
Abstract
In most studied eukaryotes, chromosomes are monocentric, with centromere activity confined to a single region. However, the rush family (Juncaceae) includes species with both monocentric (Juncus) and holocentric (Luzula) chromosomes, where centromere activity is distributed along the entire chromosome length. Here, we combine chromosome-scale genome assembly, epigenetic analysis, immuno-FISH and super-resolution microscopy to study the transition to holocentricity in Luzula sylvatica. We report repeat-based holocentromeres with an irregular distribution of features along the chromosomes. Luzula sylvatica holocentromeres are predominantly associated with two satellite DNA repeats (Lusy1 and Lusy2), while CENH3 also binds satellite-free gene-poor regions. Comparative repeat analysis suggests that Lusy1 plays a crucial role in centromere function across most Luzula species. Furthermore, synteny analysis between L. sylvatica (n = 6) and Juncus effusus (n = 21) suggests that holocentric chromosomes in Luzula could have arisen from chromosome fusions of ancestral monocentric chromosomes, accompanied by the expansion of CENH3-associated satellite repeats.
Similar content being viewed by others


Introduction
Centromeres are specialized chromosomal regions that recruit kinetochore proteins and mediate spindle microtubule binding to ensure correct chromosome segregation during mitosis and meiosis1,2. Most taxonomic groups have chromosomes with a size-restricted centromeric ___domain confined to the primary chromosome constriction, i.e., they are monocentric2. However, holocentric chromosomes lack a primary constriction and exhibit molecular and epigenetic features that allow kinetochore proteins and microtubules to bind extensively along the chromosomes2,3. Therefore, holocentric chromosomes can tolerate large-scale rearrangements, such as chromosome fusions and fissions, because the rearranged chromosomes can maintain kinetochore activity, avoiding segregation problems and preserving essential genetic information during cell divisions4,5,6.
Holocentric chromosomes have evolved repeatedly in animals and plants7,8. The lack of conclusive evidence pointing to reversions to monocentricity in any eukaryotic lineage and the sporadic distribution of holocentric versus monocentric organisms support the unidirectional transition to holocentricity7. Numerous evolutionary models have been proposed to explain the emergence of holocentricity from monocentric ancestors, which include alterations/loss/emergence of kinetochore genes or centromeric repetitive sequences during the process8,9,10,11. In the Cuscuta genus of the Convolvulaceae family, the transition to holocentricity was associated with not just massive changes in the kinetochore component localization on the chromosomes, but also with a loss/truncation/alteration of some important representatives of the KMN complex such as KNL2, KNL1, ZWINT1, MIS12 and NDC8010. The causes of the transitions, however, remain unclear, mainly because only a few holocentric species have been studied up to date and because most holocentric groups evolved a long time ago, making the factors involved in the transition difficult to determine.
In most plants, functional centromeres are epigenetically specified by the centromeric histone H3 variant (CENH3). CENH3 binding regions (hereafter CENH3 domains) in monocentric chromosomes are typically associated with extended arrays of tandemly repeated sequences (satellite DNA), which are usually highly divergent and fast evolving12,13. Although the role of these repeats in centromere function has not yet been fully elucidated, several possible advantages of centromeric repeats have been proposed. Satellites might have favorable monomer lengths stabilizing CENH3 nucleosome positioning or contain specific sequences, such as short dyad symmetries, forming non-B-DNA structures possibly aiding CENH3 nucleosome loading through interaction with CENH3 chaperone proteins, such as HJURP in the case of humans1,14,15. Nevertheless, only a few cases of holocentric species with centromeric repeats have been characterized so far. Rhynchospora Vahl. (Cyperaceae) holocentromeres are mainly composed of a 172-bp satellite called Tyba, evenly distributed along the chromosomes in ~20 kb domains and specifically colocalizing with CENH34,16. In Chionographis japonica (Willd.) Maxim. (Melanthiaceae), several large (~2 Mb) CENH3-positive domains are associated with satellite arrays of 23- and 28-bp-long monomers17. Similarly, in mulberry (Morus notabilis) few CENH3-positive domains are associated with satellite arrays of 82-bp-long monomers18. In holocentric animals, known centromeric tandem repeats are even more elusive, with only the Meloidogyne incognita root-knot showing a 19-bp sequence box conserved within diverse centromeric satellites associated with holocentromere function19.
Juncaceae Juss. (rushes/woodrushes), the sister family of Cyperaceae (sedges), is a cosmopolitan family comprising ~473 species20. Juncus L. (rush) and Luzula DC. (woodrush) represent the largest genera in the family with 332 and 124 species, respectively20. An interesting feature of this family is its variation in centromeric organization and chromosomal structure, making it an ideal model to address hypotheses about evolutionary processes during centromere-type transition. Although historically the entire Juncaceae family was thought to be holocentric, cytogenetic and genomic studies revealed that six different Juncus species are monocentric4,21,22,23. Recent chromatin immunoprecipitation sequencing revealed that J. effusus has repeat-based and CENH3-associated monocentromeres, consisting mainly of two tandem repeat families underlying one or up to three spaced cores of CENH3-enriched regions per chromosome23. On the other hand, Luzula species have been so far characterized as holocentric without specific centromeric repeats3,24,25,26. Although in Luzula species, a 178 bp satellite sharing sequence similarity with the rice centromeric repeat was discovered27, it is uncertain whether this satellite plays a centromeric role and the lack of a reference genome has made detailed studies of Luzula centromeres challenging.
Here, we perform a comprehensive (epi)genomic characterization of the chromosome-scale genome of the woodrush Luzula sylvatica, focusing on its holocentromere organization, repetitive fraction, and genome evolution. We show that L. sylvatica has a unique repeat-based centromere organization distinct from previously described holocentric species. Comparative genomic repeat profiles of 13 Luzula species reveal likely conservation of repeat-based holocentromeres in the genus, except for Luzula elegans. Further comparative genomics analysis between Juncus effusus and L. sylvatica shows footprints of extensive monocentric chromosome fusions that could have potentially played an important role in the transition to holocentricity in the lineage.
Results
Holocentromeres of L. sylvatica are predominantly repeat-based
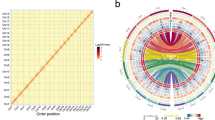
We estimated a genome size of 1 C = 476 Mb for L. sylvatica (2n = 12) based on k-mer frequencies (Supplementary Fig. 1), and assembled a chromosome-scale reference genome sequence integrating PacBio HiFi reads and a chromatin conformation capture (Hi-C) interaction dataset available at www.darwintreeoflife.org28 (Fig. 1 and Supplementary Fig. 1). The de novo genome assembly of L. sylvatica generated 1,010 contigs totaling 516.08 Mb with a GC content of 33.01%, N50 of 7.6 Mb, and Benchmarking Universal Single-Copy Orthologs (BUSCO) completeness of 93.01% (Fig. 1a–b and Supplementary Table 1). Six pseudomolecules were obtained by Hi-C scaffolding, with a total of 468.44 Mb and an N50 of 78.95 Mb (Fig. 1c and Supplementary Table 1). Similar to holocentric beak-sedges4, as the concept of chromosome arms does not apply to holocentric species, we observed no large-scale compartmentalization or telomere-to-centromere axis, as evidenced by Hi-C contact matrix (Fig. 1c). Immunolabelling of CENH3 on L. sylvatica mitotic cells confirmed the holocentricity of its chromosomes (Fig. 1d). The assembly was annotated concerning major genomic sequence types, including genes, tandem repeats and transposable elements (Fig. 1e and Supplementary Fig. 2). A dispersed but structurally heterogeneous distribution of sequences along all pseudomolecules was observed, with interstitial regions highly enriched by tandem repeats and lacking genes and transposable elements (Fig. 1e and Supplementary Fig. 2).

a Statistics of the L. sylvatica genome assembly and the final scaffolding. b BUSCO assessment for completeness of genic space with the viridiplantae_odb10 dataset, using the entire genome assembly. c Intra and inter-chromosome contact matrices of L. sylvatica. Color intensity represents contact frequency. Dark lines mark chromosomal boundaries. Boxes along the diagonal represent interactions within the same chromosome (cis), as expected for holocentric chromosomes. d Immunostaining of L. sylvatica holocentromeres using anti-CENH3 antibody (green). e L. sylvatica Chromosome 1 detailed view showing the dispersed density distribution of main genomic features: genes, CENH3, satellite DNA, LTR Ty1-copia, LTR Ty3-gypsy, H3K4me3 and H3K9me2 histone marks, and DNA methylation (CpG, CHG and CHH); as typical for holocentric chromosomes. Bin sizes of 100 kb. The distribution of features on all chromosomes is reported in Supplementary Fig. 2. f Plots of chromosome size, number of discrete CENH3 domains (units), their size (<700 kb shown), and density (units/Mb). The CENH3 unit size boxplot follows the definition in seaborn data visualization package where central lines represent median value, boxes represent 1st and 3rd quartiles, and whiskers represent the data range without outliers, defined as observations further than 1.5 of interquartile range from the respective (1st or 3rd) quartile. The number of observations (units) on each chromosome corresponds to the CENH3 unit count panel. Source data are provided as a Source Data file.
