
Abstract
Replacing petrochemicals with compounds from bio-based manufacturing processes remains an important part of the global effort to move towards a sustainable future. However, achieving economic viability requires both optimized cell factories and innovative processes. Here, we address this challenge by developing a fermentation platform, which enables two concurrent fermentations in one bioreactor. We first construct a xylitol producing Escherichia coli strain in which CRISPRi-mediated gene silencing is used to switch the metabolism from aerobic to anaerobic, even when the bacteria are under oxic conditions. The switch also decouples growth from production, which further increases the yield. The strain produces acetate as a byproduct, which is subsequently metabolized under oxic conditions by a secondary E. coli strain. Through constraint-based metabolic modelling this strain is designed to co-valorize glucose and the excreted acetate to a secondary product. This unique syntrophic consortium concept facilitates the implementation of “two fermentations in one go”, where the concurrent fermentation displays similar titers and productivities as compared to two separate single strain fermentations.
Similar content being viewed by others
Introduction
Responsible production of food and chemicals are part of the sustainable development goals set up by the United Nations1. Bio-based manufacturing is being pursued as a key technology to enable this transition, not only for food and feed applications, but also as a sustainable alternative to fossil fuels-based production of chemicals, fuels, and materials. Traditionally the development and optimization of synthetic bioprocesses focus on single-strain fermentations, however, microbial consortia-based processes have been pursued as an attractive alternative, in particular for more complex processes. Synthetic consortia that facilitate the production of complex molecules2,3 or the breakdown and further valorization of cellulose4,5,6 have successfully been engineered by dividing functional traits between the different members, and niche partitioning has been used to consume and valorize a mixture of carbon sources7,8,9.
One of the main challenges of using microbial consortia in synthetic biology is stable co-culturing. However, the development of designed niche partitioning and mutualistic syntrophic interactions have been successfully shown to enable consortia-based bioprocesses.
Another way to stabilize consortia-based bioprocesses is through growth-decoupled production, where cell growth is inhibited but production is maintained. One method to achieve this is through using CRISPR interference (CRISPRi)10, which in contrast to traditional gene deletions, can be used to target essential genes for conditional repression. This enables control of cell growth while maintaining production, through either targeting essential genes such as pyrF11, or by targeting genes in a cell that has been engineered in a way that makes a non-essential gene essential to the strain.
In this study, we aim to develop a consortia-based fermentation process, which enables concurrent aerobic and anaerobic metabolism in one bioreactor. To demonstrate the concept, we engineer a strain that can be forced to switch to anaerobic metabolism through CRISPRi-mediated gene silencing without changing the physical conditions inside the bioreactor. Because the bioreactor remains oxic, the additional strain(s) can still respire and consequently use energy-requiring pathways. To demonstrate the potential of the system, we engineer a synthetic anaerobic xylitol producing strain with inducible anaerobic physiology and an aerobic acetic acid auxotroph co-metabolizing glucose and acetic acid for the production of isobutyric acid (IBA) that operate in a syntrophic consortium process as illustrated in Fig. 1. We stabilize the consortia-based process through inducible growth-decoupled production combined with mutualistic syntrophic dependency. The overall results demonstrate the successful engineering of a microbial consortium that enables two concurrent fermentations in one bioreactor that display comparable titers and productivity as compared to two individual single strain fermentations. This highlights the potential of this consortium-based process to improve resource efficiency while minimizing bioreactor capacity requirements.

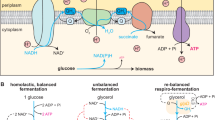
a Overall illustration of the process; one strain co-consumes xylose and glucose and produces xylitol and acetic acid, while the other strain co-consumes glucose and acetic acid and produces IBA. b Overview of knockouts in the synthetic anaerobic xylitol producing strain. c Overview of knockouts in the aerobic acetic acid auxotrophic co-metabolizing IBA producing strain. d Illustration of the anhydrotetracycline inducible CRISPRi-enabled metabolic switch to repress the expression of cytochrome BD-I.
