
Abstract
Candida albicans is the most common cause of life-threatening fungal infection in the developed world but remains a therapeutic challenge. Protein kinases have been rewarding drug targets across diverse indications but remain untapped for antifungal development. Previously, screening kinase inhibitors against C. albicans revealed a 2,3-aryl-pyrazolopyridine, GW461484A (GW), which targets casein kinase 1 (CK1) family member Yck2. Here, we report optimization of GW via two complementary approaches, synthesis of bioisosteres possessing an imidazo[1,2-a]pyridine core, and R-group substitution of GW’s pyrazolo[1,5-a]pyridine core. Characterization of compounds reveals two 6-cyano derivatives with improved pharmacological properties that retain whole-cell bioactivity and selectivity for fungal Yck2 compared to human CK1α. Efficacy studies in mice indicate both analogs possess single-agent activity against C. albicans resistant to first-line echinocandin antifungals and potentiate non-curative echinocandin treatment. Results validate Yck2 as an antifungal target and encourage further development of inhibitors acting by this previously unexploited mode of action.
Similar content being viewed by others

Introduction
Fungi kill an estimated 2.5 million individuals annually, with Candida species being a leading cause of invasive disease. Candida albicans currently reigns as the most common cause of Candida infection with mortality rates often exceeding 40% despite therapeutic intervention1. Treatment of systemic fungal infections is becoming increasingly difficult because only four front-line antifungal classes are available to treat invasive mycoses (azoles, polyenes, the pyrimidine 5-fluorocytosine, and echinocandins) with each of these classes suffering from one or more problems due to host toxicity, narrow spectrum of activity, and/or resistance development2,3,4,5,6. Furthermore, the number of individuals susceptible to fungal infections has been steadily increasing since the 1950s, as the use of cancer chemotherapeutics and other immunosuppressive agents has become more common, and the HIV-1 pandemic has left large populations severely immunocompromised1. To date, over 150,000 transplantation procedures occur per year, further increasing the global immunosuppressed population, while the widespread use of antifungal prophylaxis, in turn, drives the evolution of resistance7,8. These factors necessitate a coordinated response by researchers, clinicians, public health agencies, and the pharmaceutical industry as the burden of fungal disease continues to grow. This aligns with recent calls to action, including the World Health Organization’s (WHOs) Fungal Priority Pathogens list, which aims to drive research and policy interventions to address antifungal resistance and fungal disease9,10,11.
Fungal protein kinases are important for growth, cell signaling, proliferation, and the response to environmental stressors, making them attractive drug targets despite their broad conservation across the eukaryotic kingdom. While no kinase inhibitors are currently approved to treat fungal infection, their potential use as either single agents or potentiators of existing antifungals has been proposed12,13,14,15. Foundational work from our research group supports this approach, as a screen of public-___domain human kinase inhibitors revealed a synthetic small molecule, GW461484A (GW), with single-agent and echinocandin-potentiating activity against C. albicans and the evolutionary related and emerging fungal pathogen Candida auris12. Using chemical-genomic approaches, we established the primary target of this 2,3-aryl-pyrazolopyridine compound is the C. albicans casein kinase Yck2, a member of the eukaryotic casein kinase 1 (CK1) family12,16. Yck2 is required for the growth of C. albicans under host-relevant conditions, plays a key role in governing echinocandin resistance, and enables C. albicans virulence in both immunocompetent and immunocompromised mice12,16. GW binds to the kinase’s ATP binding pocket by interacting with multiple hydrophobic residues, while also engaging with the flexible glycine-rich loop (P-loop)12. The Yck2 P-loop is not well conserved with human CK1 orthologues12, potentially allowing for fungal target selectivity. However, in vivo testing of GW has been precluded because extensive cytochrome P450 (CYP450)-mediated oxidative metabolism by liver microsomes makes it unsuitable for systemic administration.
Here, we report the generation of GW analogs with maintained or improved antifungal potency and selectivity, while reducing susceptibility to hepatic P450 metabolism. Guided by the co-structure of GW with Yck2 and its predicted hotspots for P450 metabolism, two series of molecules were synthesized. One set involved structure-guided substitutions on the parent pyrazolo[1,5-a]pyridine scaffold while the second was based on bioisosteres containing an imidazo[1,2-a]pyridine core heterocycle. Amongst the suite of compounds synthesized, two 6-cyano-substituted compounds were found to possess markedly improved whole animal pharmacology and demonstrated promising antifungal activity in a neutropenic mouse model of systemic infection by echinocandin-resistant C. albicans.
Results
Generation of two sets of molecules to improve pharmacological properties of GW
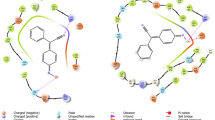
Design and synthesis of new Yck2 inhibitors was performed to overcome the metabolic liabilities of the GW parent scaffold. Initially, design efforts were guided by the co-structure of GW with Yck2 (Fig. 1a, PDB: 6U6A)12. Within the ATP-binding pocket, the nitrogen atom of the 4-pyridyl group of GW appeared responsible for a key interaction with the backbone amide of residue L120 in the hinge region of the Yck2 kinase ___domain, while the 4-fluoro substituent on the 2-phenyl group formed a hydrophobic interaction with L115 (Fig. 1a). Hence, we reasoned that modification of the 4-pyridyl or 2-fluorobenzene substituents could potentially jeopardize kinase inhibition and antifungal activity. In contrast, the pyrazolo[1,5-a]pyridine core of GW extended outwards from the ATP binding pocket into the solvent, and we reasoned that this area was more likely to tolerate modification without compromising bioactivity. The 6-methyl group on the GW core appeared important for fungal selectivity, but analysis by the SMARTCyp metabolic hotspot predictor indicated that it was also a potential site of hepatic cytochrome P450 oxidation (Fig. 1b)17. Therefore, we targeted modifications to the pyrazolo[1,5-a]pyridine core (GW series) and the bio-isosteric imidazo[1,2-a]pyridine core (GW bio-isostere series) to improve metabolic stability while maintaining kinase selectivity and antifungal activity (Fig. 1c). Ethyl, n-propyl, cyclopropyl, tertiary butyl, and trifluoromethyl modifications on the benzene ring were designed to interact with the hydrophobic region of the P-loop to optimize Yck2 fungal kinase selectivity over the human CK1 isozymes (Fig. 1a). 6-methoxy, -hydroxy, and -cyano substituents were selected to modify the polarity of the substituent to increase the potency of Yck2 enzyme inhibition by potentially engaging the carbonyl residues of the DFG loop (residues 186–188). Analogs with 6-fluoro, trifluoromethyl, cyano, and tertiary butyl substituents were predicted by SMARTCyp to improve metabolic stability by reducing cytochrome P450 oxidation17.

a Interaction map of GW-Yck2 (PDB: 6U6A) generated with Schrödinger software12. b Identification of metabolism hotspots using SMARTCyp. Yellow highlights indicate regions with the highest predicted susceptibility to oxidation by CYP450 enzymes, with a circle size proportionate to the liability score. c Proposed modifications to (i) improve Yck2 selectivity over human isoforms, (ii) improve Yck2 inhibition, and (iii) improve P450 stability. d Synthesis of GW bio-isostere analogs. e Synthesis of GW analogs. Source data are provided as Supplementary Methods: Chemical Synthesis and Spectral Details.
Initially, analogs were synthesized in the bio-isosteric imidazo[1,2-a]pyridine series due to the greater synthetic accessibility of the core using a three-step procedure (Fig. 1d). The lithiation of methylpyridine followed by a reaction with a Weinreb amide afforded a methyl ketone (Fig. 1d)18,19,20, which upon reaction with bromine in acetic acid provided an α-bromoketone. Subsequently, the GW bio-isostere core was constructed by TiCl4-assisted condensation21 of the α-bromoketone with a substituted amino pyridine, which allowed diversification of analog preparation in the final step of the synthesis (Fig. 1d).
In contrast to the bio-isoteric imidazo[1,2-a]pyridine series, synthesis of the pyrazolo[1,5-a]pyridine core in the GW series was complicated by the formation of regioisomers during the cyclization reaction (Fig. 1e). The synthesis commenced with a 3-substituted pyridine. N-amination of the pyridine followed by cycloaddition with ethyl 3-(4-fluorophenyl) propiolate resulting in a 1:2 to 1:3 mixture of regioisomeric 4- and 6-substituted pyrazolo[1,5-a]pyridines, respectively. Separation of the pyrazolo[1,5-a]pyridines regioisomers was achieved through preparative HPLC to give the pure 4- and 6-substituted analogs. Each intermediate was subjected to a sequence involving hydrolysis, decarboxylation, bromination, and Suzuki coupling, ultimately yielding two series of GW analogs (Fig. 1e)12.
