
Abstract
Skeleton editing for heteroarenes, especially pyrazoles, is challenging and remains scarce because these non-strained aromatics exhibit inert reactivities, making them relatively inactive for performing a dearomatization/cleavage sequence. Here, we disclose a cycloaddition-induced scaffold hopping of 5-hydroxypyrazoles to access the pyrazolopyridopyridazin-6-one skeleton through a single-operation protocol. By converting a five-membered aza-arene into a five-unit spine of a 6/6 fused-bicyclic, this work unlocks a ring-opening reactivity of the pyrazole core that involves a formal C = N bond cleavage while retaining the highly reactive N-N bond in the resulting product. A [4 + 2] cycloaddition of a temporarily dearomatized 5-hydroxypyrrole with an in situ generated aza-1,3-diene, followed by oxidative C-N bond cleavage, constitutes the domino pathway. A library of pyrazolopyridopyridazin-6-ones, which are medicinally relevant nitrogen-atom-rich tricyclics, is obtained efficiently from readily available materials.
Similar content being viewed by others
Introduction
Synthetic chemists and pharmaceutical researchers have been captivated by the generalized concept of “skeleton editing” for nearly one and a half centuries1,2. As eye-catching paradigms, inspiring ideas of skeletal editing3 associated with a deconstructive ring-opening process for arenes and heteroarenes are constantly emerging only recently. The state-of-the-art in this field covering single-atom editing4 (insertion5,6,7,8, deletion9,10, mutation11,12,13,14), atom-pair swapping15, deconstructive functionalization16,17,18,19,20 and so on. Despite the formidable challenge that ring-opening of specific aromatics requires a tailored dearomatisation/cleavage process to not only overcome the aromaticity but also to cleavage a σ-bond in their non-strained parent skeleton, the development of more editing modes for more types of aromatic systems is still fascinating, for achieving scaffold hoppings21 that was impossible by otherwise peripheral diversifications22.
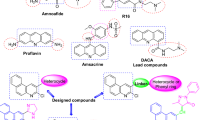
Notwithstanding elegant skeleton editing recorded for five-membered heteroarenes including furan23,24,25, imidazaole26, oxazole27, pyrrole28, thiazole29, thiophene30,31 and 1,2,4-oxadiazole32; deconstructive ring-opening of pyrazoles are relatively limited despite their significant properties as pharmacophores33,34. A remarkable work was reported in early 1966 by Pagani et al., revealing a C3 carbanion induced N-N bond dissociation to generate the N--alkyl-anthranylamides from 3-carboxy- and 3-unsubstituted pyrazoles35,36 (Fig. 1A). In 2010, Schmidt and co-workers disclosed a fascinating rearrangement of pyrazolium-3-carboxylates to 4-aminoquinolines via an in situ formed N-heterocyclic carbene intermediate37. Recently, pre-prepared pyrazolium halides and in situ generated pyrazolium ylide have been further employed to furnish the editing of pyrazole series5,38,39 (Fig. 1B). To the best of our knowledge, ring opening of pyrazoles has not yet been able to bypass the N-N bond cleavage although it has been developed for almost 60 years. Understandably, these pioneering contributions have chosen the N-N bond, with the lowest bond energy in the pyrazole ring system, as the point of penetration for the fragmentation (N-N bond, ~ 159 kJ/mol; N-C bond, ~ 305 kJ/mol; N = C bond, ~ 615 kJ/mol;)40,41. We suspected that it would be not only synthetically valuable but also mechanistically interesting to avoid breaking the N-N bond during the ring opening of the pyrazole cores, thus inheriting this difficult-to-form but easy-to-fragment linkage in the product42.

A Carbanion-induced ring opening of 3-carbonyl pyrazole series. B Pyrazolium ylide induced ring opening and rebound of 1H-pyrazole series. C Our working hypothesis of this work. D Representative biologic active compounds containing a similar scaffold of our desired product and our rational design. E Conceptual outlines for (a) ring expansion, (b) deconstructive functionalization, and (c) ring-upgrading.