Results
Construction of a xylitol producing strain with inducible anaerobic physiology under oxic conditions
To construct a xylitol producing strain with inducible anaerobic physiology under oxic conditions, we first created a strain devoid of native fermentation pathways by knocking out focA-pflB; ldhA; adhE; and frdA in order to limit potential by-product formation. To prevent xylose catabolism we deleted xylAB. We furthermore replaced the native cAMP receptor protein (CRP) with a mutated version (CRP*), to enable simultaneous uptake of different sugars12,13, and inserted a xylose reductase from Candida boidinii under the control of the constitutive promoter BBa_J23100 into the genome. The engineered strain can only oxidize glucose generated NAD(P)H by converting xylose to xylitol under anoxic conditions. To enable anaerobic fermentative physiology under oxic conditions14 we first deleted cyoB and appB, which are part of cytochromes BD-o and BD-II, respectively. This strain (xylitol base strain) can still grow by using cytochrome BD-I. However, as the natural fermentation pathways have also been deleted, this cytochrome cannot be deleted through conventional knockout technologies, as this would result in a non-growing strain. To instead facilitate inducible silencing of the last cytochrome, we first integrated dCas9 into the genome under the control of an anhydrotetracycline inducible promoter. We subsequently constructed a plasmid with a guide RNA (gRNA) against cydA to facilitate inducible repression of cytochrome BD-I (xylitol strain). The xylitol base strain had a slightly lower maximal growth rate and grew to a lower cell density as compared to E. coli K-12 MG1655 WT, whereas the growth rate was similar between the xylitol base strain and the xylitol strain (Supplementary Fig. 1).
To evaluate if the inducible CRISPRi-mediated metabolic switch worked as intended, we performed a comparative growth and production experiment with CRISPRi induced or uninduced in both minimal and richer media (minimal media with 0.5% yeast extract). The repression of cydA was remarkably effective and resulted in growth arrest after about only one doubling in cell density in minimal media and two doublings in cell density in richer media. The growth attenuation further remained stable for >30 h (Fig. 2a; Supplementary Fig. 2). The oxidation of glucose to acetate through the Embden-Meyerhof-Parnas pathway generates a maximum of 4 NADP(H), and each conversion of xylose to xylitol requires 1 NADPH, which leads to a theoretical maximum molar yield of 4 xylitol per oxidized glucose in analogous growth arrested cells under anoxic conditions15 (a theoretical carbon and redox balance of the process are shown in Supplementary Fig. 3 and Supplementary Table 1). Under oxic conditions with anaerobic physiology, we achieved a molar yield of 3.5 (±0.76) when CRISPRi was active in minimal media, which was higher than the molar yield 1.9 (±0.08) for uninduced and thus respiring cells (Supplementary Fig. 2). The molar yield in richer media decreased to 2.4 (±0.08) xylitol per oxidized glucose, which was still significantly higher than for uninduced cells during most of the experiment apart from a 4 h period (hours 12–16) (Fig. 2b).

a Growth curve of the xylitol strain with (dark blue) or without (light blue) the CRISPRi switch induced. b Molar yield of xylitol per glucose with and without the CRISPRi switch induced. c Growth of the IBA strain in the presence of glucose (light gray), acetate (dark gray) or both (yellow) in a plate reader. d Growth and metabolite levels of the IBA strain. Arrows indicate: (1) Induction of CRISPRi switch after 2 h growth, (2) induction of IBA production after 3 h growth. Error bars and shaded areas indicate mean ± s.d. (n = 4 biological replicates for (a, b), n = 6 biological replicates for (c), and n = 3 biological replicates for (d)). The statistical significance was assessed using a two-tailed unpaired t-test (Welch’s t-test). *p < 0.05. The exact p-value for the interval 2–12 h is p = 0.0338 and for 16–30 h is p = 0.0002. OD values for a and d were measured using a Jenway 6705 UV/Vis spectrophotometer, whereas the OD630 values for c were measured in the plate reader (ELx808). Source data are provided as a Source Data file.