To identify and characterize the centromeres, as well as eu- and heterochromatin regions of the L. sylvatica genome, we further performed chromatin immunoprecipitation followed by sequencing (ChIP-seq) for CENH3, H3K4me3, and H3K9me2, along with DNA methylation sequencing (see Methods, Fig. 1e–f). We detected 358 CENH3 domains distributed across the entire length of all chromosomes (Fig. 1e–f and Supplementary Fig. 2). Considering that one CENH3 ___domain is equivalent to one centromeric unit, we observed an average of 0.76 units/Mb (range 0.64–0.90 units/Mb) or 60 units (range 51-76 units) per chromosome with an average unit length of 183 kb (range 174–197 kb; Fig. 1f). Additionally, histone modification marks H3K4me3 and H3K9me2 were intermixed along the chromosomes (Fig. 1e and Supplementary Fig. 2).
The annotation of the repetitive fraction, which represents ~59% of the genome, was based on the Domain-based Annotation of Transposable Elements (DANTE), DANTE for Long terminal repeat (LTR; DANTE-LTR) and Tandem Repeat Analyzer (TAREAN) (Table 1, see Methods). Most of this fraction corresponded to satellite DNA sequences with six families representing 35.31% of the genome, where the CL1 and CL2 clusters correspond to the most abundant satellite DNAs with 25.10% and 7.06%, respectively (Supplementary Table 2 and Supplementary Fig. 3). CL1 is a 124-bp satellite, named hereafter as Lusy1 (Supplementary Table 2 and Supplementary Fig. 3b). CL2 is a satellite consisting of two variants of 174 and 175 bp sharing 62% similarity (hereafter referred to as Lusy2; Supplementary Table 2 and Supplementary Fig. 3), and only 30% similarity to Lusy1. The other five satellite DNAs have monomers with 31 to 182 bp, amounting to less than ~2% in the genome each (Fig. 2a and Supplementary Table 2). Retrotransposon elements were less abundant than satellites, making up 18% of the genome (Table 1). LTR retrotransposons of the Ty1-copia superfamily were the most represented with the Angela lineage being the most abundant (10.15%; Table 1 and Fig. 1d).

a Circos distribution of the main classes of tandem repeats and CENH3 domains with a 300 kb sliding window. b Fluorescent in situ hybridization (FISH) showing discontinuous linear-like spreading of the Lusy1 (magenta) and more disperse pattern of Lusy2 (green) repeats in the nucleus (n = 10) and metaphase chromosomes (n = 10). An arrowhead points to the chromosome enlarged in the inset. c Metaplots showing the enrichment of CENH3, H3K4me3, H3K9me2, CpG, CHH, and CGH from the start and end of different types of sequences: genes (blue), LTR transposable elements (yellow), Lusy1 (magenta) and Lusy2 repeats (green). ChIPseq signals are shown as log2 (normalized RPKM ChIP/input). Methylation signals are shown as a percentage of methylated bases in each (CpG, CHG, CHH) context. d Proportion of CENH3 domains (light blue), Lusy1 (magenta) and Lusy2 (green) arrays in 100 kb windows. Distribution on all chromosomes is reported in Supplementary Fig. 4. e Close-up view of a genomic locus showing both Lusy satellite-based and satellite-free CENH3 domains. Distribution on all chromosomes is reported in Supplementary Fig. 5. f Correlogram of genomic features in 100 kb windows (n = 4694). Gray fields indicate values on the diagonal and non-significant values of the Spearman coefficient after multiple-testing correction (see Methods). Source data are provided as a Source Data file.
Satellite DNA families’ distribution varied across the genome (Fig. 2a). Lusy1 was spread throughout all pseudochromosomes, with higher densities in interstitial regions (Fig. 2a). In contrast, the other satellites (LsylSAT3–6) were found preferentially near telomeres or irregularly distributed on the chromosomes (Fig. 2a and Supplementary Table 2). We localized in situ the two most abundant putative repeats (Lusy1 and Lusy2) to corroborate the pattern obtained in silico. We observed that Lusy1 shows a line-like distribution across the entire length of each sister chromatid (Fig. 2b), in a similar pattern to other holocentromeric repeats16. Lusy2, while also present on all chromosomes, shows a more diffuse and dispersed pattern. Lusy2 appears to be present in areas where Lusy1 is less enriched, suggesting a complementary distribution between the two satellites (Fig. 2b). Furthermore, in interphase nuclei, Lusy1 signals are more focused compared to more dispersed Lusy2 signals, with clear occurrences of co-localization as well as regions where Lusy1 and Lusy2 signals do not overlap (Fig. 2b). These results suggest that although Lusy1 and Lusy2 may occupy shared regions, they also maintain distinct territories within chromatin, further supporting the idea of their distinct roles in chromosomal organization.
To investigate whether L. sylvatica holocentromeres are repeat-based, we performed a comparison analysis of these satellites with CENH3 ChIP-seq data. ChIP-seq showed CENH3 enrichment for Lusy1 and Lusy2 repeats and depletion in LTR transposable elements throughout the L. sylvatica genome (Fig. 2c). DNA methylation was similar between Lusy1/2 sequences, being highly enriched in CpG and CHG contexts at levels comparable to those of TEs. Regulatory sequences flanking the transcribed region of genes were depleted of CpG methylation compared to intergenic regions and centers of the gene bodies (Fig. 2c). As recently reported for Rhynchospora4, CHG (but also CHH) methylation seems to increase toward the borders of Lusy satellites (Fig. 2c), reinforcing the idea of an evolutionary conserved epigenetic regulation of repeat-based holocentromeres in the cyperid clade. By overlapping the annotation of the Lusy1 and Lusy2 centromeric repeats with CENH3 domains, we observed that centromeric units are mainly composed of Lusy1 (n = 232 out of 358 domains) and/or Lusy2 sequences (n = 96) (Fig. 2d–e and Supplementary Fig. 4). Additionally, we found a small subset of CENH3 domains (n = 33) associated with satellite-free regions, which were mainly composed of low-complexity repeats (54%) and LTR-TEs (26%) (Fig. 2e and Supplementary Fig. 5). Although satellite-free CENH3 domains were depleted of genes, they often contain transposable elements (16 out of 33 domains; Fig. 2e). Athila elements belonging to Ty3-gypsy family were the most abundant, making up nearly 18% of the length of the satellite-free CENH3 domains while representing only ~3% of the genome (Supplementary Table 3). Satellite-free CENH3 domains were also positively correlated with CpG, CHG, and CHH methylation, similar to Ty3-gypsy TEs (Fig. 2f and Supplementary Fig. 6), suggesting that these two features characterize the same genomic niche. The presence of satellite-associated CENH3 domains was further confirmed by immunostaining followed by fluorescent in situ hybridization (Immuno-FISH) analyses, where satellite Lusy1 signals partially colocalize with CENH3 domains along the chromosome (Fig. 3a–b). Therefore, L. sylvatica represents a case of repeat-based holocentromeres that are mostly, but not exclusively, composed of Lusy1 repeats.

a–b Immuno-FISH showing partial colocalization of Lusy1 repeats (magenta) and CENH3 (green) in metaphase chromosomes (counterstained with DAPI, gray). c Box plot of sizes of centromeric satellites Lusy1 and Lusy2 arrays associated with CENH3 (functional) or non-centromeric (nonfunctional). Statistical significance was tested using two-tailed Mann-Whitney U test. The number of observations in each group (array count) is indicated below individual boxes. d Abundance of dyad symmetries in functional and nonfunctional arrays of centromeric satellites. Statistical significance was tested using one-tailed Mann-Whitney U test. The number of observations (array counts) is identical to Fig. 1, panel c. e Homogeneity of functional array fragments (regions overlapping CENH3 domains) and whole nonfunctional arrays of centromeric satellites Lusy1 and Lusy2. Dot plots show sequence similarity between groups of concatenated arrays from the entire genome (left), histograms show the frequency distribution of similarity values (right). Images in a and b represent single slices of 3D-SIM image stacks. Dot plots are shown proportional to their genomic abundance. Boxplots (panel c) and inner boxes of violin plots (panel d) follow the definition in seaborn data visualization package where central lines (points in violin plot) represent median value, boxes represent 1st and 3rd quartiles, and whiskers represent the data range without outliers, defined as observations further than 1.5 of interquartile range from the respective (1st or 3rd) quartile. Source data are provided as a Source Data file.