In connection with our long-term interest in cycloaddition cascade reactions43,44,45,46,47,48, we surmised that it might be possible to carry out the deconstructive ring-opening of pyrazole series by a rational designed [4 + 2] cycloaddition, although we note that the dearomative cycloadditions of azaarenes are particularly challenging due to the inherent high kinetic barrier for the aromatic system to conquer49,50,51. To address this issue, we hypothesized to introduce an auxiliary hydroxyl group at C5 for pyrazole for achieving two fundamental purposes: (A) increasing the electron density of the pyrrole core, resulting in a highly-active C4 site for incorporating a cycloaddition and (B) endowing pyrazoles with the capability to spontaneous collapsing their aromaticity by tautomerism. First reported by Knorr in 1895, three tautomers of N1, C3-disubstituted 5-hydroxypyrazole will show equilibria as III > I > II in the gas phase52,53. However, the phenol form (I) is reported favored for pyrazolones in aprotic solvents such as DMSO54. Indeed, dissolved in deuterated DMSO for 1H-NMR testing, 1-methyl-1H-pyrazol-5-ol presented only as the phenol form (Please see “Supplementary Figs. 1–3” in Supporting Information). Furthermore, we anticipate that in polar aprotic solvent, the tautomers equilibrium may partially tend towards III by 1,5-H shift, thus assisting the temporary de-aromatized pyrazole to be captured by intermolecular cycloaddition. In the presence of a tailored amphiphilic aza-diene in the reaction mixture, the cycloadduct intermediate IV would be formed and then be oxidized into V due to the high reactivity of C3-H (Fig. 1C). The pre-aromatic intermediate V would undergo an aromatization-driven C-N bond cleavage to provide VI, resulting in a pyridine core. Such oxidative elimination features a key step in Fischer indole synthesis55 but is still less explored in a fused-ring scenario56,57. Moreover, aiming at expanding the surrounding chemical space, a carbonyl group was planted at the C5 position of the aza-diene as a “hook” in the first place. A second-fold cyclization would then occur to afford a pyridazinone product VII. We note that pyrazolopyridopyridazine and related dinones are useful agents in pharmaceutical research, especially for the treatment of erectile dysfunction58,59,60 (Fig. 1D). Conventionally, the preparation of these nitrogen-atom-rich tricyclics relies on tedious synthetic steps thus limiting the development of their functionality as well as the studies of Structure-Activity Relationships61. By seeking in the I2-DMSO synthetic tool-box developed by our group62, we anticipate that using aryl methyl ketone and 5-aminopyrazole in the presence of I2-DMSO manifold would provide aza-1,3-diene-5-one straightforwardly as the ideal precursors for the proposed aza-[4 + 2] cyclization, therefore achieving a one-pot synthesis of pyrazolopyridopyridazin-6-one skeleton. It is also conceivable that I2 and DMSO would play as an oxidant63 to promote the oxidative cleavage of the C-N bond downstream. Conceptually, the planned transformation hops from a five-membered azaarene to a five-unit tether by forging with a linear partner and eventually presents as the spine of the resulting 6/6 fused ring. We believe that such skeleton editing is more than neither a well-defined ring-expansion (Fig. 1E-a)4,18,64,65 nor a deconstructive functionalization (Fig. 1E-b)16,17,66,67, but a distinct model entitled ring-upgrading (Fig. 1E-c). Here, we show a three-component reaction of acetophenones with 1H-pyrazol-5-amines and 5-hydroxypyrazoles, achieving the direct synthesis of pyrazolopyridopyridazin-6-one skeletons. An I2-DMSO reagent manifold, together with di-tert-butyl peroxide, is used as the reagents for this ring-upgrading reaction. The transformations of 5-hydroxypyrazole, a five-membered azaarene, into the 6/6 fused-bicyclic are illustrated by more than 85 diverse examples.
Results
Synthesis of pyrazolopyridopyridazin-6-one skeleton by ring opening of 5-hydroxypyrazoles
Initially, acetophenone (1a), 3-methyl-1-phenyl-1H-pyrazol-5-amine (2a), and 1-methyl-1H-pyrazol-5-ol (3a) were chosen as the model substrates to optimize the reaction conditions. After a series of screenings (Please see “Supplementary Table 1” in Supporting Information), the optimal conditions were determined as follows: 1a (1.2 equiv.), 2a (1.0 equiv.), 3a (1.0 equiv.), I2 (1.0 equiv.) and di-tert-butyl peroxide (2.0 equiv.) in a solution of DMSO at 100 °C for 4 h, affording the target product 4a in 75% yield (Table 1, entry 1). It is noteworthy that the absence of iodine or DMSO was unfavorable (Table 1, entries 2-3). The reaction was facilitated by additional peroxide such as DTBP, through the desired product was also obtained in moderate yield without DTBP input (Table 1, entry 4). A variety of peroxide/oxidants were tested as alternatives to DTBP. Potassium persulfate and PIDA both lead to a drastic decrease in yield (Table 1, entries 5-6). TBHP was not as effective as DTBP (Table 1, entry 7), while radical initiator azobisisobutyronitrile (AIBN) was also able to promote the reaction, resulting in a yield of 70% (Table 1, entry 8). Reducing or increasing the amount of oxidative agent had no positive effect on the reaction (Table 1, entries 9-10). Finally, when the reaction was carried out at 80 or 120 °C, the yields were slightly reduced (Table 1, entry 11). To our delight, we did not observe any type of ring-opening for 2a throughout the screening, illustrating the exclusive selectivity for the editing of 5-hydroxypyrazoles.
