-736R. Clones with target genes were used for standard curve construction. For Ca. Melinoarchaeum spp. 16S rRNA genes quantification, the reaction mixture included 10 μl of SYBR Premix Ex Taq (TaKaRa), 0.4 μl of ROX reference dye (50×; TaKaRa), 0.8 μM of each primer, and 1 μl of template DNA; the thermal cycling program was: initial denaturation at 95 °C for 15 min, 40 cycles at 95 °C for 15 s, 60 °C for 30 s, and 72 °C for 30 s. The abundance of each targeted gene in the DNA assemblage was determined in triplicate analyses.
Hybridization Chain Reaction-Fluorescence In Situ Hybridization (HCR-FISH)
Culture samples (1 ml) were fixed in 1% formaldehyde for 12 h at 4 °C. The fixed samples were filtered onto GTTP polycarbonate filter (0.2 µM pore size; Millipore, Darmstadt, Germany) and washed twice with 5 ml 1× PBS, then stored at −20 °C. HCR-FISH was carried out as previously described47. A universal Archaea-specific probe ARC91548, Bacteria-specific probe EUB33849 and Ca. Melinoarchaeum specific probe Melino-736 were used in this study. Hybridization of probe Melino-736 occurred at 46 °C with 25% formamide (v/v).
Synthesis of hexadecyl-CoM standards and detection of hexadecyl-CoMs
For synthesis of hexadecyl-CoM, 0.1 g of coenzyme M (sodium 2-mercaptoethanesulfonate) were dissolved in 2 ml 25% (v/v) ammonium hydroxide solution14. Twice the molar amount of each 1-iodo-hexadecane was added. Vials were heated for 12 h at 60 °C while stirring under nitrogen.1 ml of the clear upper phase was collected and stored at 4 °C. The method for hexadecyl-CoM extracting and detecting was previously described12. Cells or slurry were lysed in 1 ml of a mixture of acetonitrile: methanol: water (40:40:20, v/v/v) and 0.3 g sterile glass beads (0.1 mm diameter; Sigma-Aldrich), by shaking at 6 m/s for 50 s with a homogenizer (FastPrep-24, MP). The cell extracts were diluted in 10 ml deionized water and further purified with an HC-C18 SPE Cartridge. The standards and cell extracts were analyzed using the Q-Exactive Plus Orbitrap mass spectrometer (QE Plus Orbitrap, Thermo Fisher Scientific) and high performance liquid chromatography-tandem MS (triple quadrupole mass spectrometer, HPLC-MS/MS, AB4500, SCIEX)12. Some parameters were modified based on the previous methods12. The mode of parallel reaction monitoring (PRM) was selected for QE Plus; the gradient elute from 10% to 95% acetonitrile in water was used for HPLC-MS/MS.
Methane, carbon dioxide, hydrogen and acetate concentration measurement, and methane and carbon dioxide carbon isotope measurement
Methane and carbon dioxide were quantified by gas chromatography (Shimadzu GC-14B) using a flame ionization detector (FID) with a TDX-02 2 m × 2 mm filling column. The detection conditions were 100 °C at the inlet, 100 °C at the column temperature, 250 °C at the detector, 99.999% argon gas as carrier gas, 30 ml/min at the flow rate, and 380 °C at the methane reformer. Hydrogen was quantified by gas chromatography (Shimadzu GC-2030), the chromatography Column is Shimadzu SH-Msieve 5 A PLOT Column 0.32 mm × 30 μm × 30 m, inlet 60 °C, shimmery ratio 10:1, column temperature chamber 30 °C for 8 min. The detector was a barrier discharge ionization detector (BID) at 200 °C. For acetate concentration measurement, 400 µL supernatant of each type of incubated sediment slurry was mixed with 100 µL sulfuric acid (50%) and 200 µL methyl tert-butyl, then analyzed by gas chromatography-mass spectrometer (Agilent 7890B-7000D). The carbon isotope composition of methane and carbon dioxide were analyzed by GasBench II/PreCon-Isotope Ratio Mass Spectrometry (GB/PreCon-IRMS) 253 plus. Carbon dioxide gas samples are directly introduced into IRMS through GasBench. Methane is converted to carbon dioxide under high-temperature conditions (1000 °C) using Precon, pre-concentrated, and then introduced into IRMS for measurement.