Heterocycle-substituted GW analogs retain selective Yck2 inhibitory activity
Following the synthesis of nine GW bio-isostere analogs based on the imidazo[1,2-a]pyridine core and fifteen structure-guided R-substituent analogs of the parent GW pyrazolo[1,5-a]pyridine core, we examined the ability of each analog to inhibit the kinase activity of purified recombinant C. albicans Yck2 kinase ___domain. Purified C. albicans Yck2 kinase ___domain was incubated with ATP (at its Km of 20 μM) and each molecule of interest in a two-fold dilution series up to 1 µM. Relative ADP production in the presence of casein peptide as a substrate was measured (Table 1)12. The 50% inhibitory concentration (IC50) for each molecule was computed and compared to the GW parent and solvent controls (Table 1). We confirmed that 20 of 24 compounds inhibited the kinase activity of purified C. albicans Yck2, with an IC50 < 1 µM (Table 1). Four molecules demonstrated Yck2 IC50 values above the maximum concentration tested, 2c (6-CF3), 2h (5-CF3), 2l (4-Me), and 2o (4-CN), indicating that these modifications are poorly tolerated.
To evaluate the fungal selectivity of each molecule, we performed kinase inhibition assays against recombinant human casein kinase I alpha, CK1α. Each molecule of interest was added to CK1α reaction mixtures in a two-fold dilution series from 0–15 µM (Table 1), and IC50 values for each molecule were computed and compared to GW and solvent controls (Table 1). Where an IC50 could be computed for each kinase, C. albicans Yck2 was inhibited more potently than Homo sapiens CK1α, with selectivity values ranging from 2–41-fold (Table 1). Molecules of the imidazo[1,2-a]pyridine bio-isostere group were generally less selective for C. albicans Yck2 compared to their pyrazolo[1,5-a]pyridine counterparts, with the average fungal selectivity of bio-isosteres approximately 7-fold compared to a ~ 23-fold for the pyrazolopyridine group (Table 1). Thus, the greater ease of synthesizing bio-isosteric analogs appears to come at the cost of reduced selectivity for Yck2 in comparison to substituted analogs of the parent pyrazolo[1,5-a]pyridine core.
Growth inhibition and microsomal stability assays revealed four analogs with antifungal activity and improved metabolic stability
Having achieved promising potency and fungal selectivity for several compounds, we next sought to determine which GW analogs retained whole cell antifungal activity. To do so, we performed two-fold dose-response assays with C. albicans grown in RPMI medium at 37 °C under 5% CO2, conditions that mimic host conditions under which Yck2 is required for growth (Supplementary Fig. 1a and Table 1)12. As expected for molecules with Yck2 IC50 values > 1 µM (2c (6-CF3), 2h (5-CF3), 2l (4-Me), and 2o (4-CN)), little to no whole-cell antifungal activity was observed. For biochemically potent compounds, we found that substitution of the GW core heterocycle at the 5-position generally resulted in a marked loss of bioactivity, increasing the MIC80 of the respective analog relative to GW (Supplementary Fig. 1a, gray labeling; Table 1). In contrast, substituting the pyrazolopyridine molecules at the 6- and 4-positions had variable effects on whole-cell antifungal activity (Supplementary Fig. 1a and Table 1). Specifically, for both the 6-substituted pyrazolopyridine and imidazopyridine series, several analogs displayed robust anti-C. albicans bioactivity, including those with 6-cyano (1e, 2a), 6-fluoro (1f, 2b), and 6-methoxy (1b, 2d) substituents (Supplementary Fig. 1a and Table 1). In addition, 6-ethyl (1d) and 6-cyclopropyl (1h) modifications of the imidazopyridine core heterocycle retained bioactivity at 50 μM (Supplementary Fig. 1a and Table 1). Conversely, 6-trifluoromethyl modifications (2c and 1g) resulted in loss of bioactivity. Finally, the 4-fluoro and 4-methoxy substitutions of 2m and 2n resulted in MIC80 values of 25 μM. We also noted that the imidazopyridine GW bio-isosteres demonstrated MIC80 values 2–4-fold higher than those of their pyrazolopyridine counterparts, suggesting that changes to the GW core heterocycle yields molecules with slightly poorer target engagement and/or intracellular accumulation (Supplementary Fig. 1a and Table 1). In fact, only 2a and 2d had MIC80 values of 12.5 μM, a value comparable to the GW parent scaffold. Furthermore, biochemical inhibition of Yck2 did not always correlate with whole-cell antifungal activity. For example, despite hydroxy-substituted molecules 2e (6-OH) and 2k (5-OH) having no measurable whole-cell activity against C. albicans, both molecules demonstrated Yck2 IC50 values of 100 nM and 110 nM respectively, which were comparable to the GW parent (Supplementary Fig. 1a and Table 1). Such a disconnect suggests that certain substitutions impair the intracellular accumulation of the GW scaffold, perhaps reducing the ability of molecules to traverse the fungal cell wall and/or membrane or increasing their efflux out of the cell.
To prioritize molecules for further investigation, we determined whether any of the modifications made to the GW scaffold ameliorated the poor in vitro metabolic stability of the parent12. As mentioned previously, the 6-methyl group of the GW parent was predicted to be a hotspot for CYP450 metabolism using the SMARTCyp 3.0 server17, prompting an exploration of its modification to improve the metabolic stability of analogs. Standard mouse liver microsomal stability assays were performed to predict susceptibility to hepatic Phase I metabolism (Table 1). Molecules were categorized as metabolically ‘unstable’ if the compound concentration remaining in the microsomal suspension after incubation for 30 minutes was ≤ 50% of the starting concentration (1 μM) and metabolically ‘stable’ if the percent compound remaining was > 50% (Supplementary Fig. 1b and Table 1). As expected, the parent GW and other methyl-substituted analogs (1a, 2f, 2l), were highly unstable (Supplementary Fig. 1b and Table 1). While a majority of GW substituted analogs and bio-isosteres were also highly metabolized, molecules with fluoro-, trifluoromethyl-, and cyano-substituents had markedly improved microsomal stability (Supplementary Fig. 1b and Table 1). Of the metabolically ‘stable’ molecules, two 6-substituted pyrazolo[1,5-a]pyridines (2a (6-CN) and 2b (6-F)) and two 6-substituted imidazo[1,2-a]pyridine bio-isosteres (1e (6-CN) and 1f (6-F)) retained sufficiently promising potency at target and whole-cell bioactivity against C. albicans to be prioritized for further characterization (Supplementary Fig. 1 and Table 1).
Prioritized molecules 2a, 2b, 1e, and 1f demonstrate potent and selective inhibition of C. albicans Yck2
To further exemplify the patterns of kinase inhibition exhibited by 2a, 2b, 1e, and 1f, we compared the fungal selectivity of each inhibitor alongside GW in our biochemical kinase assays. Specifically, we demonstrated that all four analogs inhibited Yck2 with IC50 values between 80–130 nM (Table 1 and Fig. 2a). Compound 2a demonstrated the highest potency against Yck2 (IC50 = ~ 80 nM), which correlated with it being the most bioactive analog against C. albicans (MIC80 = 12.5 µM). When comparing each analog’s selectivity for Yck2 over human CK1α, the pyrazolopyridine analogs, 2a (6-CN) and 2b (6-F), were 25–fold and 21–fold more selective, respectively, values greater than the parent GW (11–fold selectivity; Table 1 and Fig. 2a). Imidazopyridine bioisosteres 1e (6-CN) and 1f (6-F), while not as fungal-selective as the pyrazolopyridine analogs, demonstrated similar fungal selectivity to GW, at 8–fold and 12–fold, respectively (Table 1 and Fig. 2a).

a Purified C. albicans Yck2 kinase ___domain (0.115 µg/reaction) and human CK1α (0.05 µg/reaction) were treated in a two-fold dose response with each Yck2 inhibitor. Reactions were supplemented with dephosphorylated casein as substrate (2 µg) and ATP at the Km of the relevant kinase (CaYck2: 20 µM (closed circles), HsCK1α: 10 µM (open circles)). Following incubation, relative kinase activity was measured using the ADP-Glo™ (Promega) bioluminescent assay (100 ms integration). Data were normalized to compound-free controls, and IC50 values computed using GraphPad Prism 9, n = 3 (technical triplicates), Error bars represent the mean ± standard deviation (SD). The experiment was performed in at least a biological duplicate with similar results. b The susceptibility of a doxycycline-repressible tetO-YCK2/yck2Δ strain of C. albicans was evaluated in the presence and absence of doxycycline (10 µg/mL). Two-fold dose-response assays were performed in RPMI at 37 °C under 5% CO2 for 48 h. Growth was determined by optical density at 600 nm (OD600) with technical duplicate measurements averaged and normalized to compound-free controls (see color bar). The experiment was performed in biological duplicate with identical results. c The ability of each GW analog to potentiate caspofungin was tested in an echinocandin-resistant C. albicans strain (DPL15; FKS1T1922C/FKS1T1922C) using a two-fold dose-response assay in YPD at 30 ˚C for 48 h in the presence or absence of a sub-inhibitory concentration of caspofungin (3 µg/mL). Growth was determined by optical density at 600 nm (OD600) with technical duplicate measurements averaged and normalized to compound-free controls (see color bar). The experiment was performed in biological duplicate with identical results. Source data are provided as a Source Data file.