Under our established conditions, the generality of the reaction substrates was then examined. As shown in Fig. 2, aryl methyl ketones bearing alkyl and alkoxy substitution on the benzene ring (4a-4e) participated in the reaction smoothly to afford desired tricyclic product 4 in good yields (70–77%). Ortho-substituted methoxy (4 f), 3,4-methylenedioxy (4 g), and functional groups such as trimethylsilyl (TMS, 4 h) and thiomethyl (4i) are all tolerated. When using halogen or dihalogen-containing substrates (F, Cl, Br, I) with different substituent positions (o, m, p) in the benzene ring (4j-4p, 65–73%), significant effects on yields were not observed. Furthermore, strong electron-withdrawing groups, such as -CF3, -OCF3, -CO2Me, -CN, -NO2, -SO2Me were suitable substitutions successfully taking part in the transformation (4q-4x). In these cases, diverse functional groups bring us the possibility of further modifying the product at a later stage. The type of aryl belonging to the methyl ketones was next tested. To our delight, 1- and 2-naphthyl (4 y, 4z), or even sterically bulkier 2-fluorenyl (5a) were competent substrates. Heterocycles such as different substitution sites of furan, thiophene, benzofuran, and benzothiophene were compatible in the reaction (5b-5h, 60–73%), thus providing inherent advantages for the study of Structure-Activity Relationship. Furthermore, structurally sensitive 1H-indole and alkaline quinoline substituents were all able to produce the desired product, albeit with slightly reduced yields (5i, 59%; 5j, 52%). Delightfully, the corresponding pyrazolopyridopyridazin-6-one products were successfully obtained using α, β-unsaturated methyl ketones as substrates (5k-5p, 56–67%). Moreover, 3,5-diene methyl ketones are also tolerated in our reaction conditions (5q, 64%; 5r, 61%). In contrast to the usage of aryl methyl ketones, the incorporation of methyl ketone substrates containing conjugated double bonds, as previously outlined, enables the further expansion of our product diversity since the double bonds in these products can be converted to saturated ones through hydrogenation. Despite the failure of the direct use of aliphatic methyl ketones in our reaction, the above experimental results serve to offset, to some extent, the impact of the inherent limitations of this methodology.

aGeneral conditions: 1 (1.2 mmol), 2 (1.0 mmol), 3a (1.0 mmol), I2 (1.0 mmol), DTBP (2.0 mmol), DMSO (5.0 mL, c = 0.2 M), 100 °C, 4 h. bGeneral conditions: 1 (1.2 mmol), 2 (1.0 mmol), 3 (1.0 mmol), I2 (1.0 mmol), DMSO (5.0 mL, c = 0.2 M), 100 °C, 2 h. cisolated yields.
Additionally, a series of 5-aminopyrazole (2) varying from N1-substituents was employed to further evaluate the substrate scope (Fig. 3). N-aryl substituents bearing electron-donating or electron-withdrawing groups, or even 2,5-disubstitions were all capable of the transformation (5s-5y). The target tricyclics were also obtained in 68% and 56% yields when 3-nitrophenyl and 2-pyridyl patterns were substituted on the N1 atom (5z, 6a). Moreover, 5-aminopyrazole with N-alkyl substitutions, including methyl, tert-butyl, cyclohexyl, benzyl, and -CH2CO2Et, all performed well in the reaction (6b-6h, 62%–83%). Exploring the different types of substituents at the C3 of 5-aminopyrazole is important since the corresponding product should be spatially crowded. Gratefully, no matter whether cyclopropyl, tert-butyl, or even phenyl groups (6i, 6 m, 6n) were all compatible in the reaction although the yields are relatively lower than the C3-free substrate (6j-6l). It is worth noting that 3-methyl-5-aminooxazole also successfully converts into the corresponding product (6o, 61%; 6p, 63%) further demonstrating the diversity of substrates. Enamine derivatives 3-amino-1-methylpyrazole were proved to be suitable candidates (6q, 32%; 6r, 57%). Notably, non-aromatic enamine motifs 6-aminouracil were also compatible in our transformation, providing, therefore, the desired 6/6/6 fused tricyclics containing a biologically significant uracil fragment (6 s, 72%; 6t, 75%). Next, for the pyrazole core, N1-H 5-hydroxypyrazole was viable to afford the desired products in 66% yield (6 u), pending further modification. Both N1-aryl and N1-alkyl substituents could be incorporated under our reaction conditions (6v-6x). For the C4 position of the pyrazole core, H and methyl substituents are applicable, while other substituents, such as phenyl could not produce the target product (6y-7b).

aGeneral conditions: 1 (1.2 mmol), 2 (1.0 mmol), 3 (1.0 mmol), I2 (1.0 mmol), DTBP (2.0 mmol), DMSO (5.0 mL, c = 0.2 M), 100 °C, 4 h. bGeneral conditions: 1 (1.2 mmol), 2 (1.0 mmol), 3 (1.0 mmol), I2 (1.0 mmol), DMSO (5.0 mL, c = 0.2 M), 100 °C, 2 h. cisolated yields.