In situ hot spring water sample geochemical analysis
The concentrations of cations from hot spring water samples were analyzed by ion chromatography (Thermofisher ICS 900) using a Dionex™ IonPac™ CS12A IC column with electrical conductivity detector. The concentrations of anions were analyzed by ion chromatography (Thermofisher ICS 5000 + ) using a Dionex™ IonPac™ AS11-HC-4µm IC column with electrical conductivity detector. For hydrocarbon analysis, 10 ml water sample was extracted with 5 ml of n-hexane by vortex for 5 min at room temperature, then analyzed by GC-MS (Thermofisher TRACE 1310-ISQ 7000). The GC was equipped with a DB-5MS column and a flame ionization detector.
Gibbs free energy calculation
Gibbs free energy calculations were made after Dolfing et al.50 under conditions with 60°C and pH = 7.0. Gibbs free energies of compounds were taken from Helgeson et al.51 and Hanselmann52 with alkanes in the liquid state, acetate in the aqueous phase, and methane, hydrogen and carbon dioxide in the gaseous phase at partial pressures of 1 atm.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The rawdata of metagenome and metatranscriptome generated in this study have been deposited in the NCBI database under BioProject PRJNA1095168. The MAGs of Ca. Melinoarchaeum fermentans DL9YTT1, Methanothermobacter and Methanothrix, as well as all high-quality MAGs (completeness>80%, contamination<5% in Supplementary Data 3) which obtained in this study were submitted under BioProject PRJNA1097016. Four MAGs belonging to the phylum Hadarchaeota, namely 7-2_LN1_denovo_Bins.032, 7-3-req_LN1_denovo_Bins.078, 7-3-req_megahit_Bins.061, and 7-4_LN1_denovo_Bins.031, obtained from the Tengchong hot spring and illustrated in Fig. 3a, have been submitted under BioProject PRJNA1061358. The rawdata of metagenome and metatranscriptome have also been deposited in NODE (The National Omics Data Encyclopedia, https://www.biosino.org/node) under accession numbers OEP005183. Other source data are provided with this paper as a Source Data file. Source data are provided with this paper.
Change history
06 January 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41467-024-55708-7
References
Jiménez, N. et al. Methanogenic hydrocarbon degradation: evidence from field and laboratory studies. J. Mol. Microbiol. Biotechnol. 26, 227–242 (2016).
Cheng, L. et al. DNA-SIP reveals that Syntrophaceae play an important role in methanogenic hexadecane degradation. PLoS ONE 8, e66784 (2013).
Gieg, L. M. et al. Syntrophic biodegradation of hydrocarbon contaminants. Curr. Opin. Biotech. 27, 21–29 (2014).
Tan, B. et al. Metagenomic analysis of an anaerobic alkane-degrading microbial culture: potential hydrocarbon-activating pathways and inferred roles of community members. Genome 56, 599–611 (2013).
Jones, D. M. et al. Crude-oil biodegradation via methanogenesis in subsurface petroleum reservoirs. Nature 451, 176–180 (2008).
Zengler, K. et al. Methane formation from long-chain alkanes by anaerobic microorganisms. Nature 401, 266–269 (1999).
Chen, S. C. et al. Anaerobic oxidation of ethane by archaea from a marine hydrocarbon seep. Nature 568, 108–111 (2019).
Hahn, C. J. et al. Candidatus Ethanoperedens,” a thermophilic genus of archaea mediating the anaerobic oxidation of ethane. mBio 11, e00600–e00620 (2020).
Laso-Pérez, R. et al. Anaerobic degradation of non-methane alkanes by “Candidatus Methanoliparia” in hydrocarbon seeps of the Gulf of Mexico. mBio 10, e01814–e01819 (2019).
Laso-Pérez, R. et al. Thermophilic archaea activate butane via alkyl-coenzyme M formation. Nature 539, 396–401 (2016).
Benito Merino, D. et al. Anaerobic hexadecane degradation by a thermophilic Hadarchaeon from Guaymas Basin. ISME J. 18, wrad004 (2024).
