
Abstract
While immune checkpoint inhibitors (ICIs) have revolutionized cancer treatment, their efficacy in high-grade serous ovarian cancer (HGSOC) remains limited. Some patients, however, achieve lasting responses, emphasizing the need to understand how tumor microenvironment and molecular characteristics influence ICI response. The phase 2 Neo-Pembro study (NCT03126812) included 33 untreated stage IV HGSOC patients, who were scheduled for 6 cycles of carboplatin-paclitaxel and interval cytoreductive surgery. Pembrolizumab (pembro) was added from cycle two and continued for one year. The primary objective was to assess intratumoral immune activation using multiplexed immunofluorescence and immune-related gene expression. Our findings show immune activation, evidenced by an increase in CD3 + , CD8 + , CD8 + /FOXP3+ ratio, TNF-α and interferon-γ signaling. Treatment was well-tolerated. We observed major pathologic responses in 9/33 patients (27%, 95%CI 14-46), with pathologic response strongly associated with immune activation and OS. At a median follow-up of 52.8 months, 8/9 major responders were alive, with 6 patients recurrence-free. In contrast, 4/24 minor responders survived, including one recurrence-free. ctDNA clearance was observed in all major responders and was associated with prolonged PFS and OS. PD-L1 expression and homologous recombination deficiency were predictive of major response and may serve as biomarkers, warranting further exploration. These results suggest major responders may benefit from neo-adjuvant pembro.
Similar content being viewed by others


Introduction
Immune checkpoint inhibitors (ICIs) have revolutionized the treatment landscape for many malignancies over the past decade1. However, their efficacy in ovarian cancer has been limited, with no approved monotherapy or combination regimens. Nevertheless, individual patients achieve durable responses2,3, highlighting the need to better understand how ICI activity depends on the tumor microenvironment and molecular characteristics to optimize their role in the treatment of ovarian cancer.
Two key concepts are pivotal for the success of ICI treatment targeting anti-programmed cell death 1 (PD-1) or its ligand (PD-L1): the tumor infiltration of effector immune cells and the predominant role of the PD-1/PD-L1 pathway in suppressing antitumor immunity. High-grade serous ovarian cancer (HGSOC) is considered immunogenic, as evidenced by its intermediate mutational burden4, which generates neo-antigens capable of evoking tumor-specific T cell responses. Notably, tumor-infiltrating lymphocytes (TILs) are present in more than half of HGSOC cases at diagnosis and are associated with a favorable prognosis5. The prevalence of PD-(L)1 expression on both tumor cells and tumor-associated immune cells in most ovarian tumors6,7,8 supports the rationale for using anti-PD-(L)1 therapies in ovarian cancer to restore the function of exhausted immune cells in the tumor microenvironment (TME).
Despite the activated immune state in ovarian cancer, recent clinical trials evaluating anti-PD-(L)1 monotherapy have shown limited objective response rates (7-20%) in pretreated patients3,9,10,11. Additionally, randomized, phase 3 trials investigating combinations of anti-PD-L1 with conventional chemotherapy regimens in both front-line and recurrent settings have failed to improve progression-free survival (PFS) and overall survival (OS)12,13,14,15, and have even shown a potential detrimental effect3. PD-L1 expression and CD8+ T cell status have been suggested as predictive biomarkers11,13,14, although findings have been inconsistent3,16,17.
The use of ICIs is gradually shifting from later-line therapy to frontline treatment and neo-adjuvant ICI induces an even stronger and broader tumor-specific T cell response compared to adjuvant administration18,19. Recent studies across various cancer types have reported remarkable pathologic responses with neo-adjuvant ICI therapy18,20,21,22,23,24. This enhanced efficacy is attributed to the higher preoperative tumor load, which correlates with increased neo-antigen exposure and subsequent T cell activation25. Additionally, conventional chemotherapeutic agents such as paclitaxel and carboplatin can promote infiltration of immune effector cells, enhance antigen presentation, induce PD-L1 expression, and eliminate immunosuppressive cells, transforming tumors from non-inflamed/“cold” into inflamed/“hot”26,27,28.