Moving forward, we sought to obtain confirmation for on-target inhibition of Yck2 by prioritized compounds using a genetic approach based on the principle that a reduction in dosage of the gene encoding a compound’s target kinase will result in hypersensitivity to the compound. We used a C. albicans strain in which one allele of YCK2 is deleted while the other allele is under the control of a doxycycline (DOX)-repressible promoter (tetO-YCK2/yck2Δ)22. To determine whether reducing the level of YCK2 in C. albicans resulted in hypersensitivity to GW analogs, we performed two-fold dose-response assays with each prioritized molecule in the presence or absence of 10 µg/mL DOX (Fig. 2b). Marked reductions in the MIC80 of each molecule in the presence of DOX compared to in the absence of DOX were observed, consistent with GW and its analogs inhibiting Yck2 as their mechanism of antifungal activity (Fig. 2b). In addition, previous work established that inhibition of Yck2 results in sensitization to cell wall perturbing agents, including echinocandins such as caspofungin12,16. To determine whether this phenotype would occur upon treatment with our prioritized Yck2 inhibitors, we used an echinocandin-resistant C. albicans strain (FKS1T1922C/FKS1T1922C) and incubated it with a two-fold gradient of GW, 2a, 2b, 1e, and 1f in the presence or absence of a sub-inhibitory concentration of caspofungin. When C. albicans FKS1T1922C/FKS1T1922C was treated with caspofungin, cells were sensitized to GW, 2a, 2b, 1e, and 1f (Fig. 2c). Specifically, reductions in MIC80 upon addition of caspofungin reached ≥ 32-fold for GW, 2a, 2b and 1e, and ≥ 4-fold for 1f (Fig. 2c).
Prioritized Yck2 inhibitors show efficacy against C. albicans when co-cultured with human cell lines in vitro
We next aimed to elucidate the therapeutic potential of each Yck2 inhibitor in vitro using mammalian cell culture. Initially, we performed co-culture experiments using luciferase-labeled HepG2 cells (HepG2-fLuc) and GFP-tagged echinocandin-resistant C. albicans (FKS1T1922C/FKS1T1922C Eno1-GFP) grown together (Fig. 3a) to determine whether each molecule was capable of clearing mammalian cells of the fungus. Co-cultures were treated with a two-fold dose-response series of GW, 2a, 2b, 1e, or 1f, and the relative viability of each cell type was quantified as a ratio of GFP fluorescence or luminescence measured in compound-treated wells relative to untreated wells. Treatment with GW or the pyrazolopyridine substituted analogs 2a (6-CN) and 2b (6-F) effectively cleared C. albicans from co-culture and allowed for HepG2 survival at 2.5, 1.25 , and 6.25 µM, respectively (Fig. 3a). Notably, 2b also resulted in a mild reduction in HepG2 viability at concentrations > 12.5 µM, which may indicate off-target toxicity in this human cancer cell line, given the fungal selectivity observed by this molecule during biochemical characterization (Table 1). 1e (6-CN) was also effective at clearing the majority of co-cultured C. albicans following treatment at 6.25 µM, while 1f (6-F) reduced C. albicans in co-culture to ~ 50% at or above 6.25 µM relative to compound-free controls, and partially rescued HepG2 viability (Fig. 3a).

a C. albicans (FKS1T1922C/FKS1T1922C Eno1-GFP) and luciferized human HepG2 (fLuc CMV) cells were grown in RPMI with 10% HI-FBS at 37 ˚C under 5% CO2. Compounds were added, and after 48 hrs, fungal burden was determined by GFP fluorescence (Ex485/Em518) (open circles), while HepG2 survival was determined by luminescence (100 ms integration; closed circles). Data were normalized to compound-free mono-cultured controls. Error bars represent the mean ± SD, n = 3 (technical triplicates). The experiment was performed in biological duplicate with identical results. b J774A.1 cells were infected with C. albicans (MOI: 2) and treated with each compound. Co-cultures were incubated in RPMI with 3% heat-inactivated FBS and propidium iodide (PI) at 37 ˚C under 5% CO2 for 24 hours. The PI area was quantified using the IncuCyte Basic Analyzer. Data was normalized to DMSO controls, and IC50 values were computed using GraphPad Prism 10, n = 3 (biological triplicate). Error bars represent the mean ± SD. c J774A.1 cells were infected with C. albicans (MOI: 2) that expresses GFP (TEF1p-GFP). After the addition of compounds, co-cultures were incubated in RPMI with 3% HI-FBS at 37 ˚C under 5% CO2 for 16 hrs. The green area was quantified using IncuCyte Basic Analyzer. Data was normalized to DMSO controls, n = 3 (biological triplicate), and error bars represent the mean ± SD. d The effects of compounds on phagocytosed C. albicans was determined by adding each inhibitor (30 µM) to co-cultures at 1 hr post-infection. Co-cultures were incubated in RPMI supplemented with 3% HI-FBS at 37˚C under 5% CO2 for 16 hrs. The green area was quantified using the IncuCyte Basic Analyzer. The average green fluorescent area from 6 images (technical triplicates from biological duplicates) is presented. e C. albicans (TEF1p-GFP) in co-culture with J774A.1 cells were supplemented with each compound and incubated in RPMI with 3% HI-FBS at 37 ˚C under 5% CO2. Images were taken using an AxioVision inverted microscope (Zeiss) at 4 h post-infection. Hoechst dye was used to stain macrophage nuclei (blue). C. albicans (TEF1p-GFP) was visualized as a green signal. Scale bar: 1 μm. Source data are provided as a Source Data file.
To investigate the potential of our Yck2 inhibitors to render C. albicans more susceptible to host-mediated immune control, we performed co-culture assays with mouse monocyte-macrophage lineage J774A.1 cells and C. albicans (SC5314). The innate immune system is the first line of defense against systemic fungal infections23,24,25, and macrophages are key effector cells within the innate immune system responsible for phagocytosis and fungal clearance. As a defense mechanism, C. albicans undergoes cell wall remodeling and morphogenesis upon phagocytosis by macrophages, which results in macrophage death and promotes fungal escape23,24,25,26,27. Given that Yck2 acts to maintain cell wall integrity and support the morphogenesis of C. albicans, we reasoned that Yck2 inhibition might improve the ability of phagocytes to survive and clear fungal infection in co-cultures16. To test this hypothesis, J774A.1 cells and C. albicans (SC5314) were co-incubated in the presence of the membrane-impermeable dye propidium iodide (PI) to quantify macrophage cell death (Fig. 3b). Treatment with GW, 2a, 2b, 1e, and 1f demonstrated dose-dependent rescue of macrophage viability, with the treatment of 30 µM for each molecule restoring full viability in co-culture (Fig. 3b). Specifically, GW, 2a, and 1e were most effective in rescuing macrophage viability with IC50 values of 2.05 µM, 2.17 µM, and 6.68 µM, respectively (Fig. 3b).
To elucidate the mechanism underlying restoration of macrophage viability in co-culture, we examined fungal growth and intracellular hyphal formation of C. albicans treated with the prioritized Yck2 inhibitors23,24,27. To quantify fungal growth in co-culture, we infected macrophages with a strain of C. albicans that constitutively expresses GFP (TEF1p-GFP) and measured the area of the green signal at 16 h post-infection. We observed that GW and 2a were the most effective treatments in reducing C. albicans growth in co-culture (Fig. 3c). Furthermore, we found that all Yck2 inhibitors reduced C. albicans’ growth in macrophages at a concentration of 30 µM (Fig. 3d). Finally, to determine whether the Yck2 inhibitors reduce phagocyte death by inhibiting the C. albicans yeast-to-filament transition, we examined the morphology of C. albicans in co-culture after a 4 h incubation, a time at which most C. albicans cells are internalized and begin to transition to filamentous growth within the phagolysosome27. Treatment with GW, 2a, 2b, 1e, and 1f inhibited filamentation of C. albicans in a dose-dependent manner, locking C. albicans in its yeast form (Fig. 3e). Collectively, these data demonstrated that the Yck2 inhibitors rescue macrophage viability in co-culture by inhibiting fungal growth and filamentation.
2a and 1e occupy a similar binding site to GW in C. albicans Yck2
To gain further insight into the structural basis of Yck2 inhibition, we resolved co-structures 1e, 1f, 2a, and 2b in complex with Yck2 (see Supplementary Table 1 for crystallographic statistics and Supplementary Fig. 2 for active site electron density maps). The compounds occupied a nearly identical position in the Yck2 ATP binding site as GW (PDB 6U6A12), with the 6-position of the pyrazolopyrimidine core positioned outwards from the core of the pocket towards the solvent-accessible region (Fig. 4). The 6-CN groups of 1e and 2a extended closer to Asp167, a highly conserved residue among CK1 orthologs, forming a favorable N-O polar interaction with this residue, an interaction that the 6-Me group of GW could not form. All four compounds also interacted with the backbone carbonyl of Asp167 via their 6-F or 6-CN groups, representing another interaction not possible with GW. Interestingly, the structures revealed that the Yck2 P-loop in the 1e- and 1f-bound structures adopted a conformation most similar to the GW bound structure, with the Glu52 residue approaching 4 to 5 Å of the 6-group and the Phe55 residue positioned > 10 Å from the compounds. Binding of 2a or 2b induced positioning of the P-loop such that the Phe55 residue approached closer to the compounds, within 4 to 6 Å, but only 2b induced positioning of the Glu52 residue < 6 Å to the 6-group (Fig. 4). These observations suggest that interactions between these compounds and the Yck2 P-loop could be transient, with potential stabilizing interactions between Glu52, Phe55, and the 6-substituent. Overall, however, each of 1e, 1f, 2a, and 2b formed additional interactions with Yck2 compared to GW.