Synthetic applications
Encouraged by the good substrate generality scope demonstrated above, we attempted to further explore the applicability of this multicomponent reaction (Fig. 4). Biological molecules such as (-).

A Pre-modification, General conditions: 1 (1.2 mmol), 2 (1.0 mmol), 3 (1.0 mmol), I2 (1.0 mmol), DTBP (2.0 mmol), DMSO (5.0 mL, c = 0.2 M), 100 °C, 4 h, isolated yields. B Gram-scale experiments. C Late-modification. D Transformations of 6i.
Menthol, ( + )-Borneol, and Probenecid (an agonist of TRPV2 channels) were installed in the acetophenone. Those pre-prepared methyl ketones were all tolerated in the reaction (7c, 7d, 7f), indicating the compatibility of our conditions with ester groups and easily oxidizable fragments. Furthermore, NH-amide, as well as lactone, were proved to be suitable in the reaction by applying substrates linked with Amantadine (7e), Camphanic acid (7 g), and lbuprofen (7 h) motifs, respectively. The easy and efficient introduction of biologically relevant compounds into the pyrazolopyridopyridazin-6-one skeleton rendered a positive effect of this synthetic methodology on the pharmaceutical study. We also conducted gram scale scaling experiments on products 4n and 6b and found that the separation yield of the target product could still be maintained at a moderate level at a reaction scale of 8.0 mmol. Treating 4n, which reserves an aryl bromide site, under Sonogashira conditions affords 8 smoothly. A palladium-catalyzed P-C coupling of 4n provides triarylphosphine oxide derivative 9 in good yield. As expected, chlorination of pyridazine using POCl3 gives chlorophthalazine 10, which could be further converted into other synthetic valuables.
Control experiments and proposed mechanism
Based on our previous study regarding the mechanism of I2-DMSO mediated C-H oxidation of methyl ketone, we anticipate that 2-iodo-1-phenylethanone (1ad) and phenylglyoxal (1ac) should act as the key intermediates. Indeed, treating 1ad and 1ac under standard conditions affords 4a in good yield, respectively (Fig. 5a and b-1). In order to figure out what conditions are required in the downstream domino process after 1ac has been generated, a series of control experiments using 1ac as the substrate were performed. It was found that DTBP could further promote the downstream domino, possibly by accelerating the oxidative aromatization step or by facilitating the regeneration of iodine (Fig. 5b-2). Without adding DTBP and I2, the control experiment gave only a 12% yield of 4a, indicating that iodine has a more important role than DTBP for the subsequent transformation (Fig. 5b-3). Finally, using HI instead of the combination of DTBP and I2 provides 4a in 65% yield. Considering that HI could be oxidized to I2 in a DMSO solution, we believe that the acid plays an important role in both cycloaddition and the C-N bond cleavage steps (Fig. 5b-4). Furthermore, radical scavengers were submitted to our model reaction (Fig. 5c), as it has been documented that the proposed C-N bond cleavage could also be triggered by a single-electron oxidation68. After a comprehensive consideration of those experimental results, we concluded that our transformation should not be a free radical-driven reaction. According to the present observations and literature existence69,70,71,72,73, a possible mechanism has been proposed in Fig. 5d, which was further supported by HRMS detection using acetophenone-D3 as a probe (Please see “Supplementary Fig. 4–9” in Supporting Information). The domino reaction is initiated by iodine substitution and the Kornblum Oxidation sequence of acetophenone. The HI generated in the former step would account for the acid-promoted 1,5-H shift of 5-hydroxypyrazole, therefore affording the temporary dearomatized tautomer pending for polar [4 + 2] cyclization. Instead of oxidizing NH into imine, HRMS results support that the resulting cycloadduct C undergoes C-H oxidation to generate pre-aromatic intermediate D with the assistance of I2 and DTBP. The fact that either the iodination of C3-H or C4-H would lead to a quick elimination to generate D. The successful HRMS detection of C and D supports the formation of C3-C4 double bonds is preferred over the formation of imines. Oxidative elimination of the C-N bond was then driven by the aromatization of the pyridine core. This step should be also promoted by acidic conditions since forming the intermediate E would further weaken the C-N bond.

Controlled experiments (a–c) and possible mechanisms supported by HRMS experiments (d).
Discussion
In summary, we report a multicomponent reaction strategically using aryl methyl ketone, 5-aminopyrazole, and 5-hydroxypyrazole to afford pyrazolopyridopyridazin-6-one scaffolds. It provides a distinct paradigm for pyrazole ring opening in which 5-hydroxypyrazole servers as a five-unit tether for the de novo generated 6/6 fused bicyclics. Enabled by the I2-DMSO reagent manifold and facilitated by DTBP, this reaction features high yield and good functional group compatibility, accessing, therefore, pharmaceutical interest nitrogen-atom-rich tricyclics in a modular fashion. Mechanistic studies supported a [4 + 2] cyclization and a cascade oxidative elimination of the C-N bond as the key to realizing the transformation. Precedential disconnection of the weakest N-N bond in the pyrazole core is completely bypassed in our conditions. We anticipate our ring-upgrading tactics will extend to other aromatic compounds.