Building on these findings, our study hypothesizes that initiation of ICI therapy with pembrolizumab (pembro, anti-PD-1) prior to surgery and preceded by one cycle of neo-adjuvant therapy with carboplatin and paclitaxel will restore tumor-specific T cell effector function.
In this work, we evaluated the changes in the tumor microenvironment induced by one cycle of neo-adjuvant chemotherapy followed by chemotherapy plus pembro and identified potential biomarkers that may predict treatment response. In addition, we evaluated the efficacy, safety, and feasibility of the combination treatment.
Results
Patients characteristics
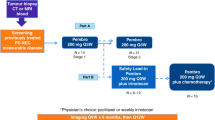
Thirty-four patients with untreated FIGO stage IV high-grade serous ovarian cancer were enrolled in the phase 2 Neo-Pembro trial between December 21, 2017 and May 2, 2022. The study design is illustrated in Fig. 1a. One patient discontinued therapy after the first cycle of chemotherapy due to severe chemotherapy-related toxicity and deteriorating physical condition (WHO performance status 3). She received no pembrolizumab dose and was excluded from subsequent analyses, leaving 33 patients evaluable for analyses (Fig. 2). Baseline patient and tumor characteristics are presented in Table 1. For comparison of efficacy endpoints, a historical cohort of 52 patients who received standard of care without pembro was assembled. Patient characteristics of the historical cohort are shown in Supplementary Table 1.

a Study flowchart. 33 previously untreated stage IV high-grade serous ovarian cancer patients received pembro after one cycle of standard neo-adjuvant carboplatin-paclitaxel. Pembro was added to subsequent chemo cycles and continued as monotherapy up to one year. Surgery was planned after three chemo cycles. ctDNA circulating tumor DNA, PBMC peripheral blood mononuclear cells, PARP Poly (ADP-ribose) polymerase. b Radiological best change (%) in target lesions during neoadjuvant treatment with carboplatin-paclitaxel plus pembrolizumab. Patients whose tumor burden was considered too extensive to achieve complete interval cytoreduction were re-evaluated after four to six cycles of neoadjuvant therapy. c Progression-free survival (PFS); median PFS was 14.2 months (95%CI 11.1-30.2); 3-year PFS was 19% (95%CI 8.8-40). d Overall survival (OS); median OS was 32.0 months (95%CI 21.5-not reached (NR)); 3-year OS was 46% (95%CI 31-68). e Kaplan–Meier of OS for major pathologic responders in the study cohort (green), major pathologic responders in the historical cohort (orange), minor pathologic responders in the study cohort (dark blue), and minor pathologic responders in the historical cohort (light blue). Source data are provided as a Source Data file.

Flowchart of patients included in the Neo-Pembro trial. HGSOC high-grade serous ovarian cancer, irAE immune-related adverse event, pembro pembrolizumab.
Changes in the tumor microenvironment after one cycle of neo-adjuvant chemotherapy and subsequent pembro-chemotherapy
To assess the primary outcome, the immune-activating effects of one cycle of neo-adjuvant chemotherapy alone and the neo-adjuvant pembro-chemotherapy combination, tissue samples were collected before and after a single cycle of chemotherapy and at interval cytoreductive surgery. These samples were analyzed using immunohistochemistry (IHC), multiplexed immunofluorescence (MIF), whole-exome sequencing, and whole-transcriptome sequencing. Supplementary Table 2 provides an overview of the samples available for translational analyses. Supplementary Tables 3 and 4 list the sample locations. No significant differences in clinical characteristics were observed between patients with available whole exome or transcriptome sequencing data and those without.