Structure of the Yck2:GW (black), Yck2:1e (light blue), Yck2:1f (dark blue), Yck2:2a (pink), and Yck2:2b (red) complexes. Protein shown in cartoon representation, and molecules shown in ball-and-stick. Key residues as well as the 6-C position of the core heterocycle of the compounds, are marked with text.
1e and 2a show the most favorable pharmacology in mice
Given the promising findings observed in co-culture experiments, we proceeded to examine the in vivo pharmacology of each analog to determine whether any would be suitable for antifungal efficacy studies in mice and if so, what would be appropriate dosing parameters. We first collected snapshot plasma pharmacokinetic (PK) profiles over 6 h in mice dosed with GW, 1e, 1f, 2a, or 2b either orally (p.o.) at 25 mg/kg or intravenously (i.v.) at 5 mg/kg (Supplementary Table 2 and Supplementary Fig. 3). Overall, the PK profiles of our prioritized analogs were greatly improved compared to GW. Importantly for an antimicrobial, the analogs demonstrated good oral bioavailability and long terminal half-lives (> 10 h) after p.o. administration (Supplementary Table 2 and Supplementary Fig. 3). The 6-cyano substituted molecules (2a and 1e) demonstrated the most promising PK profiles with extended half-lives and maximal plasma concentrations in the range of effective concentrations in vitro. After 25 mg/kg p.o., the maximal plasma concentration (Cmax) of 2a was ~ 15 µM, which is greater than its MIC80 in standard dose-response assay (Supplementary Fig. 1a, Table 1 and Supplementary Table 2), and > 11-fold the concentration required to clear C. albicans (FKS1T1922C/FKS1T1922C Eno1-GFP) co-cultured with HepG2 cells (Fig. 3a). Reflecting its greater oral bioavailability, the Cmax of 1e was ~ 36 µM, which is greater than the MIC50 of this molecule (Fig. 2b, Table 1 and Supplementary Table 2) and > 7-fold the concentration required to clear C. albicans (FKS1T1922C/FKS1T1922C Eno1-GFP) co-cultured with HepG2 cells (Fig. 3a). Dosing of analogs at 5 mg/kg IV did not provide adequate compound exposure, particularly of 1e and 2a, which did not reach concentrations consistent with effective antifungal concentrations in vitro (Supplementary Table 2 and Supplementary Fig. 3b). In addition, clearance was more rapid after i.v. administration (Supplementary Table 2 and Supplementary Fig. 3b), leading us to prioritize the oral route of administration for further studies.
Collectively, the results of our 6 h snapshot PK profiling experiments motivated us to prioritize 1e and 2a for full 24-hour PK profiling studies. 1e or 2a were administered once orally at 25 mg/kg, and samples were collected over 24 h. 1e and 2a persisted in mouse plasma over the entire 24 h period with t1/2 calculated values of 9 and 15 h, respectively, suggesting that both could be suitable for once or twice daily dosing in an infection model (Fig. 5a and b). Cmax values for both molecules mirrored those observed in the p.o. snapshot PK profiles (Fig. 5b and Supplementary Table 2). In addition, when mice were dosed with 25 mg/kg of 1e or 2a once daily over 3 days, accumulation of the compound was not observed in the plasma, and clearance was little changed suggesting minimal induction of metabolism upon repeated dosing. Importantly, no changes in animal weight or behavior were observed, highlighting that not only do the molecules demonstrate excellent oral bioavailability and plasma PK, but they are also non-toxic in uninfected mice.

a Female CD1 mice (n = 3) were dosed (25 mg/kg PO) with 1e (blue) or 2a (pink). At the indicated times post-dose, plasma was collected, and compound concentrations determined by LC-MS/MS; mean +/- SD is plotted. b) A sparse non-compartmental (NCA) model was used in Phoenix 64 WinNonlin 8.3.3.33 to calculate standard PK parameters. c) The extent of plasma protein binding by 2a or 1e (5 µM) was determined by rapid equilibrium dialysis. Binding of each compound was determined in triplicate samples using a 6-hr incubation period; fua = fraction unbound. d Female CD1 mice (n = 3) were dosed with 2a or 1e (50 mg/kg PO). At 30, 90, and 360 mins post-dose, samples were collected, and LC-MS/MS used to measure compound levels. Tissue concentrations were corrected for contamination by blood in the vasculature of the relevant organ using literature values for the volume of blood in the brain (0.03 mL/g) and kidney (0.34 mL/g). Source data are provided as a Source Data file.
To investigate additional properties of 1e and 2a that could impact their antifungal activity in vivo, we measured plasma protein binding by equilibrium dialysis in vitro and penetration of organs targeted by C. albicans (brain and kidney) in mice during systemic infection. Although both 1e and 2a exhibited relatively high plasma protein binding (91.8% and 97.7%, respectively, Fig. 5c), oral administration of compounds (50 mg/kg) yielded very good exposure in plasma, brain, and kidney (Fig. 5d). Indeed, 1e and 2a could not only cross the blood-brain barrier, but also accumulated in brain tissue at levels higher than the systemic circulation.
Prioritized Yck2 inhibitors 1e and 2a reduce fungal burden in a mouse model of invasive candidiasis
Encouraged by their antifungal activity in culture and favorable pharmacological properties in mice, we proceeded to evaluate the therapeutic potential of 1e and 2a in a neutropenic mouse model of drug-resistant candidiasis. These compounds displayed adequate single-agent activity in vitro, and the addition of caspofungin dramatically enhanced their efficacy (Fig. 2c). This biology motivated us to move forward with a 4-arm study where we assessed both the single-agent and echinocandin-potentiating activity of the inhibitors. As an initial pilot experiment, neutropenic female CD1 mice were infected intravenously with 1 × 106 CFU/mL of echinocandin-resistant C. albicans (DPL15; FKS1T1922C/FKS1T1922C) and treated one daily for a total of 4 doses with vehicle controls, 1e alone (p.o.: 25 mg/kg), a non-curative concentration of caspofungin alone (i.p.: 2 mg/kg), or the combination of 1e and caspofungin (Supplementary Fig. 4). The effects of treatments on fungal burden were evaluated by measuring colony forming units (CFUs) from kidneys resected 24 hours after the final dose of test materials. 1e alone at the dose and schedule used did not eradicate C. albicans, but reduced the fungal burden in the kidneys by approximately 1-log10 (Supplementary Fig. 4). Caspofungin alone had a similar effect, while the combination of 1e with caspofungin resulted in a greater reduction (~ 2-log10) in kidney fungal burden compared to either treatment alone (Supplementary Fig. 4). Importantly, no signs of toxicity from any compound were observed, suggesting higher concentrations of both caspofungin and Yck2 inhibitor could be used.
Encouraged by the initial results, we followed up by infecting CD1 mice once more with echinocandin-resistant C. albicans and treating animals with vehicle alone, 1e or 2a at an intensified dose and schedule (p.o: 50 mg/kg twice daily, 4 doses in total), caspofungin (i.p.: 4 mg/kg once daily, 2 doses in total), or the combination. Fungal burden was once again evaluated by obtaining CFU counts from kidneys resected 12 h after the final dose of test materials. Both Yck2 inhibitors as single agents demonstrated significant reductions in kidney CFU relative to vehicle controls (Fig. 6). Moreover, the combination of caspofungin with 1e or 2a resulted in a significant reduction in fungal burden relative to individual drug treatments, with a greater than 2 log10 reduction in CFU relative to vehicle control group (Fig. 6). While a molecule with more potent baseline fungal bioactivity and/or more favorable PK properties is likely required to demonstrate a more significant reduction in kidney fungal burden, this work presents the first evidence that targeting Yck2 shows therapeutic efficacy in vivo.

Neutropenic female CD1 mice were infected with C. albicans DPL15 (FKS1T1922C/FKS1T1922C) by lateral tail vein injection. Mice were dosed twice daily (50 mg/kg PO) for a total of four doses with either (a) 1e or (b) 2a. In other treatment groups, high dose caspofungin (4 mg/kg IP) once daily for a total of two doses, or a combination of 2a or 1e with caspofungin was given as indicated. Control groups received equal volumes of vehicles alone on the same schedule as the combination groups. Kidneys were resected 12 h after the final dose of test materials. Fungal burden from control and treatment groups is plotted as CFU per gram wet kidney weight (n = 6 mice/treatment group; Error bars represent the mean ± SD). Statistical significance was determined by one-way ANOVA with SIDAK’s multiple comparison adjustment. ****p < 0.0001; *** Vehicle vs 2a P-value = 0.0005, CF vs combination P-value = 0.0002, 2a vs combination P-value = 0.0007; ** Vehicle vs 1e P-value = 0.0082, CF vs combination P-value = 0.0013, 1e vs combination P-value = 0.0019. Source data are provided as a Source Data file.