Construction of an acetic acid auxotroph through constraint-based metabolic modeling
Acetate can act as a strong inhibitor of growth even at relatively low concentrations16,17 and is typically not utilized by microorganisms when sugars are available. To construct a compatible aerobic partner for the consortium we focused on the construction of a strain that would co-utilize glucose and the secreted secondary product acetic acid. We first made three knockouts (aceEF, focA-pflB, and poxB), which have previously been shown to enable acetate auxotrophy in E. coli18, and additionally used constraint-based metabolic modeling to identify other possible sources of acetate and acetyl-coA during growth on glucose. Three oxygen sensitive genes: tdcE (an oxygen sensitive pyruvate formate lyase), pflDC (pyruvate formate-lyase II), and pfo (an oxygen sensitive pyruvate:flavodoxin oxidoreductase), as well as deoC (a deoxyribose-phosphate aldolase; involved in purine and pyrimidine degradation) were identified as genes coding for proteins involved in pathways that, in addition to the deletions in the initial strain, could lead to sugar derived acetyl-CoA formation. These predicted pathways may potentially provide escape mechanisms from the auxotrophy due to evolution during long-term experiments, through gain-of-function or regulatory adjustments. Therefore, they were also deleted. Constraint-based metabolic modeling also identified a possible flux through serine or threonine that could theoretically lead to acetyl-CoA formation from the different sugars, but these two routes, shown in detail in Supplementary Fig. 4, were deemed highly unlikely to provide the cells with sufficient acetyl-CoA to sustain growth, and deletions to prevent these two theoretical routes were therefore not performed. To construct a glucose utilizing acetic acid auxotroph unable to catabolize C5-sugars, we further deleted xylAB and araBA, which encodes for proteins involved in xylose and arabinose catabolism, respectively. The resulting strain did not grow in minimal M9extra media when supplemented solely with glucose. It displayed some growth when supplemented with acetic acid; however, it grew rapidly when supplemented with both glucose and acetic acid (Fig. 2c). To evaluate the performance of our acetic acid auxotroph in regard to IBA production, we first did a comparison of the original strain (IBA pre-strain) and a strain with ptsG deleted (IBA strain). The performance of the two strains were not significantly different, but as there was a slight tendency of the ΔptsG to perform better (lower total OD, higher IBA titers and yield) (Supplementary Fig. 5a), our subsequent experiments were done using this strain. During our initial experiments we found that acetate was depleted after 13 h of growth (Fig. 2d). We therefore tested whether an increase in acetate concentration from 17 mM (1 g L−1) to 34 mM (2 g L−1) had an impact on growth and IBA production (Supplementary Fig. 5b). No differences were observed in growth and IBA production and subsequent experiments were conducted using 17 mM (1 g L−1) acetate.
Long-term stability and reversibility of the CRISPRi-mediated metabolic switch
To further evaluate the performance and characteristics of the growth arrested xylitol production strain, we performed a longer-term experiment to test if parts of the induced cell population were able to escape the growth arrest. No cells escaped during the 96 h that we ran the experiment (Fig. 3a). Given the longevity of the induced growth arrest, we further wanted to test the reversibility of the CRISPRi-mediated growth arrest, if the inducer was removed. We therefore performed an experiment, where part of the induced cell populations were subjected to a washing step to remove the inducer after 29 h of induction of the CRISPRi switch. After a lag phase of ~12 h, the washed cells started to grow at similar growth rates as the initially uninduced population, whereas non-washed induced cell populations continued their growth arrest (Fig. 3b). The productivity of xylitol for the washed cells was furthermore comparable (although slightly lower) to the initially uninduced cell population (Fig. 3c).

a Growth curve of the xylitol strain with (dark blue) or without (light blue) the CRISPRi switch induced. b Growth curve of the xylitol strain with (dark blue) or without (light blue) the CRISPRi switch induced. At T = 31 h a subset of induced cultures was washed to remove the inducer and re-inoculated in fresh media with glucose (black). Xylose was added to a final concentration of 133 mM xylose to these cultures 13 h after re-inoculation; ~2 h after resumption of growth. c Xylitol productivity (interval between 0−15 h after addition of xylose) for the uninduced cells at the beginning of the washing experiment (light blue) and after the washing step (gray). Arrows indicate: (1) Induction of CRISPRi switch after 2 h of growth. Error bars indicate mean ± s.d. (n = 4 biological replicates). OD600 were measured using a Jenway 6705 UV/Vis spectrophotometer. Source data are provided as a Source Data file.
Compatibility of consortium partners
To determine whether the presence of product from the other strain would have an effect on the growth and productivity in a consortia-based process, we performed single-strain fermentation experiments with addition of relevant compounds. Xylitol productivity was not affected by the presence of IBA, however, the presence of 2.5 g L−1 of IBA decreased the growth rate of the xylitol strain (Supplementary Fig. 6). To mitigate this effect during our consortia-based experiments (see below), we timed the induction of the CRISPRi-enabled switch so that we could obtain cell-arrest before the IBA concentration reached inhibitory levels (Supplementary Fig. 6). The presence of anhydrotetracycline (1 μg mL−1) had no effect on the IBA producing strain, whereas the presence of xylose (20 g L−1) or xylitol (10 g L−1) resulted in a slightly lower final cell density. IBA production was not affected by any of the three compounds (Supplementary Fig. 7).