Using in silico mapping data, we also identified arrays of satellites Lusy1 and Lusy2 that lack association with CENH3 (hereafter referred to as nonfunctional). For Lusy1, 247 out of 704 arrays (35%) overlap with CENH3 domains (functional). The length of these overlapping regions was 47 Mb out of 77 Mb in total (60%). For Lusy2, 107 out of 952 arrays (11%) contained CENH3 domains, making up 10 Mb out of 43 Mb (24%) of total length. Nonfunctional arrays tended to be smaller than the functional arrays of the same satellite family, with an average length of functional/nonfunctional arrays of 189 kb/20 kb and 94 kb/20 kb for Lusy1 and Lusy2, respectively (Fig. 3c). Functional Lusy1 arrays contained a higher abundance of dyad symmetries. In Lusy2, this difference was not significant (Fig. 3d). Functional and nonfunctional arrays also differ in their inter-array sequence similarity. Functional arrays of both Lusy1 and Lusy2 satellites had higher average similarity across discrete arrays compared to nonfunctional arrays (88.0 vs. 87.2% and 89.6 vs. 85.1% for Lusy1 and Lusy2, respectively; Fig. 3e). Interestingly, nonfunctional Lusy1 arrays show a clear bimodal distribution with one of the groups having a higher similarity than the corresponding functional arrays (Fig. 3e). Epigenetic status of the functional array chromatin also shows a striking contrast, since functional centromeric regions (i.e., Lusy1 and Lusy2 functional arrays, satellite-free centromeric units) are enriched with heterochromatin mark H3K9me2 and depleted of euchromatin mark H3K4me3, while the nonfunctional arrays are the opposite (Fig. 2f and Supplementary Fig. 6b).
Conservation of KNL1 and NDC80 kinetochore proteins
In Cuscuta, the transition to holocentricity was associated with massive changes in the localization of CENH3 and the kinetochore proteins KNL1, MIS12, and NDC80, representing the three complexes of the KMN network10,29. Unlike Cuscuta species, Luzula still possesses centromeric activity associated with CENH3 (Fig. 1d and Supplementary Fig. 7a)24,25. To test whether the kinetochore assembles along the poleward chromosome surface, as expected for holocentric chromosomes, we examined the localization of KNL1 and NDC80 in two holocentric woodrushes, L. sylvatica and Luzula nivea as well as in the related monocentric common rush J. effusus. Antibodies against KNL1 and CENH3 revealed a co-localized distribution in L. sylvatica metaphases demonstrating that KNL1 functionally integrates with CENH3 at centromeres during cell division (Fig. 4a). Furthermore, KNL1 showed a similar pattern in both L. sylvatica and L. nivea, with signals detected as multiple clusters along the poleward surface of chromosomes, where microtubules attach (Fig. 4b and Supplementary Fig. 7a and 8; Supplementary Movie 1 and 2). In addition, immuno-FISH signals from the centromeric repeat Lusy1 presented partial overlap with KNL1, where Lusy1 in a clustered pattern is contrasting the more continuous lines observed for KNL1 where microtubules attach (Fig. 4c). Although there is some centromeric association of Lusy1, this repeat is not bound exclusively to the centromere as observed at the genomic level (Fig. 4c). Unlike the KNL1 protein, NDC80 signals were observed only in L. nivea (Supplementary Fig. 7a; Supplementary Movie 3). Absence of NDC80 immunosignals in L. sylvatica could be due either to low amino acid sequence similarity with the target sequence developed in Cuscuta, or due to sensitivity of the protein during the cell fixation process, as discussed by Oliveira et al.29. In J. effusus, KNL1 and NDC80 showed a specific dot-like localization in the primary constriction region of the chromosome, also associated with microtubule attachment sites (Supplementary Fig. 7b; Supplementary Movie 4 and 5). KNL1 and NDC80 associate with spindle-binding sites detected by antibodies against α-tubulin, indicating that both proteins have a conserved kinetochore function in Luzula and Juncus.

a KNL1 (orange) and CENH3 (green) proteins co-localize and show a holocentric distribution (n = 15). b KNL1 protein localizes specifically to the centromere surface, where microtubules (gray) bind (n = 15). c Co-detection of KNL1, Lusy1 repeats (purple), and α-tubulin (n = 15). It is noteworthy that at this stage, some microtubule ends already colocalize with KNL1 proteins (indicated by arrowheads, see Supplementary Figs. 7 and 8). Maximum intensity projections of 3D-SIM image stacks.
Lusy1 and Lusy2 satellites are present across the genus Luzula
To determine whether repeat-holocentromeres are conserved in other species of the genus Luzula, both an individual and comparative analysis of the repeatome using RepeatExplorer2 was performed in 13 species, including L. sylvatica (Supplementary Table 4; Supplementary Data 1 and 2). The global genomic proportion of repetitive DNA varied from 35.24% (Luzula pilosa) to 66.29% (Luzula wahlenbergii) (Supplementary Data 1). In general, satellites were the most abundant class of repeats, comprising up to 49% of the Luzula sudetica genome. The centromeric satellite Lusy1 was one of the most abundant among all satellites, representing up to 47.41% of L. sudetica genome but only 3.33% of the L. nivea genome and entirely absent in Luzula elegans (Supplementary Data 1). Lusy2 also showed variation in abundance among species, ranging from 0.34% (Luzula multiflora subsp. frigida) to 31.03% (Luzula luzuloides), being also found in L. elegans genome (0.72%; Supplementary Data 1), a species with previously undetected holocentromeric repeats3. LTR retrotransposons revealed variable abundances among species, with the Ty1-copia superfamily being the most represented (1.21% in L. pilosa to 41.35% in L. elegans; Supplementary Data 1).
Comparative repeat analysis resulted in 166 shared clusters (Fig. 5a and Supplementary Fig. 9; Supplementary Data 2). Variants of Lusy1, the most abundant satellite family in Luzula, were found in all analyzed species, except in L. elegans, where this satellite was not detected even in an additional fine search of the raw sequencing reads (Fig. 5a). Different variants of the Ty1-copia Angela lineage were found in high abundance among the species, being more dominant in the genomes of Luzula arcuata and L. elegans (Fig. 5a; Supplementary Data 2). Ivana and SIRE Ty1-copia lineages were also shared among all species, although they exhibited lower abundance than Angela (Supplementary Data 1 and 2).

a Comparative analyses of the abundance of the main types of repetitive sequences in Luzula species. Code names correspond to Larcu: Luzula arcuata, Lcamp: Luzula campestris, Leleg: Luzula elegans, Lluzu: Luzula luzuloides, Lmtsf: Luzula multiflora subsp. frigida, Lniva: Luzula nivalis, Lnvea: Luzula nivea, Lparv: Luzula parviflora, Lpilo: Luzula pilosa, Lspic: Luzula spicata, Lsude: Luzula sudetica, Lsylv: Luzula sylvatica, Lwahl: Luzula wahlenbergii. The size of the ball is proportional to the genome abundance of that cluster for each species. The colors of the balls correspond to different repetitive sequence types (see Supplementary Table 4 for details). b FISH showing a wide-spreading localization of satellite Lusy1 (magenta) and a dispersed pattern of Lusy2 (green) repeats in mitotic metaphase chromosomes of L. nivea (N = 10). Chromosomes were counterstained with DAPI. Source data are provided as a Source Data file.
Because Lusy1 and Lusy2 were the most abundant satellites in the comparative analysis, consistent with the observation from the L. sylvatica genome, we performed FISH to confirm their distribution also in L. nivea, the species with very low abundance of Lusy1. Like L. sylvatica, the FISH signals of Lusy1 in L. nivea showed a line-like distribution along the chromosomes. However, exhibiting both enriched and depleted labeled chromosomal regions. Furthermore, Lusy2 showed clustered signals enriched at interstitial and terminal regions in a non-linear pattern (Fig. 5b). These results suggest a similar repeat-based holocentromere organization for other Luzula species as well.