Discussion
As fungal pathogens pose an ever-increasing threat to human health, there remains a great need to expand the current antifungal arsenal. To begin addressing this unmet need, we describe a multidisciplinary hit-to-lead drug development effort integrating medicinal chemistry, structural biology, biochemistry, microbiology, and pharmacology to demonstrate that targeting the stress kinase Yck2 in C. albicans provides a promising therapeutic strategy. Starting from a previously reported phenotypic screen hit, the 2,3-aryl-pyrazolopyridine Yck2 inhibitor GW, we used structural insights to guide the synthesis of metabolically stable Yck2 inhibitors with attractive, much-improved pharmacology and in vivo antifungal activity. While further development of these leads to generate compounds with sub-micromolar antifungal activity will likely be required to generate a clinical candidate, the work presented here provides proof-of-concept that small molecule inhibitors of the kinase Yck2 can exert single-agent activity in mice infected with drug-resistant C. albicans and combine with a conventional echinocandin to further improve disease control.
Although C. albicans is the focus of the studies presented here, our previous work has shown that GW itself possesses antifungal activity against other critically important fungal pathogens, including C. auris and the basidiomycete Cryptococcus neoformans12. Interestingly, no single-agent antifungal activity was observed in vitro against Nakaseomyces glabratus (formerly referred to as Candida glabrata) or Aspergillus fumigatus12. Whether this is due to lack of target engagement, lack if intracellular compound accumulation, or divergent biology for Yck2 in these species remains to be determined. A potential explanation the lack of effect of Yck2 inhibitors on N. glabratus growth is the close evolutionary relationship of this species to Saccharomyces cerevisiae, which encodes an additional member of the casein kinase I family, Yck1. Functional redundancy between Yck1 and Yck2 may require both kinases to be impaired for a reduction in cellular viability28. Complete functional characterization of A. fumigatus casein kinases has yet to be completed, however, studies in Aspergillus nidulans determined that the CK1 homolog, CkiA, is essential for survival, suggesting that druggable candidate casein kinases are present in the Aspergillus kinome29.
The co-structures defining the binding modes of 1e, 1f, 2a, and 2b within the Yck2 kinase ___domain revealed that 2a and 2b bind to Yck2 with unique P-loop conformations as compared to GW, while 1e and 1f induce P-loop conformations similar to that induced by GW binding. Upon the initial structural and functional characterization of GW, it was hypothesized that positioning of fungal-specific residue Glu52 within the P-loop, which adopted a ‘folded’ conformation, was important in facilitating inhibitor binding with the Yck2 kinase ___domain’s active site12. This may have been achieved with 1e and 1f, but overall our observations suggest that the interactions of these four compounds with the P-loop may be transient. To improve Yck2-binding affinity, additional medicinal chemistry efforts could be undertaken to design more strategic 6-substituents of the pyrazolo[1,5-a]pyridine and imidazo[1,2-a]pyridine scaffolds that would stabilize P-loop interactions while taking care to avoid the introduction of any new metabolic liabilities.
Despite the conservation of kinases across the kingdoms of life, these enzymes play critical roles in microbial growth, proliferation, virulence, and the response to xenobiotic stress, making them attractive targets for antimicrobial drug development30. Work in bacteria revealed that inhibitors of histidine kinases and serine/threonine kinases can be useful as single agents, or as potentiators of other antibiotic classes30,31,32. Our findings largely mirror those in fungal pathogens, and although no antifungal kinase inhibitors are currently licenced for clinical use, their potential for development as therapeutics remains bright12,13,14,15. Studies in C. albicans demonstrated that pharmacological inhibition of diverse kinases including TOR14 and Pkc115,33,34 results in synergy with the azoles, offering an innovative solution to cope with rising rates of drug resistance. Additionally, inhibition of Pkc1 has been associated with reductions in virulence in mammalian models of fungal infection33, and 1-acetyl-β-carboline (1-ABC), an inhibitor of the dual-specificity tyrosine phosphorylation-regulated kinase (DYRK) Yak1, represses the yeast-to-filament transition in C. albicans, a key virulence trait35. As such, this beta-carboline along with similar analogs, can block the formation of biofilms in vitro and in a rat catheter model of C. albicans infection35. While our work describes efforts to target fungal casein kinases, work in Leishmania demonstrated that pharmacological inhibition of casein kinase 1 family members blocks intracellular survival and infectivity of the parasite36,37. These studies also revealed that the ATP-binding ___domain of the abundant CK1 isoform LmCK1.2 differs in structure from that of human CK1s36,37, similar to the observations we made comparing C. albicans Yck2 with human CK1α. Such divergence suggests that the casein kinase 1 family could provide a particularly good opportunity for structure-guided development of highly selective antimicrobial agents in the future. Finally, our findings help establish protein kinases more broadly as a rich target space for the discovery and development of mechanistically distinct antifungals. Moreover, due to their activity in combination with a mainstay of the current antifungal armamentarium, the leads we report here have the potential to provide the kind of resistance-aversive combination approach to antifungal therapy that has proven key to controlling other systemic infectious diseases.
Methods
Ethical regulations
All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Wisconsin-Madison according to the guidelines of the Animal Welfare Act, The Institute of Laboratory Animals Resources Guide for the Care and Use of Laboratory Animals, and Public Health Service Policy or were conducted under Protocol 2015-100840-CORE approved by UT Southwestern Medical Center IACUC.
General synthetic procedure
All reagents and solvents used were purchased from commercial sources and were used without further purification. NMR spectra were obtained using a Bruker 850 MHz (UNC-CH) or Bruker 500 MHz (UNC-CH) or Bruker 400 MHz (UNC-CH) or Bruker 400 MHz (at Piramal Pharma) or INOVA 400 MHz spectrometers at room temperature; chemical shifts are expressed in parts per million (ppm, δ units) and are referenced to the residual protons in the deuterated solvent used. Coupling constants are given in units of hertz (Hz). Splitting patterns describe apparent multiplicities and are designated as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), and br s (broad singlet), dd (doublet of doublets), ddd (double double doublet), tt (triplet of triplets). The purity of compounds submitted for biological screening was determined to be ≥ 95% as measured by HPLC. Analytical thin layer chromatography (TLC) was performed on silica gel plates, 200 μm with an F254 indicator. Column chromatography was performed using RediSep Rf® preloaded silica gel cartridges on Isolera one Biotage automated purification systems. Samples for high-resolution mass spectrometry were analyzed with a ThermoFisher Q Exactive HF-X (ThermoFisher, Bremen, Germany) mass spectrometer coupled with a Waters Acquity H-class liquid chromatograph system. Samples were introduced via a heated electrospray source (HESI) at a flow rate of 0.3 mL/min. Electrospray source conditions were set as: spray voltage 3.0 kV, sheath gas (nitrogen) 60 arb, auxillary gas (nitrogen) 20 arb, sweep gas (nitrogen) 0 arb, nebulizer temperature 375 degrees C, capillary temperature 380 °C, RF funnel 45 V. The mass range was set to 150–2000 m/z. All measurements were recorded at a resolution setting of 120,000. Separations were conducted on a Waters Acquity UPLC BEH C18 column (2.1 × 50 mm, 1.7 μm particle size). LC conditions were set at 95 % water with 0.1% formic acid (A) ramped linearly over 5.0 mins to 100% acetonitrile with 0.1% formic acid (B) and held until 6.0 mins. At 7.0 mins the gradient was switched back to 95% (A) and allowed to re-equilibrate until 9.0 mins. The injection volume for all samples was 3 μL. Analytical LC/MS data was obtained using a Waters Acquity Ultrahigh-performance liquid chromatography (UPLC) system equipped with a photodiode array (PDA) detector using the following method: solvent A = Water + 0.2% FA, solvent B = ACN + 0.1% FA, flow rate = 1 mL/min. The gradient started at 95% A for 0.05 min. Afterward, it was ramped up to 100% B over 2 min and held for an additional minute at this concentration, before returning to the initial gradient. Compounds were purified on prep HPLC using an Agilent 1100 equipped with a Phenomenex column (Phenyl-Hexyl, 75 × 30 mm, 5 μm) using the following method: Solvent A: water + 0.05 % TFA; Solvent B: MeOH; flow rate: 70.00 mL/min. LC conditions were set at 90 % (A) ramped linearly over 8.0 mins to 100% (B) and held until 10.0 mins at 100% B. At 10.0 mins the gradient was switched back to 90% (A). Detailed synthesis notes and spectra for all molecules generated for this project can be found in Supplementary Methods.
Fungal strain and culture conditions
Archives of all strains were maintained at − 80 °C in 25% glycerol. Strains were grown in standard conditions at 30 °C in YPD (1% yeast extract, 2% peptone, 2% dextrose) unless otherwise indicated in RPMI (10.4 g/L RPMI powder with L-glutamine (Gibco), 165 mM MOPS, 2% glucose, 5 mg/mL histidine, pH 7), or SD (2% glucose, 6.7 g/L yeast nitrogen base without amino acids). All strains used in this study are listed in Supplementary Table 3.
Dose-response assays
Drug susceptibility assays were performed in 384-well plates in a final volume of 0.04 mL/well with two-fold dilutions of each compound in YPD or RPMI medium, as indicated. Plates were incubated in the dark at 30 °C or 37 °C under static conditions, and OD600 was measured after the indicated incubation times using a spectrophotometer (Molecular Devices). For assays employing strains from the GRACE (tetO) collection, strains were grown overnight in the presence and absence of the indicated doxycycline (DOX) concentration(s). Data were quantitatively displayed as heat maps using Java TreeView 1.1.6r4.
Metabolic stability assays (Analiza Inc.)