To investigate the effects of one cycle of neo-adjuvant chemotherapy, we first compared the post-chemotherapy sample to baseline. Fig 3a–h shows cell density in malignant cell areas and adjacent stromal areas assessed with MIF. Immune cells within 150 micron of the cancer cells were included in the counts. Upon one cycle of neo-adjuvant chemotherapy, a significant increase in CD8 + /FOXP3+ ratio (p = 0.003) was observed in the overall cohort (Fig. 3d), evident in both major and minor responders. At the transcriptomic level, gene-set analysis showed a number of patterns, displayed in Fig. 4a–h stratified by response, and in Supplementary Fig. 1a–h across the whole cohort. Post-chemotherapy, a trend of reduced proliferation is visible (Supplementary Fig. 1a), and significant increases in TNFα signaling (Supplementary Fig. 1b), interferon-γ (IFN-γ) signaling (Supplementary Fig. 1c), MHC class I antigen presentation genes (Supplementary Fig. 1d) and PD-L1 (Supplementary Fig. 1f). The latter was in line with the IHC assessment, showing increased PD-L1 expression on tumor and immune cells (Fig. 5). We did not observe clear changes in PD-1 gene expression in post-chemotherapy samples (Supplementary Fig. 1e). Estimates of the abundance of immune cell subtypes from the expression of marker genes indicate increased levels of CD8 + T cells post-chemotherapy (Supplementary Fig. 1g), and a trend for reduced FOXP3 + T cells (Supplementary Fig. 1h). Note that only a subset of patients had sufficient quality material available for sequencing, and therefore there is insufficient power to identify statistically significant transcriptomic differences between time points among the major responders, or between minor and major responders (Fig. 4a–h).

Pre- to post treatment changes of density of immune cell populations in the tumor microenvironment assessed through multiplexed immunofluorescence. Baseline samples available in 9 major responders and 21 minor responders. Post-chemotherapy samples available in 7 major responders and 16 minor responders. Post-pembro plus chemotherapy samples available in 7 major responders and 18 minor responders. Boxplots represent the median, and 25th and 75th percentiles; the whiskers extend from the hinge to the largest value no further than 1.5× IQR from the hinge. Pre- to post pairwise statistical significance was tested using Wilcoxon’s signed-rank test; for differences between major and minor responders, the significance was tested using Wilcoxon’s rank-sum test. All statistical tests were two sided. No adjustments were made for multiple comparisons. Only significant p-values are shown. a CD3 + T cells. b CD3 + CD8 + T cells. c CD3 + FOXP3 + T cells. d CD8 + /FOXP3 + T cell ratio. e CD20 + B cells. f CD68+ macrophages. g CD8+PD-1+ T cells. h CD8+PD-1+ /total CD8+ T cells ratio. i tertiary lymphoid structures (TLS). Source data are provided as a Source Data file.

Baseline samples were available in 7 major responders and 21 minor responders. Post-chemotherapy samples were available in 6 major responders and 10 minor responders. Surgery resection samples were available in 4 major responders and 18 minor responders. a–d single sample scoring of four genesets using singscore85. (*) indicates p < 0.05 by two-sided paired Wilcoxon test, not corrected for multiple testing. P-values: a baseline vs surgical resection: 4.5 × 10-5; b baseline vs surgical resection: 1.5 × 10-5; post-induction chemo vs surgical resection: 0.016. e, f normalized expression level of PD-1 and PD-L1 in each sample. (*) indicates p < 0.05 by two-sided paired Wilcoxon test, not corrected for multiple testing. P-values: e baseline vs surgical resection: 6.7 · 10-3; post-induction chemo vs surgical resection: 0.031; f baseline vs surgical resection: 5.6 · 10-3. g, h immune cell type abundance estimate obtained through the average expression of marker genes from Danaher et al.88. (*) indicates p < 0.05 by two-sided paired Wilcoxon test, not corrected for multiple testing. P-values: g baseline vs post-induction chemo: 2.0 × 10-3; baseline vs surgical resection: 0.045. i Mutational load as estimated by the number of somatic variants per million basepairs. Two-sided Wilcoxon test p = 0.075. j Homologous recombination deficiency signature score based on the number of large-scale state transitions in the genomic copy number profile as described in the study of Popova et al.97. Only the baseline whole exome sequencing samples were used (major responders: n = 8 and minor responders: n = 20). Horizontal gray lines indicate ploidy-specific cutoffs indicative of homologous recombination deficiency. Points are colored by BRCA1 gene mutational status, where ‘pathogenic’ BRCA1 mutations are clinically established pathogenic variants, and ‘functional damaging’ indicates a BRCA1 variant that has been found to be damaging based on three functional tests98, but has not yet been proven to be pathogenic and is clinically a variant of uncertain significance. No BRCA2 mutations were found in this cohort. Source data are provided as a Source Data file.