Consortium-based production
To follow the distribution of the two cell lines when grown together, we first integrated constitutively expressed mCherry and Yellow Fluorescent Protein (YFP) into the genomes of the xylitol and IBA producing strains, respectively. We subsequently performed some initial production experiments and found that the time of induction of the IBA strain had a significant impact on the overall performance of the consortia-based production (Supplementary Fig. 8b). During our initial experiments with the IBA strain (Fig. 2d), production was induced after 3 h of growth. However, during our consortia-based optimization experiments we found that IBA titers were significantly higher when inducing after 5 h as compared to induction after 3 and 4 h (Supplementary Fig. 8b). Induction after 5 h of growth was therefore chosen for our final production experiment. Delaying induction of the CRISPRi system in the xylitol strain for 0.5 h and inducing at 2.5 h instead of 2 h as it was done in previous experiments did result in a slightly higher OD/proportion of the strain and consequently slightly higher xylitol titer (Supplementary Fig. 8a). However, after normalizing for the increase in OD, xylitol production only increased slightly from the induction after 2 h. We therefore continued to use the 2 h induction time to enable comparisons with previous experiments.
Finally, both strains were cultivated together using these optimized induction conditions. The xylitol strain (initially inoculated in even proportions to the IBA strain) grew faster than the IBA strain, until growth was attenuated by the CRISPRi induced metabolic switch. The continuously growing IBA strain subsequently increased in proportion, and after the growth phase the distribution was about 80% IBA strain and 20% xylitol strain, after which it remained stable to the end of the experiment (Fig. 4a, Supplementary Fig. 9). Acetate concentration initially increased, reaching a maximum concentration of 21.8 mM after 6 h of growth, after which it was fully consumed by the IBA strain (Fig. 4b). A small accumulation of pyruvate (5.5 mM) was furthermore detected after 4 h of growth, but was not detectable after 8 h. The titer of IBA and xylitol after 24 h was 44 mM and 22 mM, respectively, which was comparable to titers achieved using single strain inoculations, with IBA and xylitol titers of 54 mM and 34 mM, respectively. The difference is largely attributed to a higher cumulative cell density in single cell cultures; this is reflected when comparing the specific productivity of the strains in the consortia-process to the respective single strain-processes (Fig. 4c). Here we observed a 43% increase in xylitol productivity, going from 0.67 (±0.01) to 0.96 (±0.02) mM xylitol/OD600/h, whereas IBA productivity only decreased from 0.30 (±0.0004) to 0.29 (±0.01) mM IBA/OD600/h.

a Distribution of the synthetic anaerobic xylitol producing strain (blue) and the aerobic IBA strain (yellow) during the consortia-based fermentation (see also Supplementary Fig. 9). b Substrate, product and byproduct concentrations during the experiment. Arrows indicate: (1) induction of the CRISPRi-mediated metabolic switch in the synthetic anaerobic xylitol producing strain and (2) induction of IBA production in the aerobic auxotrophic IBA strain. c The specific productivity of the single consortia-based fermentation (gray) compared to two single strain fermentations (xylitol strain (blue), IBA strain (yellow)). Error bars indicate mean ± s.d. (n = 4 (consortium) or 2 (single strains) biological replicates). Source data are provided as a Source Data file.