Zhou, Z. et al. Non-syntrophic methanogenic hydrocarbon degradation by an archaeal species. Nature 601, 257–262 (2022).
Borrel, G. A microbe that uses crude oil to make methane. Nature 601, 196–197 (2022).
Zehnle, H. et al. Candidatus alkanophaga archaea from guaymas basin hydrothermal vent sediment oxidize petroleum alkanes. Nat. Microbiol. 8, 1199–1212 (2023).
Kompanichenko, V. N. et al. Organic compounds in thermal water: the Mutnovskii area and the Uzon caldera. J. Volcano Seismol. 10, 305–319 (2016).
Song, Z. Q. et al. Bacterial and archaeal diversities in Yunnan and Tibetan hot springs, China. Environ. Microbiol. 15, 1160–1175 (2013).
Evans, P. N. et al. Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics. Science 350, 434–438 (2015).
Hua, Z. S. et al. Insights into the ecological roles and evolution of methyl-coenzyme M reductase-containing hot spring Archaea. Nat. Commun. 10, 4574 (2019).
Wang, Y. et al. Expanding anaerobic alkane metabolism in the ___domain of Archaea. Nat. Microbiol. 4, 595–602 (2019).
Wang, Y. et al. Methyl/alkyl-coenzyme M reductase‐based anaerobic alkane oxidation in archaea. Environ. Microbiol. 23, 530–541 (2020).
Wang, Y. Z. et al. A methylotrophic origin of methanogenesis and early divergence of anaerobic multicarbon alkane metabolism. Sci. Adv. 7, eabd7180 (2021).
Parks, D. H. et al. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 50, D785–d794 (2022).
Aeckersberg, F. et al. Anaerobic oxidation of saturated hydrocarbons to CO2 by a new type of sulfate-reducing bacterium. Arch. Microbiol. 156, 5–14 (1991).
Kniemeyer, O. et al. Anaerobic oxidation of short-chain hydrocarbons by marine sulphate-reducing bacteria. Nature 449, 898–901 (2007).
Park, C. & Park, W. Survival and energy producing strategies of alkane degraders under extreme conditions and their biotechnological potential. Front. Microbiol. 9, 1081 (2018).
Wang, Y. et al. Diverse anaerobic methane- and multi-carbon alkane-metabolizing archaea coexist and show activity in Guaymas Basin hydrothermal sediment. Environ. Microbiol. 21, 1344–1355 (2019).
Dolfing, J. et al. Thermodynamic constraints on methanogenic crude oil biodegradation. ISME J. 2, 442–452 (2008).
Cheng, L. et al. Enrichment and dynamics of novel syntrophs in a methanogenic hexadecane-degrading culture from a Chinese oilfield. FEMS Microbiol. Ecol. 83, 757–766 (2013).
McKay, L. J. et al. Occurrence and expression of novel methyl-coenzyme M reductase gene (mcrA) variants in hot spring sediments. Sci. Rep. 7, 7252 (2017).
Chernova, T. G. et al. Geochemical features of hydrocarbon distribution in the bottom sediments of Andaman Basin (the northeastern part of the Indian Ocean). Geochem. Int. 39, 793–804 (2001).
Teece, B. L. et al. Biogeochemistry of Recently Fossilized Siliceous Hot Spring Sinters from Yellowstone, USA. Astrobiology https://doi.org/10.1089/ast.2022.0012 (2022).
Capaccioni, B. et al. Source conditions and degradation processes of light hydrocarbons in volcanic gases: an example from El Chichón volcano (Chiapas State, Mexico). Chem. Geol. 206, 81–96 (2004).
Gurgey, K. et al. Origin of petroliferous bitumen from the Buyuk Menderes-Gediz geothermal graben system, Denizli - Saraykoy, western Turkey. Appl. Geochem. 22, 1393–1415 (2007).
Widdel, F. & Bak, F. Gram-negative mesophilic sulphate-reducing bacteria. in The Prokaryotes 4th edn, vol. 4 (eds Balows, A., Truper, H. G., Dworkin, M., Harder, W. & Schleifer, K.-H.), (Springer, New York, 1998).