Chromosome fusions drive karyotypic evolution in Luzula
Chromosomes from some grasses and several holocentric species have undergone extensive karyotypic rearrangements through fusions4,5,30. To investigate the possible association between holocentricity and chromosome fusions, we analyzed synteny between the genomes of the holocentric L. sylvatica and the monocentric J. effusus species (Fig. 6 and Supplementary Fig. 10). Considering n = 20 as the putative ancestral karyotype for the family Juncaceae31, the synteny analysis between the two genomes revealed that the chromosomes of L. sylvatica consist of fused blocks from J. effusus chromosomes (dysploid with n = 21), resulting in a descending dysploidy to n = 6 (Fig. 6a). Despite their high chromosome number and centromere-type differences, small arrangements and large syntenic blocks were identified between both genomes, indicating well conserved genomic structures in this family (Fig. 6b and Supplementary Fig. 10; Supplementary Data 3). A total of 86.4% (23,016 gene pairs) of the J. effusus genome is syntenic with L. sylvatica. Within this fraction, fine-scale synteny analysis revealed several large centromeric units that appear to be conserved between J. effusus and L. sylvatica genomes, despite the extensive variation of centromere locations and the divergence of >60 Myr of these genomes (Supplementary Figs. 11 and 12). This evidence of fusions and chromosomal rearrangements was also found using synteny of individual (unscaffolded) contigs of L. sylvatica and J. effusus (Supplementary Fig. 13).

a Genome synteny patterns showing macro-conserved blocks of J. effusus that were fused into Luzula chromosomes. b L. sylvatica chromosome 1 and 6 (Lsyl) showing the fusion of syntenic blocks of J. effusus (Jef) 1, 11, 18 and 21. Genes, CENH3 and telomere domains are annotated as blue-purple, green and black stripes, respectively. c FISH of telomeric DNA showing chromosomes with remnant interstitial telomeric sites, suggesting ancestral fusion events (n = 5). d Colocalization between major genomic and epigenomic features and fusion regions (columns) based on comparison of overlap with simulated regions (rows). Heatmap values show the rank of real overlap values among a distribution of overlaps with simulated regions. Asterisk points to exclusion of satellite array positions from permitted simulated region locations, for the rest of the features, the whole genome was used for simulated regions. e Examples of fusion sites on Chr1 and Chr6 in L. sylvatica. Source data are provided as a Source Data file.
We have recently shown that the holocentromeric repeat Tyba can be involved in facilitating end-to-end chromosome fusions in Rhynchospora species4. To assess the possible role of Lusy1 and Lusy2 in the fusion regions observed in Luzula genomes, we looked for specific enrichment of these repeats and telomeric repeats at the fusion regions. We found evidence for interstitial telomeric sites (ITS) using FISH experiments (Fig. 6c). Looking at the telomere annotation of L. sylvatica genome assembly, we observe three instances of possible interstitial telomeric sites localized in or near a fusion region (Supplementary Fig. 14). Further, the size of the fusion regions in L. sylvatica, defined as the space between syntenic blocks, revealed a size range from 10 kb to 8 Mb. At the block boundaries (flanking regions of 50 kb), we observed a positive association with genes (Fig. 6d and Supplementary Fig. 15). Large fusion regions (>100 kb) also contained satellites and/or transposable elements (Fig. 6e). However, the enrichment of the large fusion regions with repeats is not prominent enough to be recognized at the scale of genome-wide colocalization between features (except possible enrichment for functional Lusy1 arrays; Fig. 6d).
Discussion
Holocentromeres have evolved from a monocentric ancestor multiple times during the evolution of eukaryotes, and despite the convergent appearance of extended centromere, each of these events results in specific genome organization and adaptation of the kinetochore protein machinery4,8,11,32. In Cuscuta, holocentricity co-occurs with CENH3 independent mitotic spindle attachments and extensive changes in kinetochore structural and regulatory protein genes10,29. In insects, transitions to holocentricity are associated with the loss of CENH3, while the inner kinetochore complex remained relatively conserved9,33. In cases where the kinetochore proteins’ function is maintained, their localization can present a spectrum between continuous line-like and discrete cluster-like distribution along the chromosome2. While previously studied Luzula species (L. elegans and L. nivea) display a line-like distribution of CENH324,25, we detected signals more resembling a cluster-like distribution for CENH3 and KNL1 proteins on chromosomes in a less decondensed state in L. sylvatica, suggesting a more discontinuous and dynamic holocentromere organization in this species.
We show that functional holocentromeres in L. sylvatica are mainly made of Lusy1 and partially Lusy2 satellite repeats organized as kilobase-scale, non-uniformly spaced CENH3-positive centromeric units. However, the presence of several repeat-less centromere units suggests a more complex determination of centromere function in this species that needs to be further examined. These units display heterochromatin-typical characteristics and alternate with euchromatin, rich in coding regions, along the chromosome. Previous studies have identified a potential 178 bp centromeric tandem repeat in L. nivea and other Luzula species with similarity to the centromeric satellite RCS2 from rice27. Indeed, the 178 bp satellite shares 87% similarity with the Lusy2 satellite, which we found in all species. Lusy2 partially enriches centromeric regions but does not cover entire chromosomes like 124 bp Lusy1, which encompasses the holocentromere of L. sylvatica and, along with its abundance in most analyzed Luzula species, suggest that Lusy1 is the primary centromeric satellite. An exception is the early diverging species L. elegans, where Lusy1 is absent and Lusy2 only represents <1% of the genome. None of the 20 previously analyzed satellite repeats in L. elegans exhibited a centromeric pattern, despite repetitive sequences making up ~60% of the genome3,34. Repeat-based holocentromeres have been previously reported in Chionographis japonica17, Rhynchospora4,16,35, Eleocharis36,37, in the nematode Meloidogyne incognita19, and in mulberry18. Luzula elegans lacks Lusy1, mirroring the absence of the holocentromeric repeat Tyba in early diverging Rhynchospora lineages, which instead have diverse satellite arrays arranged in block-like patterns38,39. In both of these genera, the colonization of holocentromeres by contemporary genus-specific centromeric satellite families occurred only after the process of transition to holocentricity began39.
Our results raise questions about the expansion and functional role of satellites in holocentromeres. A process similar to the establishment of neocentromeres as found in monocentrics could be taking place. Neocentromeres can arise in heterogeneous genomic regions that become subject to rapid cycles of invasion and purification of repetitive sequences through satellite homogenization40. In L. sylvatica, we have identified several satellite-free centromeric units reminiscent of maize de novo centromeres41 in their gene-poor region targeting, CHG and CHH methylation, and possible association with Ty3-gypsy elements. These units could then become a subject of competition for centromere dominance between Lusy satellites driven by satellite homogenization and evolutionary selection pressure for centromere stabilizing effects such as advantages in CENH3 loading15 or nucleosome formation and positioning1. An analogous process of acquisition of heterochromatin epigenetic modifications, accumulation of transposable elements, and invasion of satellite repeats has been described in monocentrics as evolutionary new centromere maturation42,43.
Furthermore, holocentric chromosomes can also originate from monocentromere spreading or chromosome rearrangements. The transition to holocentricity in Luzula could have been initiated earlier than the split of the Luzula/Juncus genus, since the repeat-based centromeres of J. effusus were recently described as an atypical monocentromere, with up to three centromere cores and different types of centromeric organization, resembling an almost widespread metapolycentric organization23. In Luzula, dramatic karyotype changes took place, resulting in reduced chromosome number. Although phylogenetic relationships are poorly resolved in the genus, descending dysploidy has been observed in species from different Luzula clades (L. elegans in the Marlenia clade and L. purpureo-splendens in the Nodulosae clade), indicating that this process of chromosomal evolution has occurred independently at least twice during the evolution of the genus26. Our results support dysploidy as the main driver of karyotype evolution in holocentric organisms (Supplementary Fig. 11), since fission and fusion events have been observed in sedges, leading to dysploid karyotypes in the holocentric genera Rhynchospora4,6 and Carex 5,44, as well in some holocentric butterflies45,46. However, the fusion of ancestral chromosomes that resemble chromosomes from the sister genus Juncus resulting in the dysploid L. sylvatica is intriguing, since it involves a simultaneous shift of centromere organization. The 21 putative Juncus ancestral-like chromosomes merged into six L. sylvatica chromosomes while undergoing additional chromosome rearrangements, genomic reshuffling, and repetitive DNA turnover in the past ~60 million years of divergence47. Unlike the repeat-mediated chromosome fusions observed in Rhynchospora4, fusion sites in L. sylvatica lack genomic footprints, suggesting another mechanism of chromosome fusions during karyotype evolution in Luzula and/or a masking over time by additional chromosomal rearrangements and dynamic repeat turnover.