Methods
General procedure for nitrogen-atom-rich tricyclics
General conditions a (4a-5j, 5s-6x, 7c-7h): The reactions did not require the protection of inert gases. In a 35 mL sealed tube were added arylmethyl ketones 1 (1.2 mmol), iodine (254 mg, 1.0 mmol), and dimethyl sulfoxide (5 mL), and the resulting mixture was stirred at 100 °C (heating block), the reaction tube was removed after about 1 h. Then additional enamines 2 (1.0 mmol), 5-hydroxypyrazoles 3 (1.0 mmol), and DTBP (292 mg, 2.0 mmol) were added at room temperature, followed by reaction at 100 °C for 4 h until substrate conversion was almost complete by TLC analysis. After the reaction stopped and cooled at room temperature, the reaction mixture was quenched with saturated Na2S2O3 solution (50 mL) and NaCl solution (200 mL). The mixture was then extracted with EtOAc (150 mL × 2), and the organic layers were separated and merged. The mixture was dried with anhydrous Na2SO4 and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (200–300 mesh) to afford the product.
General conditions b (5k-5r, 6y-7b): The reactions did not require the protection of inert gases. In a 35 mL sealed tube were added methyl ketones 1 (1.2 mmol), iodine (254 mg, 1.0 mmol), and dimethyl sulfoxide (5 mL), and the resulting mixture was stirred at 100 °C (heating block), the reaction tube was removed after about 1 h. Then, additional enamines 2 (1.0 mmol), 5-hydroxypyrazoles 3 (1.0 mmol) were added at room temperature, followed by reaction at 100 °C for 2 h until substrate conversion was almost complete by TLC analysis. After the reaction stopped and cooled at room temperature, the reaction mixture was quenched with saturated Na2S2O3 solution (50 mL) and NaCl solution (200 mL). The mixture was then extracted with EtOAc (150 mL × 2), and the organic layers were separated and merged. The mixture was dried with anhydrous Na2SO4 and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (200–300 mesh) to afford the product.
Data availability
The X-ray crystallographic coordinates for structures reported in this Article have been deposited at the Cambridge Crystallographic Data Center (CCDC), under deposition numbers CCDC 2344553 and CCDC 2344669. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via https://www.ccdc.cam.ac.uk/structures/. All other data supporting the findings of the study, including experimental procedures and compound characterization, are available within the paper and the Supplementary Information. Correspondence and requests for materials should be addressed to A.X.W., all data are available from the corresponding author upon request.
References
Ciamician, G. L. & Dennstedt, M. Über die einwirkung des chloroforms auf die kaliumverbindung pyrrols. Ber. Dtsch. Chem. Ges. 14, 1153–1163 (1881).
Joynson, B. W. & Ball, L. T. Skeletal editing: Interconversion of arenes and heteroarenes. Helv. Chim. Acta 106, e202200182 (2022).
Peplow, M. Almost magical’: chemists can now move single atoms in and out of a molecule’s core. Nature 618, 21–24 (2023).
Jurczyk, J. et al. Single-atom logic for heterocycle editing. Nat. Synth. 1, 352–364 (2022).
Hyland, E. E., Kelly, P. Q., McKillop, A. M., Dherange, B. D. & Levin, M. D. Unified access to pyrimidines and quinazolines enabled by N–N cleaving carbon atom insertion. J. Am. Chem. Soc. 144, 19258–19264 (2022).
Dherange, B. D., Kelly, P. Q., Liles, J. P., Sigman, M. S. & Levin, M. D. Carbon atom insertion into pyrroles and indoles promoted by chlorodiazirines. J. Am. Chem. Soc. 143, 11337–11344 (2021).
Reisenbauer, J. C., Green, O., Franchino, A., Finkelstein, P. & Morandi, B. Late-stage diversification of indole skeletons through Nitrogen atom insertion. Science 377, 1104–1109 (2022).
Boudry, E., Bourdreux, F., Marrot, J., Moreau, X. & Ghiazza, C. Dearomatization of pyridines: Photochemical skeletal enlargement for the synthesis of 1,2-diazepines. J. Am. Chem. Soc. 146, 2845–2854 (2024).
Woo, J. et al. Scaffold hopping by net photochemical carbon deletion of azaarenes. Science 376, 527–532 (2022).
Bartholomew, G. L., Carpaneto, F. & Sarpong, R. Skeletal editing of pyrimidines to pyrazoles by formal carbon deletion. J. Am. Chem. Soc. 144, 22309–22315 (2022).
Pearson, T. J. et al. Aromatic nitrogen scanning by ipso-selective nitrene internalization. Science 381, 1474–1479 (2023).