Pre- to post treatment changes of density of immune cell populations in the tumor microenvironment assessed through multiplexed immunofluorescence. Baseline samples available in 9 major responders and 20 minor responders. Post-chemotherapysamples available in 7 major responders and 13 minor responders. Boxplots represent the median, and 25th and 75th percentiles; the whiskers extend from the hinge to the largest value no further than 1.5× IQR from the hinge. Pre- to post pairwise statistical significance was tested using Wilcoxon’s signed-rank test; for differences between major and minor responders, the significance was tested using Wilcoxon’s rank-sum test. All statistical tests were two sided. No adjustments were made for multiple comparisons. Only significant p-values are shown. a Combined positive score. b Immune proportion score (%). c Tumor proportion score (%). d–f representing delta (Δ) expression values in minor pathologic responders (blue) and minor pathologic responders (green), showing post-chemo minus baseline. Source data are provided as a Source Data file.
To further investigate the effect of adding pembro to chemotherapy, we next compared the resection sample to the baseline sample and to the post-chemotherapy sample. Following the combination of chemotherapy and pembro, a significant difference in Δ values of CD8 + T cells between major and minor responders (p = 0.001, Fig. 6b) was observed with MIF, resulting in a significantly higher CD8 + T cell infiltration in major responders than in minor responders (p = 0.008, Fig. 3b). In addition, a significantly higher CD3+ density was observed in major responders (p = 0.008, Fig. 3a). The CD8 + /FOXP3+ ratio increased in major responders. In contrast, it decreased in minor responders, consistent with a significant difference in Δ values (p = 0.009, Fig. 6d) and a significant higher ratio in major responders at the time of resection (p = 0.033, Fig. 3d). Minor responders showed a significant decrease in the density CD68+ macrophages (p = 0.009, Fig. 3f). This trend was also observed in the major responders, although not reaching significance (p = 0.156). The density of tertiary lymphoid structures (TLSs) in the resection samples did not differ between major and minor responders, nor was CXCL13 expression (Fig. 3i). At the transcriptomic level, proliferation was strongly decreased during pembro-chemotherapy combination (Supplementary Fig. 1a), and TNFα signaling was further increased compared to both the pre-treatment and post-chemotherapy samples (Supplementary Fig. 1b). PD-1 expression at the transcriptomic level was markedly higher after pembro-chemotherapy combination treatment when compared to both the pre-treatment and post-chemotherapy samples (Supplementary Fig. 1e). Finally, transcriptomic estimates of the number of CD8+ cells did not further change after combination treatment (Supplementary Fig. 1g), while the estimates of the number of FOXP3+ cells were lower after combination treatment than at baseline (Supplementary Fig. 1h).

a–f Dynamics of cell density of immune cell populations in the tumor microenvironment assessed through multiplexed immunofluorescence represented as delta (Δ) expression values in minor pathologic responders (blue) and minor pathologic responders (green), showing post-chemo minus baseline (delta 1; minor, n = 15; major, n = 7), resection minus post-chemo (delta 2; minor, n = 12; major, n = 5), and resection minus baseline (delta 3; minor, n = 18; major, n = 5). Boxplots represent the median, and 25th and 75th percentiles; the whiskers extend from the hinge to the largest value no further than 1.5× IQR from the hinge. The difference between major and minor responders was tested using a two-sided Wilcoxon Rank-sum test. Only significant p-values are shown. a CD3 + T cells. b CD3 + CD8 + T cells. c CD3 + FOXP3 + T cells. d CD8 + /FOXP3 + T cell ratio. e CD20 + B cells. f CD68+ macrophages. Source data are provided as a Source Data file.
Together, these data suggest that neo-adjuvant pembro-chemotherapy leads to immune activation in the TME of HGSOC. Major pathologic responders had more pronounced CD3+ and CD8 + T cell influx, and an increased CD8 + /FOXP3+ ratio than minor responders. Negative effectors of the immune response such as regulatory T cells and macrophages are depleted during neo-adjuvant treatment.