Based on our findings, which are supported by the few ancestral centromeric regions found in both J. effusus and L. sylvatica, we propose a multistep model of evolutionary transition to holocentricity in the genus Luzula (Fig. 7). At first, a hypothesized change of the genomic processes responsible for centromere maintenance enables centromere spreading without inactivation, as observed in Juncus. Next, stepwise chromosome fusions of Juncus-like chromosomes could generate a ‘protopolycentric’ chromosome, which could gradually transition to a more diffused holocentric organization with tens to hundreds of discrete CENH3 domains through genome rearrangements and CENH3 spreading or seeding11. Subsequent centromere maturation can culminate with satellites invading these loci by a combination of satellite DNA library diversification and concerted evolution, as observed for Tyba repeats39. From this point of view, the presence of satellite-free centromeric units, the uneven distribution of centromeric satellite repeats and the cluster-like distribution of CENH3 and outer kinetochore proteins in L. sylvatica can be interpreted as intermediate stages of ongoing holocentromere maturation. An alternative model, with the first transition to holocentricity followed by stepwise fusion of chromosomes, cannot be discarded4. Further research can provide insight into the molecular mechanisms’ adaptation taking place during the holocentric transition and its triggers.

After several fusions of whole atypical monocentric chromosomes ( Juncus-like type), centromeric domains were initially conserved in the larger chromosomes (hypothetical intermediate state), forming polycentric chromosomes. Subsequently, expansion of the centromeric ___domain and genome rearrangement gave rise to the holocentric condition. Later colonization of Lusy-type satellites allowed the maintenance of functional centromeres. M monocentric, H holocentric. Divergence time was obtained from the Timetree of Life (https://timetree.org/).
Methods
Plant material
For cytogenetic analyses, plants from natural populations of L. sylvatica were collected in Cologne, Germany, and further cultivated under controlled greenhouse conditions (16 h daylight, 26 °C, >70% humidity). The ornamental plant L. nivea was commercially obtained (Dingers Gartencenter) and cultivated under controlled greenhouse conditions (16 h daylight, 20 °C).
Genome assembly and Hi-C scaffolding
HiFi and Hi-C reads obtained through the Darwin Tree of Life database (www.darwintreeoflife.org)28 were assembled using Hifiasm48, available at https://github.com/chhylp123/hifiasm, following the command: “hifiasm -o output.asm -t 40 reads.fq.gz”. Preliminary assemblies were evaluated for contiguity and completeness with BUSCO49 and QUAST50.
Hi-C reads were first mapped to the primary contigs file obtained from the Hifiasm assembler using BWA51 following the hic-pipeline (https://github.com/esrice/hic-pipeline). Hi-C scaffolding was performed using SALSA2 (https://github.com/marbl/SALSA)52 with default parameters using ‘GATC, GAATC, GATTC, GAGTC, GACTC’ as restriction sites. After testing several minimum mapping quality values of bam alignments, the final scaffolding was performed with MAPQ10. Following the automated scaffolding by SALSA2, several rounds of visual assembly correction guided by Hi-C heatmaps were performed. When regions showed multiple contact patterns, manual re-organization of the scaffolds was performed with Juicebox53 and 3D-DNA assembly pipeline54 to correct position/orientation and to obtain the six pseudomolecules.
Genome size estimate was obtained from HiFi reads using findGSE55. First, a histogram of k-mers was created using jellyfish56, and then the findGSE R package was used for model fitting according to package documentation (https://github.com/schneebergerlab/findGSE).
Chromatin immunoprecipitation (ChIP-seq) sequencing and analysis
ChIP experiments were performed following Hofstatter et al.4. In brief, L. sylvatica leaves were harvested and frozen in liquid nitrogen until sufficient material was obtained. The samples were fixed in 4% formaldehyde for 30 min and the chromatin was sonicated to enrich for 300 bp fragments. Then, 40 ng of sonicated chromatin was incubated with 2 ng of antibody overnight. Immunoprecipitation experiments were carried out for the rabbit anti-L. elegans CENH3 (LeCENH3)57, rabbit anti-H3K4me3 (abcam, ab8580), and mouse anti-H3K9me2 (abcam, ab1220). Anti-LeCENH3 that was originally developed against 3-RTKHFSNRKSIPPKKQTPAK-23 peptide from Luzula elegans bears 65% similarity to the corresponding sequence 3-RTKHFSLRSRHPKKQRTAA-22 from Luzula sylvatica CENH3 (GenBank: KJ934236.1). Recombinant rabbit IgG (abcam, ab172730) and no-antibody inputs were used as controls. Two experimental replications were also maintained for all the combinations. ChIP DNA was quality-controlled using the NGS-assay on a FEMTO-pulse (Agilent); next, an Illumina-compatible library was prepared for all immunoprecipitants with the Ovation Ultralow V2 DNA-Seq library preparation kit (Tecan Genomics) and single-end 1 ×150-bp reads were sequenced on a NextSeq 2000 (Illumina) device. For each library, an average of 20 million reads was obtained.
The raw sequencing reads were trimmed by Cutadapt58 to remove low-quality nucleotides (with quality score less than 20) and adapters. Trimmed ChIPed 150-bp single-end reads were mapped to the respective reference genome with bowtie259, where all read duplicates were removed and only the single best-matching read was kept on the final alignment BAM file. ChIP vs input signal was calculated as the log2 ratio of read coverages normalized by reads per kilobase per million mapped reads (RPKM) using the bamCompare tool from deepTools package60. Averaged signal from both replicates was visualized using pyGenomeTracks61.
Metaplots obtained by the plotProfile function from deepTools were used to compare the distribution with other genomic features60. To concretize enriched domains, we performed peak-calling by MACS362 and epic263 and filtered only the peaks identified by both tools in both replicates. This high stringency peak filtering approach was chosen to reduce the risk of including false positive CENH3 domains in subsequent analyses. Based on analysis of CENH3 peak clustering, peaks closer than 150 kb were merged to obtain uninterrupted centromeric units (code available on GitHub at https://github.com/437364/Repeat-based-holocentromeres-of-Luzula-sylvatica).
Methylation sequencing and analysis
To analyze DNA methylation level, a sequencing library was prepared using NEBNext® Enzymatic Methyl-seq Kit (NEB; catalog number E7120S). Library sequencing was performed using NextSeq 2000 (Illumina) platform, obtaining ~20 M paired-end reads. Sequencing data was analyzed using the Bismarck pipeline64 according to the toolkit documentation (https://felixkrueger.github.io/Bismark/bismark/). Coverage files for CpG, CHG, and CHH methylation contexts were converted to bigwig.
Repeat characterization
Available Illumina reads from ENA was filtered by quality with 95% of bases equal to or above the quality cut-off value of 10 using RepeatExplorer2 pipeline (https://repeatexplorer-elixir.cerit-sc.cz/)65. The clustering was performed using the default settings of 90% similarity over 55% of the read length. For the comparative analyses, we performed an all-to-all similarity comparison across all species following the same approach. Because the genome size is unknown for some analyzed species, each set of reads was down-sampled to 1,000,000 for each species. Additionally, a subsample of eight species with known genome size were analyzed to compare the results. Samples from each species were identified with the four-letter prefixes shown in Table 1, and concatenated to produce datasets as input for RepeatExplorer2 graph-based clustering.
The automatic annotation of repeat clusters obtained by RepeatExplorer2 was manually inspected and reviewed, and was followed by recalculation of the genomic proportion of each repeat type when appropriate. DANTE and DANTE-LTR retrotransposon identification (Galaxy Version 3.5.1.1) pipeline was used to identify full-length LTR retrotransposons in the assembled genome, using a set of protein domains from REXdb66. All complete LTR-RTs contain GAG, PROT, RT, RH and INT domains, including some lineages encoding additional domains, such as chromodomains (CHD and CHDCR) from chromoviruses67 or ancestral RNase H (aRH) from Tat elements68. DANTE_LTR retrotransposon filtering (Galaxy Version 3.5.1.1) was used to search for good quality retrotransposons, those with no cross-similarity between distinct lineages. This tool produced a GFF3 output file with detailed annotations of the LTR-RTs identified in the genome and a summary table with the numbers of the identified elements66. Overall repeat composition was calculated, excluding clusters of organelle DNA (chloroplast and mitochondrial DNA). Tandem sequences were identified using TAREAN69. All putative tandem sequences were compared for homology with DOTTER70. All tandem sequences were individually mapped to the genome by BLAST with 95% similarity using Geneious71. The mapped sequence files were converted to BED and used as an input track for a genome-wide overview with ShinyCircos using a 300 kb window72. Interstitial telomere sequences (ITS) were annotated on two stringency levels by searching for regions longer than 200 bp with at least 75 or 90% similarity to consensus Arabidopsis-type telomere arrays (monomer TTTAGGG) using Geneious71, arrays closer than 10 kbp were merged.