Discussion
Microbial cell factories that can produce value-added fuels and chemical building blocks have received extensive attention in order to enable more sustainable manufacturing processes19. Most microbial manufacturing processes are being pursued as single strain fermentations, however, in the last decade a number of consortia-based approaches have been developed2,3,4,5,6,7,8,9. An important parameter for synthetic multi-strain bioprocess development is population control and stability. Here we used a CRISPRi-mediated metabolic switch to enable a growth-decoupled synthetic anaerobic production process under oxic conditions and coupled it with a mutualistic syntrophic consortia partner that was designed to be auxotrophic for acetic acid and co-consume acetic acid and glucose. The inducible growth arrest in the xylitol strain along with co-dependency of acetic acid for growth but not for production for the IBA strain enables population control and stability during the production phase. CRISPRi-mediated gene silencing has previously been used for CRISPRi aided strain development20,21,22, and facilitation of growth-decoupled production of biochemicals11. CRISPRi libraries have furthermore been generated to elucidate both functional screening of essential genes23,24 and novel targets for biotechnological applications25. Here we used CRISPRi to silence the last functioning cytochrome oxidase, which in the engineered cells enables anaerobic physiology under oxic conditions14. As the cells were further engineered to be devoid of natural fermentation pathways, the generated cells can only oxidize glucose generated redox equivalents by reducing xylose to xylitol15. Given that the last functioning cytochrome oxidase is an essential gene in the engineered strain, gene silencing is the only way to obtain this desired phenotype. To facilitate co-valorization of the excreted byproduct, acetic acid, we engineered an acetic acid auxotroph starting with three genes previously shown to facilitate acetic acid auxotrophy in E. coli18. Using constrained-based metabolic modeling we found additional nodes, which could be possible sources of acetyl-CoA and consequently decided to delete most of these genes as well. Apart from enabling acetic acid auxotrophy, blocking the pyruvate to acetyl-coA node has also previously been shown to aid in the production of both pyruvate and other pyruvate derived bioproducts such as lactate26 and isobutyl acetate27.
Bioprocesses (both single and multi-strain) are traditionally run with either oxic, microoxic or anoxic conditions, depending on the organism, the product, and the biosynthetic pathway in question. This type of bioprocess is similar to what microbes experience when growing in well mixed aquatic conditions. The interactions in our consortia-based process rather resembles the interactions in layered microbial communities with physico-chemical gradients.
In conclusion, in this study we designed an innovative consortium-based process that enables both anaerobic and aerobic physiology in the same bioreactor. By limiting the possibility of respiration by one strain to the growth phase, we were able to concomitantly run an aerobic and a synthetic anaerobic fermentation process in the same reactor without loss in titers and yields as compared to when the fermentations were run with single strains in two reactors. This highlights the potential of this process to increase efficiency of resource utilization and to lower bioreactor capacity requirements.
Methods
Strains, plasmids, and media
All strains were routinely grown in LB broth or on LB agar plates supplemented, when needed, with appropriate antibiotics (ampicillin 100 µg mL−1 (amp), kanamycin 30–50 µg mL−1 (km), chloramphenicol 10–25 µg mL−1 (cm)). Cells were grown at either 30 °C or 37 °C. 75 µL liquid LB-amp supplemented with 1 g L−1 sodium acetate (NaOAc) was spread on LB-km or LB-cm plates for plasmid maintenance during the construction of the deletion strains, when using pSIJ828. Strains and plasmids used in this study are listed in Supplementary Tables 2 and 3, respectively, and oligoes and reagents and supplies are listed as Supplementary Data 1 and Supplementary Data 2, respectively. Cultivation experiments were performed with modified M9 minimal media called M9extra (6.8 g L−1 of Na2HPO4, 3 g L−1 of KH2PO4, 0.5 g L−1 of NaCl, 1 g L−1 NH4Cl, 2 mM MgSO4, 0.1 mM CaCl2) supplemented with 50 μM FeCl3, a trace element solution (12.5 μM MnCl2·4H2O, 2.1 μM CoCl2·6H2O, 8.5 μM ZnSO4 · 7H2O, 0.6 μM CuCl2 · 2H2O, 0.8 μM H3BO3, 1.05 μM NiCl2 · 6H2O, 1.25 μM Na2MoO4 · 2H2O). Yeast extract was added to a final concentration of 5 g L−1 to relevant cultivations. For cultivations that included the IBA strain, 1.39 g L−1 of sodium acetate (1 g L−1 acetate) was added unless otherwise specified.
Deletion and insertion of genes
PCR reactions were performed using standard PCR conditions. PCR templates for gene deletions were generated by amplifying gDNA extracted from previously generated single deletion strains or by using extended oligoes with 50 bp homology arms. Amplified FRT-flanked antibiotic cassettes were used to delete native E. coli genes by the combined action of lambda Red recombineering and flippase recombinase using either the procedure for using pKD4629 and pCP2030, where the lambda Red recombineering plasmid is located on pKD46, and the flippase recombinase is present in pCP20, or the temperature sensitive plasmid pSIJ828, where the lambda Red recombineering genes and the flippase recombinase are located in the same plasmid. Constitutively expressed fluorescent proteins (YFP and mCherry) were PCR amplified from plasmids and ligated together with an FRT-flanked kanamycin cassette by USER-ligation-PCR. The spliced cassettes were subsequently integrated into the genomes of the specific strains (9 base pairs downstream of the glmS gene) after which the integrated FRT-flanked kanamycin cassettes were removed.