Neo-adjuvant pembrolizumab plus chemotherapy induces 27% major pathological responses
The majority of patients (n = 22, 66.7%) were treated with three to four neo-adjuvant cycles before undergoing surgery (Table 2). In five patients (15.2%), surgery was postponed until after six cycles of neo-adjuvant therapy, and six patients (18.2%) did not undergo surgery. Of the six patients who did not undergo cytoreductive surgery, one patient was considered ineligible for surgery due to disease progression during neo-adjuvant therapy, and five patients due to a high residual tumor burden. A complete cytoreduction was achieved in 19 patients (57.6%).
Nine (27% [95%CI 14-46%]) of 33 patients had (near) complete pathologic response after three cycles of neo-adjuvant treatment (chemotherapy response score; CRS 3) and were considered major responders. Among these nine, two patients had a complete response, with no residual tumor found on histopathological assessment. One of these complete responders had surgery postponed until after six cycles of neo-adjuvant chemotherapy due to a cerebrovascular accident requiring anticoagulation without interruption. A biopsy after three cycles, however, showed CRS 3. Fifteen (45% [95%CI 29-63%]) patients had appreciable pathologic response (CRS 2), and five (15% [95%CI 6-33%]) patients had minimal to no pathologic response (CRS 1). Patients with CRS 1 or 2 were considered minor responders. In four patients, no tissue was available for pathologic response assessment because they did not undergo surgery, and a substitute biopsy was not feasible. These patients were considered minor responders, as interval cytoreductive surgery was not feasible due to the high residual tumor burden observed after neo-adjuvant treatment. Baseline and treatment characteristics of the major and minor responders are shown in Supplementary Table 5.
Radiological response to neo-adjuvant treatment was evaluated by comparing CT scans at baseline and after two treatment cycles. Figure 1b shows the best change in tumor volume (%) according to RECIST v1.1 after two neo-adjuvant cycles. We observed an objective response rate (ORR) of 63.6% [95%CI 48.2-82.0], including radiologic complete response (CR) in one patient and partial response (PR) in 20 (60.6%) patients. Stable disease (SD) was observed in 11 (33.3%) patients and progressive disease (PD) in one (3.0%) patient. The objective response was significantly associated with PFS (log-rank p = 0.011) but not with OS (log-rank p = 0.230).
The association between radiological and pathological response was significant (Fisher two-sided p = 0.030). Among the nine patients exhibiting a major pathologic response, one was classified as a radiological CR and eight as radiological PR. Eleven minor pathologic responders showed PR, eight had SD, and one had PD (Fig. 1b, Supplementary Table 6).
Pathological response is strongly associated with survival outcomes
At the clinical data cutoff of April 1, 2024, median follow-up was 52.8 (IQR 34.5-62.6) months. Follow-up was available for all patients. Twenty-six (79%) of 33 patients had an event of disease recurrence or progression, and 20 (61%) of 33 patients had died at the time of data cutoff. In all patients who died, the cause of death was progressive disease. Median PFS was 14.2 months (95%CI 11.1-30.2) (Fig. 1c) and median OS was 32.0 months (95%CI 21.5-not reached (NR)) (Fig. 1d). 3-year PFS was 19% (95%CI 8.8-40), and 3-year OS was 46% (95%CI 31-68).
Pathologic response was strongly associated with outcome, as demonstrated by a significantly longer PFS and OS in major responders than in minor responders (median PFS NR [95%CI 30.3-NR] vs 11.4 months [95%CI 9.4-14.9], hazard ratio (HR) 0.12 [95%CI 0.04-0.42], p = 0.001); median OS NR vs 23.5 months [95%CI 15.7-40.2], HR 0.07 [95%CI 0.01-0.51], p = 0.009) (Fig. 1e). To account for potential confounding factors, we performed Cox regression analyses, adjusting individually for key prognostic variables. These adjustments did not meaningfully affect the results, with HRs below 0.15 for PFS and below 0.09 for OS that remained significant across models. Six of nine patients with a major response were progression-free at data cutoff, and eight of nine were alive. In contrast, only one of 24 minor responders remained progression-free, and four of 24 patients were alive.