Characterization of centromeric units
Centromeric units from ChIP-seq analysis were grouped by chromosome and their size, count, and density were calculated. Next, centromeric units were overlapped with locations of satellites to obtain locations and extract sequences of functional array fragments (precise regions where centromeric satellites Lusy1/Lusy2 and CENH3 domains overlap), nonfunctional arrays (whole Lusy1/Lusy2 arrays not overlapping CENH3 domains), and satellite-free units (CENH3 domains not overlapping any satellites). Sequences of discrete arrays of each type were concatenated and their homogeneity assessed using ModDotPlot (https://github.com/marbl/ModDotPlot)73. Dyad symmetries were identified in each array using the EMBOSS palindrome tool15 with nummismatches parameter set to 0. Statistical significance of the increase of dyad symmetry abundance for functional arrays was tested using one-tailed Mann-Whitney U test from scipy package74. Proportions of functional and nonfunctional arrays as well as other genetic and epigenetic features in 100 kb windows were correlated using Spearman’s rank correlation from scipy package and resulting correlation coefficients were plotted in a heatmap (code available on GitHub at https://github.com/437364/Repeat-based-holocentromeres-of-Luzula-sylvatica). Additional packages were used for data handling and visualization75,76,77,78,79.
Synteny analysis
The synteny analysis between L. sylvatica and J. effusus (as well as additional synteny of contig-level L. sylvatica assembly, see Supplementary Fig. 13) was performed with CoGe SynMap platform (https://genomevolution.org/coge/SynMap.pl)80 and SyMAP v. 5.0.681. For this analysis, CDS sequences, centromeric and telomeric repeats of both species were used. Synteny plots were obtained with GENESPACE82. Orthologs were identified following the steps: (1) using the BlastZ tool; (2) synteny analysis was performed using DAGChainer, using 25 genes as the maximum distance between two matches (-D) and 20 genes as the minimum number of aligned pairs (-A); (3) Quota Align Merge was used to merge syntenic blocks, with 50 genes as the maximum distance between them; and (4) orthologous and paralogous blocks were differentiated according to the synonymous substitution rate (Ks) using CodeML (where 2 was the maximum value of log10), and represented with different colors in the dot plot (Supplementary Fig. 10).
For the characterization of the regions involved in fusions, we followed Hofstatter et al.4. The synteny alignment between L. sylvatica and J. effusus genomes obtained in SyMAP allowed us to pin the putative regions around the borders of the fusion events. In order, to identify the underlying sequences at the fusion regions, we loaded annotation features for genes, TEs, and tandem repeats on SyMAP alignments. This allowed us to detect the sequence types in the putative fused regions. Further inspection and characterization of such regions were done by checking the genome coordinates and annotation features with Geneious71.
To estimate the position of ancestral centromeres in L. sylvatica genome based on synteny with J. effusus (Supplementary Fig. 12e), we projected the position of the closest synteny blocks on both sides of the J. effusus centromere onto the L. sylvatica genome using the “2D” visualization of syntenic blocks in SyMAP. The region between these two projected coordinates was designated as a possible ___location of the ancestral centromere. However, due to numerous chromosomal rearrangements, the position of some ancestral centromeres could not be projected precisely, resulting in dramatically larger projected regions than the size of the original centromere (chromosome 6). Projected ancestral centromere regions were visualized using RIdeogram R package83.
To verify that the chromosomal rearrangements discovered by synteny analysis are not a result of technical errors during the genome assembly scaffolding stage, we generated a synteny plot between J. effusus and individual large contigs of L. sylvatica (> 1 Mb) using GENESPACE82.
To further analyze colocalization of genomic features, epigenetic marks, and fusion regions; we selected 50 kb regions upstream and downstream of syntenic block edges facing the fusion regions and also fusion regions where the space between two syntenic blocks was larger than 100 kb. To analyze whether these regions are enriched or depleted of specific features, 1000 rounds of random region distribution or random region distribution excluding satellite array locations were simulated (this was done to improve the reliability of the null distribution for features that are defined as not overlapping with satellite arrays, e.g. satellite-free CENH3 domains and genes)84 using bedtools shuffle. Then, overlap with all other studied features was calculated for simulated and real regions as a proportion of overlapping bases to all bases covered by the feature. The percentage of real overlap proportion in the distribution of simulated values was reported (code available on GitHub at https://github.com/437364/Repeat-based-holocentromeres-of-Luzula-sylvatica).
Cytogenetic and immunostaining of CENH3 protein
Mitotic preparations were made from root meristems fixed in 4% paraformaldehyde and Tris buffer (10 mM Tris, 10 mM EDTA, 100 mM NaCl, 0.1% Triton, pH 7.5) for 30 min on ice in vacuum and for another 20 min only on ice. After washing twice in 1 x PBS for 10 min, the roots were digested in a cellulase-pectinase (2% w/v /20% v/v solution) containing PBS buffer and squashed in PBS. The coverslips were removed in liquid nitrogen and the slides were air-dried and stained in 2 µg/mL DAPI/Vectashield mounting medium for slide selection under the epifluorescence microscope. The slides with the highest number of cells in division were incubated in 3% (w/v) bovine serum albumin (BSA) containing 0.1% Triton X-100 in PBS. Immunostaining was performed using the primary antibodies rabbit anti-LeCENH3 (dilution 1:100)57, rabbit anti-KNL1 (dilution 1:1000, GenScript, NJ, USA), rabbit anti-NDC80 (dilution 1:1000, Biomatik, ON, Canada)29 and mouse anti-α-tubulin (dilution 1:100, Sigma-Aldrich, St. Louis, MO; catalog number T6199). Antibodies against KNL1 and NDC80 were originally developed using respective peptides EDHFFGPVSPSFIRPGRLSDC and EQGINARDAERMKRELQALEG from Cuscuta sp. These epitopes have a respective 55.6% and 57.1% similarity to peptides DDNFFGPVSAKFLKSGRFSDT and EQEVNLRDVDRMKREMQLIER identified by tblastn similarity search of Cuscuta europaea KNL1 and NDC80 protein sequences in L. sylvatica genome. As the secondary antibody, goat anti-Rabbit IgG antibody conjugated with Alexa Fluor 488 (Invitrogen; catalog number A27034), goat anti-rabbit conjugated with Rhodamine Red X (Jackson ImmunoResearch, catalog number: 111-295-144) or goat anti-mouse conjugated with Alexa Fluor 488 ( Jackson ImmunoResearch; catalog number 115-545-166) were used in a 1:500 dilution. Slides were incubated overnight at 4 °C, washed 3 times in 1×PBS and then the secondary antibody was applied, incubated at room temperature for 3 h and washed 3 times in 1×PBS. The slides were counterstained with 2 µg/mL DAPI/Vectashield mounting medium. Microscopic images were recorded using a Zeiss Axiovert 200 M microscope equipped with a Zeiss AxioCam CCD. Images of at least 5 cells were analyzed using the ZEN software (Carl Zeiss GmbH). Immuno-FISH was performed following Dias et al.23, the immunostained slides were washed with 1xPBS for 15 min, postfixed in 4% paraformaldehyde in 1xPBS for 5 min, and then probed with the satellite Lusy1 for 24 hours at 37 °C. Stringent washes were performed with 2× and 0.1× SSC at 42 °C to give a final stringency of ~76%.
Oligo probes from the most abundant tandem repeats Lusy1 (GATCTCAAGAACACGTTATTTAGACTCGTCAAAGCA) and Lusy2 (AATTAATGACTAACACGATGCGAATTTCAATTTTTT) and the Arabidopsis telomeric sequence (TTTAGGG) were used for fluorescent in situ hybridization (FISH). Mitotic chromosomes from roots pretreated with 2 mM 8-hydroxyquinoline for 24 h at 4 °C and fixed with ethanol:acetic acid (3:1 v/v) for 2 h, were prepared using the air-drying method38. FISH was performed in denatured chromosomes at 75 °C for 5 min. The hybridization mixture contained formamide 50% (v/v), dextran sulphate 10% (w/v), 2 × SSC, and 50 ng/μL of each labelled probe. The slides were hybridized with this mixture for at least 24 hours at 37 °C23. Stringent washes were performed with 2× and 0.1× SSC at 42 °C to give a final stringency of ~76%. The slides were counterstained with 2 µg/mL DAPI in Vectashield (Vector) mounting medium. The images of at least 10 cells were captured as described above.