Mouse liver microsomal stability assays were completed by Analiza Inc. Compounds were supplied as 10 mM DMSO stocks and diluted serially to 2.5 mM (DMSO) and 0.5 mM (acetonitrile). Reaction plates were prepared with 691.25 μL, pre-warmed (37 °C) mouse liver micosomes (0.63 mg/mL in 100 mM KPO4 + 1.3 mM EDTA) to an empty well of a 96 well plate and maintained at 37 °C. Test compounds were added to microsomes in the reaction plate and mixed. Solutions were pre-incubated for 5 m at 37 °C. Compound at t = 0 was determined by aliquoting a sample of each reaction solution and adding MeOH and NADPH regeneration solution immediately. The remaining reaction solutions were incubated in the presence and absence of NADPH for 30 m at 37 °C before adding MeOH to quench the reactions. LC-TOFMS was used to quantify each test compound. Data acquisition was completed using Agilent 6538 Ultra High Accuracy TOF MS (m/z 100–1000) using generic conditions in positive mode. Exact mass and peak integration was determined using MassHunter (Agilent). Data were expressed as the percent remaining of the unchanged parent compound at each time point.
Kinase inhibition assays
Kinase assays were performed using the ADP-Glo kinase assay kit (Promega) in solid white 384 well plates (Corning). Assays with were performed in kinase buffer (1x: 2 mM NaHEPES pH 7.5, 650 mM KCl, 50 mM MgCl2, 25 mM b-glycerophosphate) supplemented with 2 µg casein kinase I peptide substrate (SignalChem) per reaction, and ~ 0.115 μg purified recombinant CaYck2 kinase ___domain or 0.05 µg purified HsCK1a (Abcam) per reaction as indicated. Each kinase inhibitor of interest was added in a two-fold dilution series at the concentrations indicated, followed by the addition of ATP at 20 µM (CaYck2 KmATP) or 10 µM (HsCK1a). Assays were performed in 10 μL reactions (n = 3) and incubated for 30 minutes at 30 °C. ADP-Glo kinase assay reagents (Promega) were applied to assay wells per the manufacturer’s instructions. Luminescence was measured with a TECAN Spark® multimode microplate reader, and background luminescence was subtracted from reaction wells from control wells incubated with all reaction reagents except for the relevant kinase enzymes. IC50 values were calculated, and data plotted using GraphPad Prism 9’s Nonlinear fit function.
Co-culture assays
HepG2 cells (ATCC, male, CAT# HB-8065) infected with a lentiviral vector expressing firefly luciferase from a CMV promoter (HepG2-fLuc) were grown in DMEM medium (Gibco) with 10% fetal bovine serum (FBS). Experiments were performed in cells within 12 passages post-recovery from stocks stored in liquid nitrogen and confirmed to be PCR negative for mycoplasma contamination. HepG2-fLuc cells were seeded at 1 × 105 cells/mL in black-walled, clear-bottom 384-well plates. Following overnight incubation at 37 °C with 5% CO2 continuous infusion, GFP-labeled C. albicans DPL15 (Eno1-GFP; CaLC6194) was added to assay wells seeded with HepG2-fLuc cells at a concentration of 2.5 × 104 cells/mL. Two-fold dilutions of each test compound were added to assay wells at the concentrations specified in the text. Control plates containing monocultures of HepG2-fLuc cells or C. albicans were prepared using equal compound titrations. All assay plates were incubated for 48 h at 37 °C with 5% CO2. The relative fluorescence of each well was measured using a TECAN Spark® multimode microplate reader at Ex.485/Em.535. Following fluorescence readings, 5 µL Steady-Glo® Luciferase Assay reagent was added to each assay well prior to incubation at room temperature for 10 min. Relative luminescence was read using a TECAN Spark® multimode microplate reader. Reported results are representative of two biological replicates, each of which was performed in technical triplicate.
Macrophage killing assay
To quantify macrophage death in co-culture with C. albicans, mouse monocyte-macrophage lineage J774A.1 cells were seeded in 96-well tissue culture-treated plates by adding 100 µL of cell suspension (4 × 105 cells/mL) into each well. Cell culture was grown in RPMI medium supplemented with 3% HI-FBS for 18 h at 37 °C + 5 % CO2. On the following day, overnight culture of wild-type C. albicans (SC5314) was washed threes time with PBS and diluted to 8 × 105 cells/mL in RPMI medium supplemented with 3% HI-FBS and 2 µg/mL of propidium iodide (PI, Sigma, P417). Infection was performed by adding 100 μL of fungal cell suspension to each well previously seeded with J774A.1 cells. Co-cultures were treated with drugs of interest in a two-fold serial dilution, starting at 30 µM. Equal volume of DMSO to Yck2 inhibitor was used as a vehicle control. Co-cultures were incubated for 24 h at 37 °C + 5% CO2. Co-cultures were imaged using the IncuCyte® S3 Live-Cell Analysis System. The red area was quantified using the IncuCyte Base Analysis Software. Data was normalized to DMSO controls.
Quantification of C. albicans growth in co-culture with macrophages
To examine the growth of C. albicans co-culture with macrophage in the presence of drug treatment, J774A.1 cells were seeded in a 96-well plate by adding 100 μL of 4 × 105 cells/mL of cell suspension to each wall. Plates were incubated for 18 h at 37 °C + 5 % CO2. On the following day, C. albicans (pTEF1-GFP) overnight cultures were washed three times with PBS and diluted to 8 × 105 cells/mL in RPMI medium supplemented with 3% HI-FBS. C. albicans cell suspension (100 μL/well) was added to the wells previously seeded with J7 cells and incubated for 16 h at 37 °C + CO2. Co-cultures were treated with each drug of interest in two-fold serial dilutions, starting at 30 µM. Equal volume of DMSO to Yck2 inhibitor was used as a vehicle control. Data was normalized to DMSO controls.
To determine the effects of the Yck2 inhibitors on the growth of intracellular C. albicans, J774A.1 cells, and phagocytosed C. albicans co-cultures were washed with PBS to remove extracellular C. albicans and treated with 30 µM of each drug of interest dissolved in RPMI medium supplemented with 3% HI-FBS at 1 h post-infection. Equal volume of DMSO to Yck2 inhibitor was used as a vehicle control. 2 µg/mL amphotericin B was used as a positive control38. Co-cultures were imaged using an IncuCyte® S3 Live-Cell Analysis System, and green area was quantified using the IncuCyte Base Analysis Software.
Visualization of intracellular C. albicans in macrophages
To monitor C. albicans filamentation in J774A.1 cells, J774A.1 cells were seeded in a 96-well plate by adding 100 μL of cell suspension (4 × 105 cells/mL) to each wall. Plates were incubated for 18 h at 37 °C + 5 % CO2. On the next day, overnight C. albicans (pTEF1-GFP) cultures were washed three times with PBS and diluted to 8 × 105 cells/mL in RPMI medium supplemented with 3% HI-FBS. 100 µL of fungal cell suspension was added to the J774A.1-seeded 96-well plate. Co-cultures were treated with either 0.94 µM, 7.5 µM, or 30 µM of GW, YK-I-02, YK-I-03, MN-I-157, or MN-I-158 and incubated in RPMI + 3% HI- FBS at 37 °C + 5% CO2 for 4 h. Macrophage nuclei was stained with 5 µg/mL of Hoeschst stain (Invitrogen, Hoechst 33342) and visualized via blue signal. C. albicans (pTEF1-GFP) were visualized via green signal. Images were obtained with an AxioVision inverted microscope (Carl Zeiss) using phase contrast optics, white light illumination, and an X-cite series 120 light source for fluorescence excitation.
Pharmacokinetic (PK) profiling
All mouse PK experiments were conducted under Protocol 2015-100840-CORE approved by UT Southwestern Medical Center IACUC. PK profiles were generated for GW and prioritized analogs using female CD1 mice (n = 3) from Charles River (Wilmington, MA) dosed orally (PO) at 25 mg/kg or intravenously (IV) at 5 mg/kg and monitored for 6 or 24 hours, as indicated. The drug was administered in the following vehicles: 10% DMSO + 90% of 0.5% Methocel A4M/0.2% Tween 20 for P.O. administration and 10% DMSO + 90% of 20% solution of captisol for I.V. administration. At the indicated times post-dose, mice were submandibular bled or overdosed with CO2 and bled by cardiac puncture. Plasma was obtained by centrifugation of blood for 10 min at 9600 × g at 4 °C. When analyzed, tissues were collected, rinsed to remove surface adhering blood, weighed and snap frozen. Upon thawing, tissues were homogenized in 3X weight by volume PBS. Vendor supplied K2EDTA plasma from BioIVT (Westbury, NY) or blank tissue homogenate spiked with known concentrations of each compound was used to prepare standards and quality control samples. 100 µL samples or standards were crashed with 300 µL of acetonitrile containing formic acid (0.1% in the final mixture) and n-benzylbenzamide internal standard (50 ng/mL in the final mixture). Tubes were vortexed, incubated at room temperature (RT) for 10 min, and spun for 5 min at 16,100 × g. The supernatant was transferred to a second tube and again spun for 5 min at 16,100 × g. The supernatant was analyzed by LC-MS/MS on a Sciex (Framingham, MA) 4500 Triple Quad™ system coupled to a Shimadzu (Columbia, MD) Prominence LC. An Agilent (Santa Clara, CA) C18 XDB column (5 micron packing 50 × 4.6 mm size) was used for chromatography under gradient conditions with water (Buffer A) and methanol (Buffer B) both containing 0.1% formic acid. Compounds were detected in multiple reaction monitoring (MRM) mode using ESI. Transitions monitored were as follows: GW 304.057/208.0; 1e 315.112/185.2; 1f 308.271/185.2; 2a 314.787/219.0; 2b 308.011/212.0.:. Tissue concentrations were determined after subtraction of the drug in tissue vasculature using measured plasma concentrations and literature values39 or the volume of blood in the brain (0.03 mL/g) and kidney (0.34 mL/g). Compounds were assumed to partition equally between plasma and red cells. Phoenix 64 WinNonlin 8.3.3.333 (Certara, Corp, Radnor, PA) was used to calculate PK parameters for half-life, the area under the curve (AUC), Tmax, Cmax, clearance, volume of distribution, and mean residence time. A sparse non-compartmental (NCA) model was used.