Constraint based metabolic modeling
Cameo (Computer Aided Metabolic Engineering and Optimization) and Escher were used for simulation and visualization of metabolic models31,32. Gene deletions necessary for complete acetic acid auxotrophy were determined through testing of the designs with the E. coli metabolic model, iJO136633, with a minor modification: Tryptophanase reaction catalyzed by tnaA gene was considered irreversible under physiological conditions. The knockouts of the initial design were projected to iJO1366 reactions as follows: aceEF/PDH; focA/FORt2pp; pflB/PFL and poxB/POX. Further deletions necessary to ensure acetic acid auxotrophy were determined by examining the flux profiles generated for utilized pathways leading to acetate/acetyl-CoA and iteratively eliminating them.
Shake flask cultivations of engineered E. coli strains
A colony was picked from an agar-plate and cultivated for 6–8 h in 1 mL LB broth. 500 µL of this culture was added to 50 mL of the M9extra media in a 250 mL shake flask with 111 mM (20 g L−1) glucose and cultured over night at 37 °C, 250 rpm. The cells were spun down at 5000 × g for 5 min and resuspended in M9extra media. 250 mL shake flasks with 50 mL media were inoculated to OD600 0.1 (or 0.05 in experiments with the xylitol strain with yeast extract). Experiments with both strains were inoculated to OD600 0.1 of each strain. Glucose concentrations were as follows: xylitol strain without yeast extract, 30 mM (5.4 g L−1); xylitol strain with yeast extract, 60 mM (10.8 g L−1); IBA strain, 111 mM (20 g L−1); consortia, 111 mM (20 g L−1). Xylose was added to a final concentration of 133 mM (20 g L−1) to cultivations with the xylitol strain present 2 h after inoculation unless otherwise stated. The cultures were grown at 37 °C and 250 rpm shaking until induction, after which the temperature was lowered to 30 °C. The CRISPRi switch was induced to a final concentration of 1 μg mL−1 anhydrotetracycline (aTc), and IBA production was induced by adding IPTG to a final concentration of 0.1 mM. If both were induced, the temperature was lowered after induction of IBA production. Samples were taken for measurements of OD600, extracellular metabolites, and the distribution of the strains in 2–4 h intervals during growth phase and 6–12 h intervals after the growth phase.
For experiments with washed cells 10 mL of induced culture was centrifuged at 5000 × g for 5 min then resuspended in the same amount of fresh media. The OD600 of the resuspension was adjusted to 1 and used to a inoculate 50 mL fresh media to a start OD600 of 0.05. After cells reached an OD600 of at least 0.1 xylose was added to a final concentration of 133 mM (20 g L−1).
Quantification of cell density
The cell densities at selected time-points were measured during the cultivations using optical density at a wavelength of 600 nm using a Jenway 6705 UV/Vis spectrophotometer (for shake-flask experiments) or was automatically measured every 20 min at either OD600 (Epoch 2) or OD630 (ELx808) (for plate-reader experiments).
Flow cytometry
A total of 900 µL sample was spun down at 6500 × g for 5 min. After collection of the supernatant for extracellular metabolite quantification (see below), the pellet was fixed by re-suspending in 2% paraformaldehyde in 1x phosphate-buffered saline (PBS) and incubated for a minimum of 30 min. After fixation, the cells were spun down at 6500 × g for 5 min, re-suspended in 1x PBS and stored at 4 °C until further analysis, which was performed within 2 weeks. The relative abundance of the cells in the consortium was determined by flow cytometry using a MacsQUANT VYB cytometer (Miltenyi Biotec, Germany). Gating in the forward scatter and side scatter channels was fine-tuned to ensure that single cells were obtained, and that all cells in a given sample were represented in the measurement. mCherry fluorescence was measured with a 561 nm laser; 615/20 nm band-pass filter, whereas YFP fluorescence was measured with a 488 nm laser; 525/50 band-pass filter. Gating of the channels was performed using single strain controls, and pre-determined mixtures of the strains were used to ensure that cells were not present in both of the gatings, and that the number of cells in the two gates corresponded to the total number of cells counted.