Patel, S. C. & Burns, N. Z. Conversion of aryl azides to aminopyridines. J. Am. Chem. Soc. 144, 17797–17802 (2022).
Uhlenbruck, B. J. H., Josephitis, C. M., de Lescure, L., Paton, R. S. & McNally, A. A deconstruction-reconstruction strategy for pyrimidine diversification. Nature 631, 87–93 (2024).
Conboy, A. & Greaney, M. F. Synthesis of benzenes from pyridines via N to C switch. Chem 10, 1940–1949 (2024).
Cheng, Q. et al. Skeletal editing of pyridines through atom-pair swap from CN to CC. Nat. Chem. 16, 741–748 (2024).
Qiu, X. et al. Cleaving arene rings for acyclic alkenylnitrile synthesis. Nature 597, 64–69 (2021).
Cheng, Z. et al. Carbene-assisted arene ring-opening. J. Am. Chem. Soc. 146, 16963–16970 (2024).
Wang, H. et al. Dearomative ring expansion of thiophenes by bicyclobutane insertion. Science 381, 75–81 (2023).
Roque, J. B., Kuroda, Y., Göttemann, L. T. & Sarpong, R. Deconstructive diversification of cyclic amines. Nature 564, 244–248 (2018).
Qin, H., Guo, T., Lin, K., Li, G. & Lu, H. Synthesis of dienes from pyrrolidines using skeletal modification. Nat. Commun. 14, 7307 (2023).
Hu, Y., Stumpfe, D. & Bajorath, J. Recent advances in scaffold hopping. J. Med. Chem. 60, 1238–1246 (2017).
Levin, M. D. Retrosynthetic simplicity. Synlett 35, 1471–1474 (2024).
Clerc, M. et al. Promoting the furan ring-opening reaction to access new donor-acceptor Stenhouse adducts with hexafluoroisopropanol. Angew. Chem. Int. Ed. 60, 10219–10227 (2021).
Feng, Q., Wang, S., Ma, X., Rao, C. & Song, Q. Design, synthesis, and applications of stereospecific 1,3-diene carbonyls. Sci. China Chem. 65, 912–917 (2022).
Hazra, C. K., Gandhamsetty, N., Park, S. & Chang, S. Borane catalysed ring opening and closing cascades of furans leading to Silicon functionalized synthetic intermediates. Nat. Commun. 7, 13431 (2016).
Pratt, R. & Kraus, K. Ring opening and closing reactions of omidazoles and other 1,3-diazaheterocycles with vinyl chloroformate and phenyl chloroformate. Tetrahedron Lett. 22, 2431–2434 (1981).
Zeinali, N., Oluwoye, I., Altarawneh, M. & Dlugogorski, B. Z. Kinetics of photo-oxidation of oxazole and its substituents by singlet oxygen. Sci. Rep., 10, 3668 (2020).
Wang, Z. et al. Four-component ring-opening reaction of pyrroles via C–N bond ceavage under multiple functions of elemental Sulfur. Org. Lett. 25, 3094–3098 (2023).
Zeidan, M. A., Othman, D. I., Goda, F. E. & Mostafa, A. S. Thiazole ring-cleavage: Versatile products obtained in the course of synthesis of certain sulfonamide derivatives. J. Mol. Struct. 1279, 135018 (2023).
Dailey, K. M. K., Rauchfuss, T. B., Rheingold, A. L. & Yap, G. P. A. A new pathway for thiophene ring opening by transition metals. J. Am. Chem. Soc. 117, 6396–6397 (1995).
Ehret, F. et al. Metal-induced thiophene ring opening and C-C bond formation to produce unique hexa-1,3,5-trienediyl-coupled non-innocent ligand chelates. Chem. Eur. J. 21, 15163–15166 (2015).
Piccionello, A. P., Pace, A., Buscemi, S., Vivona, N. & Giorgi, G. Synthesis of fluorinated 1,2,4-oxadiazin-6-ones through ANRORC rearrangement of 1,2,4-oxadiazoles. Tetrahedron Lett. 50, 1472–1474 (2009).
Xu, Y. L. et al. Pyrazolone–quinazolone Hybrids: A novel class of human 4-hydroxyphenylpyruvate dioxygenase inhibitors. Bioorg. Med. Chem. 22, 5194–5211 (2014).
Xu, Y. L. et al. Synthesis and bioevaluation of pyrazole-benzimidazolone hybrids as novel human 4-Hydroxyphenylpyruvate dioxygenase inhibitors. Eur. J. Med. Chem. 92, 427–438 (2015).
Fusco, R., Rosnati, V. & Pagani, G. Ring opening in the pyrazole series. Tetrahedron Lett. 8, 4541–4544 (1967).
Fusco, B., Rosnati, V. & Pagani, G. Ring opening of 3-carboxy-and 3-unsubstituted pyrazoles. Tetrahedron Lett. 7, 1739–1744 (1966).