To analyze the chromatin ultrastructure, we applied super-resolution spatial structured illumination microscopy (3D-SIM) using a 63x/1.40 Oil Plan-Apochromat objective of an Elyra PS.1 microscope system and the ZENBlack software (Carl Zeiss GmbH)85. Maximum intensity projections from image stacks were calculated from 3D-SIM image stacks. Zoom-in sections were presented as single slices to indicate the subnuclear chromatin structures at the super-resolution level.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The sequencing data generated in this study have been deposited in the NCBI database under the BioProject ID PRJNA1135980. The processed reference genomes, sequencing data, annotations and all tracks data generated in this study are available at Zenodo [https://zenodo.org/records/14007621]. The REXdb database Viridiplantae v.3.0 is publicly available at Github [https://github.com/repeatexplorer/rexdb]. Source data are provided with this paper.
Code availability
The original code used in this study is available at GitHub [https://github.com/437364/Repeat-based-holocentromeres-of-Luzula-sylvatica]86.
References
Talbert, P. B. & Henikoff, S. What makes a centromere? Exp. Cell Res. 389, 111895 (2020).
Schubert, V. et al. Super-Resolution Microscopy Reveals Diversity of Plant Centromere Architecture. IJMS 21, 3488 (2020).
Heckmann, S. et al. The holocentric species Luzula elegans shows interplay between centromere and large‐scale genome organization. Plant J. 73, 555–565 (2013).
Hofstatter, P. G. et al. Repeat-based holocentromeres influence genome architecture and karyotype evolution. Cell 185, 3153–3168.e18 (2022).
Escudero, M., Marques, A., Lucek, K. & Hipp, A. L. Genomic hotspots of chromosome rearrangements explain conserved synteny despite high rates of chromosome evolution in a holocentric lineage. Mol. Ecol. https://doi.org/10.1111/mec.17086 (2023).
Mata-Sucre, Y. et al. Oligo-barcode illuminates holocentric karyotype evolution in Rhynchospora (Cyperaceae). Front. Plant Sci. 15, 1330927 (2024).
Escudero, M., Márquez-Corro, J. I. & Hipp, A. L. The Phylogenetic Origins and Evolutionary History of Holocentric Chromosomes. Syst. Bot. 41, 580–585 (2016).
Senaratne, A. P., Cortes-Silva, N. & Drinnenberg, I. A. Evolution of holocentric chromosomes: Drivers, diversity, and deterrents. Semin. Cell Dev. Biol. 127, 90–99 (2022).
Drinnenberg, I. A., deYoung, D., Henikoff, S. & Malik, H. S. Recurrent loss of CenH3 is associated with independent transitions to holocentricity in insects. eLife 3, e03676 (2014).
Neumann, P. et al. Disruption of the standard kinetochore in holocentric Cuscuta species. Proc. Natl Acad. Sci. USA. 120, e2300877120 (2023).
Kuo, Y.-T., Schubert, V., Marques, A., Schubert, I. & Houben, A. Centromere diversity: How different repeat-based holocentromeres may have evolved. BioEssays 46, e202400013 (2024).
Plohl, M., Meštrović, N. & Mravinac, B. Centromere identity from the DNA point of view. Chromosoma 123, 313–325 (2014).
Šatović-Vukšić, E. & Plohl, M. Satellite DNAs—From Localized to Highly Dispersed Genome Components. Genes 14, 742 (2023).
Hobza, R. et al. An accumulation of tandem DNA repeats on the Y chromosome in Silene latifolia during early stages of sex chromosome evolution. Chromosoma 115, 376–382 (2006).
Kasinathan, S. & Henikoff, S. Non-B-Form DNA Is Enriched at Centromeres. Mol. Biol. Evol. 35, 949–962 (2018).
Marques, A. et al. Holocentromeres in Rhynchospora are associated with genome-wide centromere-specific repeat arrays interspersed among euchromatin. Proc. Natl Acad. Sci. USA. 112, 13633–13638 (2015).
Kuo, Y.-T. et al. Holocentromeres can consist of merely a few megabase-sized satellite arrays. Nat. Commun. 14, 3502 (2023).
Ma, B. et al. The gap-free genome of mulberry elucidates the architecture and evolution of polycentric chromosomes. Horticult. Res. 10, uhad111 (2023).
Despot-Slade, E. et al. The Centromere Histone Is Conserved and Associated with Tandem Repeats Sharing a Conserved 19-bp Box in the Holocentromere of Meloidogyne Nematodes. Mol. Biol. Evol. 38, 1943–1965 (2021).
POWO. Plants of the World Online. Facilitated by the Royal Botanic Gardens, Kew. Published on the Internet; http://www.plantsoftheworldonline.org/ (2024).
Guerra, M., Ribeiro, T. & Felix, L. P. Monocentric chromosomes in Juncus ( Juncaceae) and implications for the chromosome evolution of the family. Bot. J. Linn. Soc. 191, 475–483 (2019).
Mata-Sucre, Y. et al. Repeat-based phylogenomics shed light on unclear relationships in the monocentric genus Juncus L. ( Juncaceae). Mol. Phylogenet. cs Evol. 189, 107930 (2023).
Dias, Y. et al. How diverse a monocentric chromosome can be? Repeatome and centromeric organization of Juncus effusus ( Juncaceae). Plant J. tpj.16712 https://doi.org/10.1111/tpj.16712 (2024).
Nagaki, K., Kashihara, K. & Murata, M. Visualization of Diffuse Centromeres with Centromere-Specific Histone H3 in the Holocentric Plant Luzula nivea. Plant Cell 17, 1886–1893 (2005).
Heckmann, S. et al. Holocentric Chromosomes of Luzula elegans Are Characterized by a Longitudinal Centromere Groove, Chromosome Bending, and a Terminal Nucleolus Organizer Region. Cytogenet Genome Res 134, 220–228 (2011).
Bozek, M., Leitch, A. R., Leitch, I. J., Záveská Drábková, L. & Kuta, E. Chromosome and genome size variation in Luzula ( Juncaceae), a genus with holocentric chromosomes: Chromosome and C-Value Evolution in L uzula. Bot. J. Linn. Soc. 170, 529–541 (2012).
Haizel, T., Lim, Y. K., Leitch, A. R. & Moore, G. Molecular analysis of holocentric centromeres of Luzula species. Cytogenet Genome Res 109, 134–143 (2005).
Goodwin, Z. A. et al. The genome sequence of great wood-rush, Luzula sylvatica (Huds) Gaudin. Wellcome Open Res 9, 124 (2024).
Oliveira, L. et al. KNL1 and NDC80 represent new universal markers for the detection of functional centromeres in plants. Chromosome Res 32, 3 (2024).
Wang, J. et al. A high-quality chromosome-scale assembly of the centipedegrass [Eremochloa ophiuroides (Munro) Hack.] genome provides insights into chromosomal structural evolution and prostrate growth habit. Hortic. Res 8, 201 (2021).
Drábková, L. Z. A Survey of Karyological Phenomena in the Juncaceae with Emphasis on Chromosome Number Variation and Evolution. Bot. Rev. 79, 401–446 (2013).
Lucek, K., Augustijnen, H. & Escudero, M. A holocentric twist to chromosomal speciation? Trends Ecol. Evol. 37, 655–662 (2022).
Cortes-Silva, N. et al. CenH3-Independent Kinetochore Assembly in Lepidoptera Requires CCAN, Including CENP-T. Curr. Biol. 30, 561–572.e10 (2020).
Jankowska, M. et al. Holokinetic centromeres and efficient telomere healing enable rapid karyotype evolution. Chromosoma 124, 519–528 (2015).
Castellani, M. et al. Meiotic recombination dynamics in plants with repeat-based holocentromeres shed light on the primary drivers of crossover patterning. Nat. Plants 10, 423–438 (2024).
Souza, T. B. D. et al. Distinct patterns of satDNA distribution in holocentric chromosomes of spike-sedges (Eleocharis, Cyperaceae). Genome gen-2024-0089 https://doi.org/10.1139/gen-2024-0089 (2024).
Liu, H. et al. The genome of Eleocharis vivipara elucidates the genetics of C 3 –C 4 photosynthetic plasticity and karyotype evolution in the Cyperaceae. JIPB jipb.13765 https://doi.org/10.1111/jipb.13765 (2024).
Ribeiro, T. et al. Centromeric and non-centromeric satellite DNA organisation differs in holocentric Rhynchospora species. Chromosoma 126, 325–335 (2016).
Costa, L., Marques, A., Buddenhagen, C. E., Pedrosa-Harand, A. & Souza, G. Investigating the diversification of holocentromeric satellite DNA Tyba in Rhynchospora (Cyperaceae). Ann. Bot. 131, 813–825 (2023).