Quantification of extracellular metabolites
A total of 900 μL of culture was spun down at 6500 × g for 5 min, and the supernatant was collected and stored at −20 °C until further analysis. The thawed supernatant was analyzed by high performance liquid chromatography (HPLC) using an Ultimate 3000 HPLC equipped with a Biorad Aminex HPX-87H column. Samples (run for 33 min) were eluted using 5 mM sulfuric acid as mobile phase at a flow rate of 0.6 mL min−1, and a column temperature of 30 °C. Glucose, xylose, xylitol, acetic acid, pyruvic acid, and IBA were detected using a refractive index detector (RI) and/or a tunable absorbance detector set at 210 nm.
Statistical analysis
All samples represent biological replicates. Data are presented as mean ± s.d. Statistical analysis was performed using the GraphPad Prism 10 software. If applicable the statistical significance between groups was assessed using a two-tailed unpaired t-test (Welch’s t-test). Values of p < 0.05 were considered significantly different.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data supporting the findings of this work are available within the paper and its Supplementary Information files. Biological materials are available from the corresponding author upon request. Source data are provided with this paper.
Code availability
A model repository of the genome-scale metabolic analysis is available on Github [https://github.com/biosustain/consortia_synthetic_anaerobe] and Zenodo34.
References
UN General Assembly. Transforming Our World: the 2030 Agenda for Sustainable Development (United Nations Division for Sustainable Development, 2015).
Zhou, K., Qiao, K., Edgar, S. & Stephanopoulos, G. Distributing a metabolic pathway among a microbial consortium enhances production of natural products. Nat. Biotechnol. 33, 377–383 (2015).
Zhang, H., Pereira, B., Li, Z. & Stephanopoulos, G. Engineering Escherichia coli coculture systems for the production of biochemical products. Proc. Natl. Acad. Sci. USA 112, 8266–8271 (2015).
Shahab, R. L., Luterbacher, J. S., Brethauer, S. & Studer, M. H. Consolidated bioprocessing of lignocellulosic biomass to lactic acid by a synthetic fungal-bacterial consortium. Biotechnol. Bioeng. 115, 1207–1215 (2018).
Minty, J. J. et al. Design and characterization of synthetic fungal-bacterial consortia for direct production of isobutanol from cellulosic biomass. Proc. Natl. Acad. Sci. USA 110, 14592–14597 (2013).
Zuroff, T. R., Xiques, S. B. & Curtis, W. R. Consortia-mediated bioprocessing of cellulose to ethanol with a symbiotic Clostridium phytofermentans/yeast co-culture. Biotechnol. Biofuels 6, 59 (2013).
Eiteman, M. A., Lee, S. A. & Altman, E. A co-fermentation strategy to consume sugar mixtures effectively. J. Biol. Eng. 2, 3 (2008).
Bernstein, H. C., Paulson, S. D. & Carlson, R. P. Synthetic Escherichia coli consortia engineered for syntrophy demonstrate enhanced biomass productivity. J. Biotechnol. 157, 159–166 (2012).
Xia, T., Eiteman, M. A. & Altman, E. Simultaneous utilization of glucose, xylose and arabinose in the presence of acetate by a consortium of Escherichia coli strains. Microb. Cell Fact. 11, 77 (2012).
Qi, L. S. et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013).
Li, S. et al. Enhanced protein and biochemical production using CRISPRi-based growth switches. Metab. Eng. 38, 274–284 (2016).
Khankal, R., Chin, J. W., Ghosh, D. & Cirino, P. C. Transcriptional effects of CRP* expression in Escherichia coli. J. Biol. Eng. 3, 13 (2009).
Harman, J. G., McKenney, K. & Peterkofsky, A. Structure-function analysis of three cAMP-independent forms of the cAMP receptor protein. J. Biol. Chem. 261, 16332–16339 (1986).
Portnoy, V. A., Herrgard, M. J. & Palsson, B. O. Aerobic fermentation of D-glucose by an evolved cytochrome oxidase-deficient Escherichia coli strain. Appl. Environ. Microbiol. 74, 7561–7569 (2008).
Akinterinwa, O. & Cirino, P. C. Anaerobic obligatory xylitol production in Escherichia coli strains devoid of native fermentation pathways. Appl. Environ. Microbiol. 77, 706–709 (2011).
Luli, G. W. & Strohl, W. R. Comparison of growth, acetate production, and acetate inhibition of Escherichia coli strains in batch and fed-batch fermentations. Appl. Environ. Microbiol. 56, 1004–1011 (1990).