Schmidt, A., Münster, N. & Dreger, A. Functionalized 4‐aminoquinolines by rearrangement of pyrazole N‐Heterocyclic Carbenes. Angew. Chem. Int. Ed. 49, 2790–2793 (2010).
Chen, Q., Liu, X., Guo, F. & Chen, Z. An unexpected rearrangement of pyrazolium halides based on N–N bond cleavage: Synthesis of 1,2-dihydropyrimidines. Chem. Commun. 53, 6792–6795 (2017).
Koronatov, A. N., Rostovskii, N. V., Khlebnikov, A. F. & Novikov, M. S. Rh(II)-Catalyzed ring expansion of pyrazoles with diazocarbonyl compounds as a method for the preparation of 1,2-Dihydropyrimidines. J. Org. Chem. 83, 9210–9219 (2018).
Sanderson, R. Chemical Bonds and Bonds Energy. 21, (Elsevier, 2012).
Luo, Y. R. Comprehensive Handbook of Chemical Bond Energies. (CRC press, 2007).
Wang, H. et al. Nitrene-mediated intermolecular N–N coupling for efficient synthesis of hydrazides. Nat. Chem. 13, 378–385 (2021).
Zhou, Y. et al. I2-DMSO mediated tetra-functionalization of enaminones for the construction of Novel furo[2‘,3‘:4,5]pyrimido[1,2-b]indazole skeletons via in situ capture of ketenimine cations. Chin. Chem. Lett. 36, 109799 (2025).
Lei, S. G. et al. I2-DMSO Mediated dual α,β-C(sp2)–H functionalization/bicyclization of o-hydroxyphenyl enaminones to construct C2,C3-disubstituted chromone derivatives: Chromeno[2,3-b]pyrrol-4(1H)-ones. Org. Chem. Front. 10, 4843–4847 (2023).
Tang, B. C. et al. Quadruple C-H activation coupled to Hydrofunctionalization and C-H Silylation/borylation enabled by weakly coordinated palladium catalyst. Nat. Commun. 11, 5662 (2020).
Geng, X. et al. Iodine-promoted N–H/α,β-C(sp3)-trifunctionalization of L-proline: Access to 3,4-dihydrobenzo[b][1,7]naphthyridines via consecutive decarboxylation/ring opening/dicyclization. Org. Lett. 21, 4939–4943 (2019).
Zhao, P., Zhou, Y., Wang, C. & Wu, A. X. Iodine-promoted thioylation and dicarbonylation of enaminone Α-C sites: Synthesis of fully substituted thiazoles via C=C bond cleavage. J. Org. Chem. 89, 2505–2515 (2024).
Zhang, J. et al. Diamination/oxidative cross-coupling/bicyclization of anilines and methyl ketones: Direct I2-promoted Synthesis of 1,2-fused oxindoles. Org. Lett. 19, 408–411 (2017).
Ma, J. et al. Photochemical intermolecular dearomative cycloaddition of bicyclic azaarenes with alkenes. Science 371, 1338–1345 (2021).
Zhu, M. et al. Photo-induced intramolecular dearomative [5 + 4] cycloaddition of arenes for the construction of highly strained medium-sized-rings. Nat. Commun. 15, 2462 (2024).
Li, M. et al. Gd(III)-Catalyzed regio-, diastereo-, and enantioselective [4 + 2] photocycloaddition of naphthalene derivatives. J. Am. Chem. Soc. 146, 16982–16989 (2024).
Knorr, L. Ueber abkömmlinge der phenolform des 1-Phenyl-3-methyl-5-pyrazolons. Berichte der deutschen chemischen. Ber. Dtsch. Chem. Ges. 28, 706–714 (1895).
Parchment, O. G., Green, D. V. S., Taylor, P. J. & Hillier, I. H. The prediction of tautomer equilibria in hydrated 3-hydroxypyrazole: a challenge to theory. J. Am. Chem. Soc. 115, 2352–2356 (1993).
Zhang, Y. et al. Arylazanylpyrazolone derivatives as inhibitors of mutant superoxide dismutase 1 dependent protein aggregation for the treatment of amyotrophic lateral sclerosis. J. Med. Chem. 56, 2665–2675 (2013).
Gribble, G. W. Indole Ring Synthesis: From Natural Products to Drug Discovery. (John Wiley & Sons, 2016).
Guarnieri-Ibáñez, A., Aguirre, A., Besnard, C., Poblador-Bahamonde, A. I. & Lacour, J. Regiodivergent synthesis of pyrazino-indolines vs. triazocines via α-imino carbenes addition to imidazolidines. Chem. Sci. 12, 1479–1485 (2021).
Fan, Y. X., Cao, X. L., Chen, L., Chen, Y. H. & Yan, S. J. Multicomponent cascade reactions of HKAs: synthesis of highly functionalized 5H-chromeno[4,3-d]pyrimidines. Org. Chem. Front. 8, 4508–4513 (2021).