Wlodzimierz, P. et al. Cycles of satellite and transposon evolution in Arabidopsis centromeres. Nature 618, 557–565 (2023).
Hiatt, E. N., Kentner, E. K. & Dawe, R. K. Independently Regulated Neocentromere Activity of Two Classes of Tandem Repeat Arrays. Plant Cell 14, 407–420 (2002).
Piras, F. M. et al. Uncoupling of Satellite DNA and Centromeric Function in the Genus Equus. PLoS Genet 6, e1000845 (2010).
Cappelletti, E. et al. Robertsonian Fusion and Centromere Repositioning Contributed to the Formation of Satellite-free Centromeres During the Evolution of Zebras. Mol. Biol. Evol. 39, msac162 (2022).
Ning, Y. et al. The chromosome-scale genome of Kobresia myosuroides sheds light on karyotype evolution and recent diversification of a dominant herb group on the Qinghai-Tibet Plateau. DNA Res. 30, dsac049 (2023).
Wright, C. J., Stevens, L., Mackintosh, A., Lawniczak, M. & Blaxter, M. Comparative genomics reveals the dynamics of chromosome evolution in Lepidoptera. Nat. Ecol. Evol. https://doi.org/10.1038/s41559-024-02329-4 (2024).
Augustijnen, H. et al. A macroevolutionary role for chromosomal fusion and fission in Erebia butterflies. Sci. Adv. 10, eadl0989 (2024).
Elliott, T. L. & Davies, T. J. Phylogenetic attributes, conservation status and geographical origin of species gained and lost over 50 years in a UNESCO Biosphere Reserve. Biodivers. Conserv 28, 711–728 (2019).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: Assessing Genome Assembly and Annotation Completeness. in Gene Prediction (ed. Kollmar, M.) 1962 227–245 (Springer New York, New York, NY, 2019).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Ghurye, J. et al. Integrating Hi-C links with assembly graphs for chromosome-scale assembly. PLoS Comput Biol. 15, e1007273 (2019).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 3, 99–101 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Sun, H., Ding, J., Piednoël, M. & Schneeberger, K. findGSE: estimating genome size variation within human and Arabidopsis using k -mer frequencies. Bioinformatics 34, 550–557 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k -mers. Bioinformatics 27, 764–770 (2011).
Ma, W. et al. The distribution of α-kleisin during meiosis in the holocentromeric plant Luzula elegans. Chromosome Res 24, 393–405 (2016).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet j. 17, 10 (2011).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Ramírez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–W165 (2016).
Lopez-Delisle, L. et al. pyGenomeTracks: reproducible plots for multivariate genomic datasets. Bioinformatics 37, 422–423 (2021).
Zhang, Y. et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Stovner, E. B. & Sætrom, P. epic2 efficiently finds diffuse domains in ChIP-seq data. Bioinformatics 35, 4392–4393 (2019).
Krueger, F. & Andrews, S. R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572 (2011).
Novák, P., Neumann, P. & Macas, J. Global analysis of repetitive DNA from unassembled sequence reads using RepeatExplorer2. Nat. Protoc. 15, 3745–3776 (2020).
Novák, P., Hoštáková, N., Neumann, P. & Macas, J. DANTE and DANTE_LTR: lineage-centric annotation pipelines for long terminal repeat retrotransposons in plant genomes. NAR Genom. Bioinforma. 6, lqae113 (2024).
Neumann, P. et al. Plant centromeric retrotransposons: a structural and cytogenetic perspective. Mob. DNA 2, 4 (2011).
Neumann, P., Novák, P., Hoštáková, N. & Macas, J. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mob. DNA 10, 1 (2019).
Novák, P. et al. TAREAN: a computational tool for identification and characterization of satellite DNA from unassembled short reads. Nucleic Acids Res. 45, e111 (2017).
Sonnhammer, E. L. L. & Durbin, R. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene 167, GC1–GC10 (1995).
Kearse, M. et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Yu, Y., Ouyang, Y. & Yao, W. shinyCircos: an R/Shiny application for interactive creation of Circos plot. Bioinformatics 34, 1229–1231 (2018).
Sweeten, A. P., Schatz, M. C. & Phillippy, A. M. ModDotPlot—rapid and interactive visualization of tandem repeats. Bioinformatics 40, btae493 (2024).
Virtanen, P. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272 (2020).
Hunter, J. D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 9, 90–95 (2007).
McKinney, W. pandas: a foundational Python library for data analysis and statistics. Python high. Perform. Sci. Comput. 14, 1–9 (2011).
Dale, R. K., Pedersen, B. S. & Quinlan, A. R. Pybedtools: a flexible Python library for manipulating genomic datasets and annotations. Bioinformatics 27, 3423–3424 (2011).
Harris, C. R. et al. Array programming with NumPy. Nature 585, 357–362 (2020).
Waskom, M. seaborn: statistical data visualization. JOSS 6, 3021 (2021).
Lyons, E., Pedersen, B., Kane, J. & Freeling, M. The Value of Nonmodel Genomes and an Example Using SynMap Within CoGe to Dissect the Hexaploidy that Predates the Rosids. Trop. Plant Biol. 1, 181–190 (2008).
Soderlund, C., Bomhoff, M. & Nelson, W. M. SyMAP v3.4: a turnkey synteny system with application to plant genomes. Nucleic Acids Res. 39, e68 (2011).
Lovell, J. T. et al. GENESPACE tracks regions of interest and gene copy number variation across multiple genomes. eLife 11, e78526 (2022).
Hao, Z. et al. RIdeogram: drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput. Sci. 6, e251 (2020).
Kanduri, C., Bock, C., Gundersen, S., Hovig, E. & Sandve, G. K. Colocalization analyses of genomic elements: approaches, recommendations and challenges. Bioinformatics 35, 1615–1624 (2019).
Weisshart, K., Fuchs, J. & Schubert, V. Structured Illumination Microscopy (SIM) and Photoactivated Localization Microscopy (PALM) to Analyze the Abundance and Distribution of RNA Polymerase II Molecules on Flow-sorted Arabidopsis Nuclei. BIO-PROTOCOL 6, (2016).
Krátká, M. Repeat-based-holocentromeres-of-Luzula-sylvatica. Zenodo https://doi.org/10.5281/ZENODO.13945236 (2024).
Acknowledgements
We thank Dra. Magdalena Vaio for her fruitful comments on the manuscript. We acknowledge the excellent technical assistance of Ursula Pfordt and Christina Philipp. This work was funded by the Max Planck Society (core funding to A.M.), by the Deutsche Forschungsgemeinschaft (DFG, grant no. MA 9363/3-1 to A.M.) and by the European Union (European Research Council Starting Grant, HoloRECOMB, grant no. 101114879 to A.M.). DFG founded this work under Germany’s Excellence Strategy—EXC 2048/1–390686111 (A.M.). We thank the International Cooperation Program PROBRAL (CAPES/DAAD project number 88881.144086/2017-01) for the scholarship offered to Y.M.S. We thank the Darwin Tree of Life Project at the Wellcome Sanger Institute for making the data available (https://www.darwintreeoflife.org/project-resources). Computational resources for RepeatExplorer analysis were provided by the ELIXIR-CZ project (LM2023055), part of the international ELIXIR infrastructure. e-INFRA CZ project (ID:90254), supported by the Ministry of Education, Youth and Sports of the Czech Republic provided computational resources for the analysis of ChIP-seq data. The work of M.K. was supported by the Czech Science Foundation, grant no. GA24-11400S. E.K. was supported by the grant 21-00580S from the Czech Science Foundation. L.O. was supported by grant 2025440S from the Czech Science Foundation. A.P.H. and G.S. received productivity fellowship from CNPq (process numbers PQ-312852/2021-5 and PQ-312852/2021-5, respectively).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Y.M.S.: Investigation, Validation, Formal analysis, Data Curation, Writing- Original draft preparation. M.K.: Validation, Formal analysis, Data Curation, Writing- Reviewing and Editing. L.O., P.N. and J.M.: Investigation, Formal analysis, Resources, Writing- Reviewing and Editing. B.H., V.S. and A.H.: Resources, Writing- Reviewing and Editing. E.K, A.P.H. and G.S.: Supervision, Resources, Writing- Reviewing and Editing. A.M.: Conceptualization, Supervision, Resources, Funding acquisition, Writing- Reviewing and Editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mata-Sucre, Y., Krátká, M., Oliveira, L. et al. Repeat-based holocentromeres of the woodrush Luzula sylvatica reveal insights into the evolutionary transition to holocentricity. Nat Commun 15, 9565 (2024). https://doi.org/10.1038/s41467-024-53944-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-53944-5