Bolger, A. M. et al. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, D. et al. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinforma. 11, 119 (2010).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Wu, Y. et al. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607 (2016).
Parks, D. H. et al. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Hug, L. A. et al. A new view of the tree of life. Nat. Microbiol. 1, 16048 (2016).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Capella-Gutiérrez, S. et al. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Nguyen, L. T. et al. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Jia, Z. et al. Optimizing the hybridization chain reaction-fluorescence in situ hybridization (HCR-FISH) protocol for detection of microbes in sediments. Mar. Life Sci. Tech. 3, 529–541 (2021).
Amann, R. I. et al. Fluorescent-oligonucleotide probing of whole cells for determinative, phylogenetic, and environmental studies in microbiology. J. Bacteriol. 172, 762–770 (1990).
Amann, R. I. et al. Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl. Environ. Microbiol. 56, 1919–1925 (1990).
Dolfing, J. Protocols for Calculating Reaction Kinetics and Thermodynamics. In Hydrocarbon and Lipid Microbiology Protocols, Springer Protocols Handbooks, pp. 155–163 (2015).
Helgeson, H. C., Owens, C. E., Knox, A. M. & Richard, L. Calculation of the standard molal thermodynamic properties of crystalline, liquid, and gas organic molecules at high temperatures and pressures. Geochim Cosmochim Acta 62, 985–1081 (1998).
Hanselmann, K. W. Microbial energetics applied to waste repositories. Experientia 47, 645–687 (1991).
Acknowledgements
We thank David Benito Merino (Max Planck Institute for Marine Microbiology) for supplying MAGs of Hadarchaeota; Jialin Hou (Shanghai Jiao Tong University) and Ruize Xie (Shanghai Jiao Tong University) for advice in the metagenome analysis, Lewen Liang (Shanghai Jiao Tong University) and Maoyu He (Shanghai Jiao Tong University) for assisting in the HCR-FISH; Zhuo Zhou (Biogas Institute of Ministry of Agriculture and Rural Affairs) for advice in isotope experiments; Xiaobo Xie (Analytical & Testing Center, Sichuan University) for assisting in detection of metabolites; Lili Feng (School of Environmental Science and Engineering, Shanghai Jiao Tong University) for assisting in detection of methane and carbon dioxide; Fan Yang, Yu Gao and Yumin Liu (Instrumental Analysis Center, Shanghai Jiao Tong University) for technical assistance. This work was supported financially by the National Natural Science Foundation of China Grant (92251303, 42276139, 42230401, 32325002) to F.W., T.Y., and L.C., Shanghai Jiao Tong University 2030 initiative (Grant WH510244001) to F.W., Southern Marine Science and Engineering Guangdong Laboratory (Zhuhai) (SML2023SP237) to F.W., the Agricultural Science and Technology Innovation Project of the Chinese Academy of Agriculture Science (CAAS-ASTIP2016-BIOMA) to L.C., and the German Research Foundation (DFG) through the Clusters of Excellence EXC 2077 “The Ocean Floor-Earth’s Uncharted Interface” (project no. 390741601) to G.W.
Author information
Authors and Affiliations
Contributions
T.Y., F.W. and L.C. designed research. T.Y. collected samples; performed culture experiments; designed Q-PCR primers and HCR-FISH probes; nucleic acid isolation, Q-PCR; detected acetate; calculated Gibbs free energy; analyzed of metagenomic and metatranscriptomic data. T.Y. and Y.D. performed HCR-FISH. T.Y. and Y.C. detected methane, carbon dioxide and hydrogen. L.F. synthesized alkyl-CoM standards and detected alkyl-CoM; Y.W. constructed phylogenetic trees. T.Y. wrote the initial draft, T.Y., L.F., Y.W., G.W., L.C. and F.W. reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Daisuke Mayumi, Florin Musat and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yu, T., Fu, L., Wang, Y. et al. Thermophilic Hadarchaeota grow on long-chain alkanes in syntrophy with methanogens. Nat Commun 15, 6560 (2024). https://doi.org/10.1038/s41467-024-50883-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50883-z