Yu, G. et al. Substituted pyrazolopyridopyridazines as orally bioavailable potent and selective PDE5 inhibitors: Potential agents for treatment of erectile dysfunction. J. Med. Chem. 46, 457–460 (2003).
Tseng, C. C. et al. Selective synthesis and photoluminescence study of pyrazolopyridopyridazine diones and N-aminopyrazolopyrrolopyridine diones. Molecules 25, 2409 (2020).
Chung, C. Y. et al. Structural identification between phthalazine-1,4-diones and N-aminophthalimides via vilsmeier reaction: Nitrogen cyclization and tautomerization study. Molecules 26, 2907 (2021).
Mason, H. J., Wu, X., Schmitt, R., Macor, J. E. & Yu, G. Synthesis of fused pyridopyrrolidine dione derivatives using hetero diels–alder reactions. Tetrahedron Lett. 42, 8931–8934 (2001).
Yang, D. S., Chen, X. L. & Wu, A. X. Review of application of the I2 and dimethyl sulfoxide combined reagent system to aryl methyl ketones for diverse transformations. Org. Chem. Front. 11, 2665–2692 (2024).
Singhal, R., Choudhary, S. P., Malik, B. & Pilania, M. I2/DMSO-mediated oxidative C–C and C–heteroatom Bond Formation: a Sustainable approach to chemical synthesis. RSC Adv. 14, 5817–5845 (2024).
Biletskyi, B. et al. Small rings in the bigger picture: Ring expansion of three- and four-membered rings to access larger all-carbon cyclic systems. Chem. Soc. Rev. 50, 7513–7538 (2021).
Donald, J. R. & Unsworth, W. P. Ring‐expansion reactions in the synthesis of macrocycles and medium‐sized rings. Chem. Eur. J. 23, 8780–8799 (2017).
Xu, C., Yin, G., Jia, F. C., Wu, Y. D. & Wu, A. X. Merging annulation with ring deconstruction: Synthesis of (E)-3-(2-Acyl-1H-benzo[d]imidazol-4-yl)acrylaldehyde derivatives via I2/FeCl3-promoted dual C(sp3)–H amination/C–N bond cleavage. Org. Lett. 23, 2559–2564 (2021).
Tan, Z. et al. Catalytic conversion of N-heteroaromatics to functionalized arylamines by merging hydrogen transfer and selective coupling. ACS Catal. 10, 5243–5249 (2020).
Miao, H. J. et al. Aromatization-driven deconstructive functionalization of spiro dihydroquinazolinones via dual Photoredox/nickel catalysis. Chem. Sci. 15, 8993–8999 (2024).
Gao, Q. et al. Selective access to dipyrazolo-fused pyridines via formal [3 + 2 + 1] heteroannulation of methyl ketones with pyrazol-5-amines. Org. Chem. Front. 5, 765–768 (2018).
Zhou, Y. et al. I2-Promoted site-selective C–C bond cleavage of aryl methyl ketones as C1 synthons for constructing 5-acyl-1H-pyrazolo[3,4-b]pyridines. Org. Chem. Front. 9, 4416–4420 (2022).
Zuo, W. et al. Visible-light-induced domino cyclization to access Pyrido[2, 3-d] pyrimidine-2, 4-diones via a radical-polar crossoverreaction. Chin. J. Chem. 42, 2346–2350 (2024).
Chen, T. et al. Modular andselective synthesis of Pyrazolo-azepino-centred polycyclic aromatic and non-aromatic architectures. Org. Chem. Front. 10, 4122–4130 (2023).
Zhou, Y. et al. I2-Promoted gem-Diarylethene involved aza-diels–alder reaction and wagner–meerwein rearrangement: Construction of 2,3,4-trisubstituted pyrimido[1,2-b]indazole skeletons. Org. Lett. 25, 3386–3390 (2023).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 22171098 for A.X.W., 22301035 for J.C.X.). This work was supported by Chengdu Guibao Science & Technology Co., Ltd (for A.X.W.) and the 111 Project B17019 (for A.X.W.). This work was also supported by the Jiangsu Provincial Natural Science Foundation of China (BK20210202 for J.C.X.).
Author information
Authors and Affiliations
Contributions
Y.Z., J.C.X., and A.X.W. conceived and designed the experiments. Y.Z. and S.G.L. carried out the experiments and completed the data collation. B.A., L.S.W., and Z.C.Y. participated in substrate preparation and completed the organization of supporting information. Y.Z., J.C.X., and A.X.W. interpreted the results and co-wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, Y., Lei, SG., Abudureheman, B. et al. Transforming an azaarene into the spine of fusedbicyclics via cycloaddition-induced scaffold hopping of 5-Hydroxypyrazoles. Nat Commun 15, 10907 (2024). https://doi.org/10.1038/s41467-024-55312-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55312-9