Protein binding
The extent of nonspecific binding of 1e and 2a in murine plasma was determined using rapid equilibrium dialysis. Briefly, commercial mouse K2EDTA plasma and PBS were warmed to 37 °C, 5% CO2, and 75% RH in a CO2 incubator. The pH was measured and adjusted to pH 7.4 as necessary. Plasma was diluted 20x with PBS and spiked with compounds in DMSO at a final concentration of 5 µM. Two hundred microliters of sample was added to the red chamber of a Pierce Rapid Equilibrium Dialysis (RED) device (ThermoFisher Scientific, Waltham, MA) and 400 µL of PBS to the white chamber in triplicate. Chambers were placed in a plate which was sealed with a semipermeable membrane and incubated for six hours at 37 °C, 5% CO2, and 75% RH on an orbital shaker. At the incubation end, samples (50 µL) were removed from each chamber (PBS side first) and matrix-matched prior to the addition of an equal volume of methanol containing 0.2% formic acid and 50 ng/ml N-benzylbenzamide) to precipitate protein. Samples were centrifuged twice and evaluated as described above by LC-MS/MS. The extent of protein binding in each matrix was determined as:
Where CR is the total drug concentration in plasma in the red chamber,
CW is free drug concentration in the white chamber.
All concentrations are approximated by the peak area ratio of analytes/IS in the LCMS. To account for the impact of dilution, the following correction was applied40
Where D is the fold dilution, and fu2 is the unbound fraction determined using a diluted matrix.
X-ray crystallography and structural analysis
The corresponding region of Yck2 residues 37–345 were PCR amplified from C. albicans genomic DNA and subcloned into the vector pMCSG53, which codes for a N-terminal His6 tag, TEV protease site, followed by the Yck2 protein. E. coli BL21 LOBSTR competent cells were transformed with this plasmid and the Yck2 protein was purified using methodology previously described41. Crystallization was performed at RT using the sitting drop method. Crystals of apo Yck2 were grown with 1 μL protein at 20 mg/mL and reservoir solution 0.1 M Tris pH 8, 25 mM magnesium chloride, and 20% (w/v) PEG3350. To obtain the Yck2-1e, 1 f, 2a, and 2b complexes, 1 μL of the inhibitors dissolved in DMSO were soaked into apo Yck2 crystals for 60 min. All crystals were cryoprotected in Paratone oil. X-ray diffraction data at 100 K was collected at beamline CMCF-ID, Canadian Macromolecular Crystallography Facility, Canadian Light Source. Diffraction data was reduced using xia242. The structures were solved by the Molecular Replacement (MR) method using Phenix.phaser43 and the Yck2-GW structure (PDB: 6u6a)12. Refinement was completed with Phenix.refine and Coot44. All B-factors were refined, and TLS parameterization was included in the final rounds of refinement. X-ray crystallographic statistics are provided in Supplementary Table 1. Structural analyses and visualization were completed using PyMOL (Schrödinger).
To generate the Yck2-GW interaction map, The protein structure (PDB: 6U6A) was prepared using the Protein Preparation Wizard in Schrödinger Maestro. Standard preparation protocols were employed, removing waters beyond 5 Å from the co-crystallized ligand, and preparing the structure at a physiological pH (7.4), as well as constraining the convergence of heavy atoms to within 0.3 Å relative to the crystal structure. Notable conformational shifts within the active site following hydrogen-bond network optimization and preparation convergence include the LYS73 residue which shifted to participate in a hydrogen bonding interaction with HOH514. All crystal structures have been deposited in the Worldwide Protein Databank (PDB ID: 9EDV (1e), 9EDW (1f), 9EDX (2a), and 9EDY (2b)).
Mouse therapeutic efficacy studies
Female CD-1 mice were rendered neutropenic (neutrophils, < 100/mm3) by injecting them with cyclophosphamide (Mead Johnson Pharmaceuticals, Evansville, IN) subcutaneously 4 days (150 mg/kg) and 1 day (100 mg/kg) before infection and 2 days after infection (100 mg/kg). Organisms were subcultured on SDA 24 h prior to infection. The inoculum was prepared by placing three to five colonies into 5 mL of sterile pyrogen-free 0.9% saline that had been warmed to 35 °C. The final inoculum was adjusted to a 0.6 transmittance at 530 nm. Disseminated infection was achieved by injection of 1 × 106 CFU C. albicans CaLC990 suspended in 0.1 mL inoculum via the lateral tail vein 2 h prior to the start of drug therapy. Six infected animals per treatment group (n = 6) were utilized alongside a treatment period of 48–96 h with each molecule at the dose specified in text. At the end of the study period, the animals were sacrificed by CO2 asphyxiation. After sacrifice, the kidneys of each mouse were removed and placed in sterile 0.9% saline at 4 °C. The homogenate was then serially diluted 1:10, and aliquots were plated on SDA for viable fungal colony counts after incubation for 24 h at 35 °C. The lower limit of detection was 100 CFU/ml. Results were expressed as the mean number of CFU per kidney for three mice. No-treatment and zero-hour controls were included in all experiments. All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Wisconsin-Madison according to the guidelines of the Animal Welfare Act, The Institute of Laboratory Animals Resources Guide for the Care and Use of Laboratory Animals, and the Public Health Service Policy. The approved animal protocol number is DA0042. All mice were maintained at 22.2 °C, 45% humidity, and with light/dark alternating every 12 h.
Statistics and reproducibility
The sample size was always n = 3 or greater when statistical analysis was required. In cases of fungal growth assessment, assays were performed in technical duplicates, which were averaged. In all other cases, experiments were performed in technical triplicate as indicated in the figure legend and methods. Each experiment was performed in at least biological duplicate, with both replicates showing similar results. All attempts at replication were successful. The selection of these sample sizes was sufficient to observe statistically significant and reproducible results in all experiments. Randomization was not relevant to the type of experimentation reported. All assays had a quantitative output where statistical analysis was performed, rather than qualitative observations, and therefore, randomization was not required to eliminate user bias. Blinding was not relevant to this study as this was not an observational study with no opportunity for bias to factor into quantitative results. All assays had quantitative output, rather than qualitative, and therefore, blinding was not required to eliminate user bias. All data is available in the Source data file.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All raw data used to generate the main manuscript and Supplementary Figures can be found in the Source Data file. Crystallographic data for the structures reported in this article have been deposited in the Worldwide Protein Databank (PDB ID: 9EDV (1e), 9EDW (1f), 9EDX (2a), and 9EDY (2b)). Additional data that supports the findings of this study are available from the corresponding author upon reasonable request. Source data are provided in this paper.
References
Fisher, M. C. et al. Threats posed by the fungal kingdom to humans, wildlife, and agriculture. mBio 11, 1–17 (2020).
Brown, G. D. et al. Hidden killers: Human fungalinfections. Sci. Transl. Med. 4, 165rv13 LP-165rv13 (2012).
Pfaller, M. A., Diekema, D. J., Turnidge, J. D., Castanheira, M. & Jones, R. N. Twenty years of the SENTRY antifungal surveillance program: Results for Candida species from 1997-2016. Open Forum Infect. Dis. 6, S79–S94 (2019).
Pfaller, M. A. & Diekema, D. J. Epidemiology of invasive mycoses in North America. Crit. Rev. Microbiol. 36, 1–53 (2010).
Lockhart, S. R. et al. Species identification and antifungal susceptibility testing of Candida bloodstream Isolates from population-based surveillance studies in two U.S. Cities from 2008 to 2011. J. Clin. Microbiol. 50, 3435–3442 (2012).
Perlin, D. S., Rautemaa-Richardson, R. & Alastruey-Izquierdo, A. The global problem of antifungal resistance: Prevalence, mechanisms, and management. Lancet Infect. Dis. 17, e383–e392 (2017).
Papon, N. et al. Fungal infections in transplant recipients: Pros and cons of immunosuppressive and antimicrobial treatment. Lancet Microbe 2, e6–e8 (2021).
Casadevall, A. Fungal diseases in the 21st century: The near and far horizons. Pathog. Immun. 3, 183–196 (2018).
Burki, T. WHO publish fungal priority pathogens list. Lancet Microbe 4, e74 (2023).
Fisher, M. C. et al. Tackling the emerging threat of antifungal resistance to human health. Nat. Rev. Microbiol 20, 557–571 (2022).