Lennen, R. M. & Herrgard, M. J. Combinatorial strategies for improving multiple-stress resistance in industrially relevant Escherichia coli strains. Appl. Environ. Microbiol. 80, 6223–6242 (2014).
Zelic, B. et al. Fed-batch process for pyruvate production by recombinant Escherichia coli YYC202 strain. Eng. Life Sci. 3, 299–305 (2003).
Sheldon, R. A. & Woodley, J. M. Role of biocatalysis in sustainable chemistry. Chem. Rev. 118, 801–838 (2018).
Liu, J. et al. CRISPR-assisted rational flux-tuning and arrayed CRISPRi screening of an L-proline exporter for L-proline hyperproduction. Nat. Commun. 13, 891 (2022).
Yao, L. et al. Pooled CRISPRi screening of the cyanobacterium Synechocystis sp PCC 6803 for enhanced industrial phenotypes. Nat. Commun. 11, 1666 (2020).
Fang, L. et al. Genome-scale target identification in Escherichia coli for high-titer production of free fatty acids. Nat. Commun. 12, 4976 (2021).
Peters, J. M. et al. A comprehensive, CRISPR-based functional analysis of essential genes in bacteria. Cell 165, 1493–1506 (2016).
Bosch, B. et al. Genome-wide gene expression tuning reveals diverse vulnerabilities of M. tuberculosis. Cell 184, 4579–4592.e4524 (2021).
Li, S. et al. Genome-wide CRISPRi-based identification of targets for decoupling growth from production. ACS Synth. Biol. 9, 1030–1040 (2020).
Zhu, Y., Eiteman, M. A., DeWitt, K. & Altman, E. Homolactate fermentation by metabolically engineered Escherichia coli strains. Appl. Environ. Microbiol. 73, 456–464 (2007).
Tashiro, Y., Desai, S. H. & Atsumi, S. Two-dimensional isobutyl acetate production pathways to improve carbon yield. Nat. Commun. 6, 7488 (2015).
Jensen, S. I., Lennen, R. M., Herrgard, M. J. & Nielsen, A. T. Seven gene deletions in seven days: fast generation of Escherichia coli strains tolerant to acetate and osmotic stress. Sci. Rep. 5, 17874 (2015).
Datsenko, K. A. & Wanner, B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl Acad. Sci. USA 97, 6640–6645 (2000).
Cherepanov, P. P. & Wackernagel, W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158, 9–14 (1995).
Cardoso, J. G. R. et al. Cameo: A Python Library for Computer Aided Metabolic Engineering and Optimization of Cell Factories. ACS Synth. Biol. 7, 1163–1166 (2018).
King, Z. A. et al. Escher: a web application for building, sharing, and embedding data-rich visualizations of biological pathways. PLoS Comput. Biol. 11, e1004321 (2015).
Orth, J. D. et al. A comprehensive genome-scale reconstruction of Escherichia coli metabolism-2011. Mol. Syst. Biol. 7, 535 (2011).
Özdemir, E. biosustain/consortia_synthetic_anaerobe: Publication release (v1.0.0). Zenodo. https://doi.org/10.5281/zenodo.11058262 (2024).
Acknowledgements
This work was funded by the Novo Nordisk Foundation through a grant to DTU Biosustain (Grant no. NNF10CC1016517 & NNF20CC0035580), the Novo Nordisk Foundation within the framework of the Fermentation-based Biomanufacturing Initiative (FBM, Grant no. NNF17SA0031362 (A.T.N., S.I.J.)), the Novo Nordisk Foundation Copenhagen Bioscience PhD Program (Grant no. NNF21SA0069783 (A.F.)), and the Independent Research Foundation Denmark (Grant no. 7017-00321B (S.I.J.)).
Author information
Authors and Affiliations
Contributions
S.I.J. conceived the study. Y.R., A.F., A.T.N., and S.I.J. designed the experiments. Y.R., A.F., E.Ö., A.S.M.L., S.L., and S.I.J. performed the experiments. Y.R., A.F., and S.I.J. analyzed the data and wrote the manuscript. All authors have read, corrected, and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests
Peer review
Peer review information
Nature Communications thanks Kechun Zhang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rong, Y., Frey, A., Özdemir, E. et al. CRISPRi-mediated metabolic switch enables concurrent aerobic and synthetic anaerobic fermentations in engineered consortium. Nat Commun 15, 8985 (2024). https://doi.org/10.1038/s41467-024-53381-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-53381-4