World Health Organization. WHO Fungal Priority Pathogens List to Guide Research, Development and Public Health Action. Geneva: World Health Organization, 1−48 (2022).
Caplan, T. et al. Overcoming fungal echinocandin resistance through inhibition of the non-essential stress kinase Yck2. Cell Chem. Biol. 27, 269–282.e5 (2020).
Perfect, J. R. The antifungal pipeline: A reality check. Nat. Rev. Drug Discov. 16, 603–616 (2017).
Shekhar-Guturja, T. et al. Dual action antifungal small molecule modulates multidrug efflux and TOR signaling. Nat. Chem. Biol. 12, 867–875 (2016).
Blankenship, J. R., Fanning, S., Hamaker, J. J. & Mitchell, A. P. An extensive circuitry for cell wall regulation in Candida albicans. PLoS Pathog. 6, e1000752 (2010).
Jung, S. I. et al. Yeast casein kinase 2 governs morphology, biofilm formation, cell wall integrity, and host cell damage of Candida albicans. PLoS ONE 12, 1–20 (2017).
Rydberg, P., Gloriam, D. E. & Olsen, L. The SMARTCyp cytochrome P450 metabolism prediction server. Bioinformatics 26, 2988–2989 (2010).
Miwatashi, S. et al. Synthesis and biological activities Of 4-Phenyl-5-Pyridyl-1,3-Thiazole derivatives as p38 MAP kinase inhibitors. Chem. Pharm. Bull. 53, 410–418 (2005).
Bhavsar, D. et al. Chemistry & biology interface. Chem. Biol. 1, 251–262 (2011).
Patel, H. S. et al. New synthetic approaches to estrogen receptor modulators: Imidazo[1,2-a]pyridines. Tetrahedron Lett. 44, 4077–4080 (2003).
Cai, L., Brouwer, C., Sinclair, K., Cuevas, J. & Pike, V. W. All raw data used to generate the main manuscript and Supplementary Figures can be found in the Source Data file provided with this paper. Synthesis 1, 133–145 (2006).
Roemer, T. et al. Large-scale essential gene identification in Candida albicans and applications to antifungal drug discovery. Mol. Microbiol. 50, 167–181 (2003).
Miramón, P., Kasper, L. & Hube, B. Thriving within the Host: Candida spp. interactions with phagocytic cells. Med. Microbiol. Immunol. 202, 183–195 (2013).
Jiménez-López, C. & Lorenz, M. C. Fungal immune evasion in a model host–pathogen interaction: Candida albicans versus macrophages. PLoS Pathog. 9, e1003741 (2013).
Netea, M. G., Joosten, L. A. B., van der Meer, J. W. M., Kullberg, B.-J. & van de Veerdonk, F. L. Immune defence against Candida fungalinfections. Nat. Rev. Immunol. 15, 630–642 (2015).
McKenzie, C. G. J. et al. Contribution of Candida albicans cell wall components to recognition by and escape from murine macrophages. Infect. Immun. 78, 1650–1658 (2010).
Uwamahoro, N. et al. The pathogen Candida albicans hijacks pyroptosis for escape from macrophages. mBio 5, e00003–e00014 (2014).
Robinson, L. C. et al. Yeast casein kinase I homologues: An essential gene pair. Proc. Natl Acad. Sci. USA 89, 28–32 (1992).
Apostolaki, A. et al. Aspergillus nidulans CkiA is an essential casein kinase I required for delivery of amino acid transporters to the plasma Membrane. Mol. Microbiol. 84, 530–549 (2012).
King, A. & Blackledge, M. S. Evaluation of small molecule knase inhibitors as novel Antimicrobial and Antibiofilm Agents. Chem. Biol. Drug Des. 98, 1038–1064 (2021).
Schaenzer, A. J. et al. A screen For kinase inhibitors identifies antimicrobial imidazopyridine aminofurazans as specific Inhibitors of the Listeria monocytogenes PASTA kinase PrkA. J. Biol. Chem. 292, 17037–17045 (2017).
Shakya, T. et al. A small molecule discrimination map of the antibiotic resistance kinome. Chem. Biol. 18, 1591–1601 (2011).
Lafayette, S. L. et al. PKC signaling regulates drug resistance of the fungal pathogen Candida albicans via circuitry comprised of Mkc1, calcineurin, and Hsp90. PLoS Pathog. 6, 79–80 (2010).
Sussman, A. et al. Discovery of cercosporamide, a known antifungal natural product, as a selective Pkc1 kinase inhibitor through high-throughput screening. Eukaryot. Cell 3, 932–943 (2004).
MacAlpine, J. et al. A small molecule produced by Lactobacillus species blocks Candida albicans filamentation by inhibiting A DYRK1-family kinase. Nat. Commun. 12, 6151 (2021).
Rachidi, N. et al. Pharmacological assessment defines Leishmania donovani casein kinase 1 as a drug target and reveals important functions in parasite viability and intracellular infection. Antimicrob. Agents Chemother. 58, 1501–1515 (2014).
Durieu, E. et al. From drug screening to target deconvolution: A target-based drug discovery pipeline using Leishmania casein kinase 1 isoform 2 to identify compounds with antileishmanial activity. Antimicrob. Agents Chemother. 60, 2822–2833 (2016).
Van’t Wout, J. W. et al. Effect of amphotericin B, fluconazole and itraconazole on intracellular Candida albicans and germ tube development in macrophages. J. Antimicrob. Chemother. 25, 803–811 (1990).
Kwon, Y. Handbook of Essential Pharmacokinetics, Pharmacodynamics and Drug Metabolism for Industrial Scientists. (Kluwer Academic Publishers, Boston, 2002).
Cory Kalvass, J. & Maurer, T. S. Influence of nonspecific brain and plasma binding on CNS exposure: implications for rational drug discovery. Biopharm. Drug Dispos. 23, 327–338 (2002).
Stogios, P. J. et al. Substrate recognition by a colistin resistance enzyme from Moraxella catarrhalis. ACS Chem. Biol. 13, 1322–1332 (2018).
Winter, G. Xia2: An expert system for macromolecular crystallography data reduction. J. Appl Crystallogr. 43, 186–190 (2010).
Adams, P. D. et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 (2010).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Acknowledgements
We thank all past and current Cowen lab members and Dr. Pascal Marchand and the team from the University of Nantes for helpful discussions. L.E.C. is supported by a National Institutes of Health (NIH) R01 grant (1R01AI162789), and is a Canada Research Chair (Tier 1) in Microbial Genomics & Infectious Disease and co-Director of the CIFAR Fungal Kingdom: Threats & Opportunities program. We acknowledge the effort and resources provided by the institutionally supported UT Southwestern Preclinical Pharmacology Core. The Structural Genomics Consortium (SGC) is a registered charity (no: 1097737) that receives funds from Bayer AG, Boehringer Ingelheim, Bristol Myers Squibb, Genentech, Genome Canada, through Ontario Genomics Institute [OGI-196], EU/EFPIA/OICR/McGill/KTH/Diamond Innovative Medicines Initiative 2 Joint Under-taking [EUbOPEN grant 875510], Janssen, Merck KGaA (also known as EMD in Canada and the US), Pfizer, and Takeda. The research reported in this publication was supported by in part by the NC Biotech Center Institutional Support Grant 2018-IDG-1030 and by NIH grant S10OD032476 for upgrading the 500 MHz NMR spectrometer in the UNC Eshelman School of Pharmacy NMR Facility. This work has been funded in whole or in part with U.S. Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. 75N93022C00035 (Center for Structural Biology of Infectious Diseases, CSBID) to A.S. Part or all of the research described in this paper was performed using beamline CMCF-ID at the Canadian Light Source, a national research facility of the University of Saskatchewan, which is supported by the Canada Foundation for Innovation (CFI), the Natural Sciences and Engineering Research Council (NSERC), the National Research Council (NRC), the Canadian Institutes of Health Research (CIHR), the Government of Saskatchewan, and the University of Saskatchewan. We thank Sofia Lemak, Meenakshi Venkatesan, and Abigayle Williams for Yck2 purification assistance.
Author information
Authors and Affiliations
Contributions
E.P., N.R., L.W., T.M.W., and L.E.C. conceptualized the study. L.E.C. N.S.W., A.S., D.R.A., and T.M.W. obtained funding and provided resources for the study. E.P., M.N., B.Y., P.J.S., B.G.S., R.Z., and X.W. conducted investigations. E.P., M.N., B.Y., P.J.S., N.S.W., and N.R. performed analysis and visualization. N.R., L.W., A.S., N.S.W., D.R.A., T.M.W., and L.E.C. supervised the study. E.P., N.R., and L.W. wrote the draft manuscript, and all authors reviewed and edited the final manuscript.
Corresponding author
Ethics declarations
Competing interests
L.E.C. and L.W. are co-founders and shareholders in Bright Angel Therapeutics, a platform company for the development of novel antifungal therapeutics. L.E.C. is a Science Advisor for Kapoose Creek, a company that harnesses the therapeutic potential of fungi. All other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Puumala, E., Nandakumar, M., Yiu, B. et al. Structure-guided optimization of small molecules targeting Yck2 as a strategy to combat Candida albicans. Nat Commun 16, 2156 (2025). https://doi.org/10.1038/s41467-025-57346-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57346-z