We compared pathologic response and survival of the study cohort to a historical cohort of 52 patients with stage IV high-grade serous ovarian cancer, all of whom received at least two cycles of neo-adjuvant chemotherapy and met the key eligibility of the trial. Patients in the historical cohort were significantly older (median age 69.0 vs 64.0 years, p = 0.023) and had a significantly shorter interval between neo-adjuvant chemotherapy and surgery (median 31 vs. 38 days, p < 0.001) (Supplementary Table 1). The use of poly (ADP-ribose) polymerase inhibitors (PARPi) as front-line maintenance treatment was similar in the historical and intervention cohort (11.5% vs. 9.1% [p = 0.626], respectively). In the historical cohort, we observed a major pathological response in nine out of 52 patients treated without pembro (17% [95%CI 8-30]). Compared to the study cohort, this difference was not statistically significant (Fisher’s exact test, two-sided p = 0.290). Notably, OS of major responders in the study cohort was significantly better than OS of major responders in the historical cohort (Fig. 1c, median OS NR [95%CI NR-NR]) vs 30.7 months [95%CI 24.6-NR], HR 0.12 [95%CI 0.01-0.97], p = 0.047), while PFS was not (median PFS NR [95%CI 30.3-NR] vs 24.6 months [95%CI 12.9-NR], HR 0.31 [95%CI 0.08-1.25], p = 0.098). Among non-responders, PFS and OS were similar in the study and historical cohorts (median PFS 11.4 vs 13.4 months, HR 1.16 [95%CI 0.69-1.95], p = 0.584; and median OS was 23.5 vs 30.1 months, HR 1.08 [95%CI 0.62-1.89], p = 0.789). To account for potential confounding factors, we performed Cox regression analyses, adjusting individually for key prognostic variables. These adjustments did not meaningfully affect the results, with hazard ratios and statistical significance remaining consistent across all models.
Safety and feasibility
Toxicity was manageable and there were no new safety signals. Grade ≥2 immune-related adverse events (irAEs) were observed in 15 patients (45.5%), of which five patients (15.2%) developed grade 3-4 irAEs (Table 3). No grade 5 irAEs occurred. In two patients (6.1%), irAEs led to discontinuation of pembro. One patient developed a grade 3 bullous toxicodermia after two cycles of pembro, requiring high-dose intravenous steroids and antibiotics during an eight-day hospitalization. Symptoms resolved after steroid tapering. Another patient developed recurrent organizing pneumonia with episodes of fever, malaise and respiratory symptoms. The symptoms improved with oral prednisone which was continued in a low-dose for several months.
Twenty-seven patients (81.8%) completed standard therapy with six cycles of chemotherapy. Seventeen patients (48.5%) completed pembro treatment, including one year of maintenance therapy with pembro monotherapy (Table 2). Reasons for discontinuation are listed in Table 2. The median relative dose intensity (RDI) was 89% (IQR 70-95) for carboplatin and 77% (IQR 66-90) for paclitaxel. Based on previously published studies, an RDI < 85% is generally considered to be a clinically significant reduction from planned therapy29. In this cohort, 14 patients (42.4%) had an RDI < 85% for carboplatin and 20 patients (60.6%) had an RDI < 85% for paclitaxel, which is in line with previous studies in patients with ovarian cancer receiving only NACT30,31. Grade 3-4 chemotherapy-related adverse events were observed in 29 patients (87.9%) but were not unexpected (Supplementary Table 7).
The median time from the last neo-adjuvant cycle to surgery was 38 days (IQR 32-45). Delay in surgery (>42 days after the last neo-adjuvant chemotherapy cycle) was not caused by treatment-related adverse events, but by logistical constraints. Surgery-related grade 3-4 AEs were observed in 7 patients (25.9%) (Supplementary Table 8). No unexpected surgical adverse events or surgery-related deaths were observed.
Biomarker analysis of neo-adjuvant ICI response in HGSOC
As an exploratory outcome, we conducted further comparisons between major and minor responders to identify biomarkers of response. Based on evidence from various studies showing a positive correlation between PD-L1 expression and response to ICIs in different cancer types, we examined PD-L1 expression as a potential biomarker. We used the immune proportion score (IPS), tumor proportion score (TPS), and combined positive score (CPS) determined by IHC and explored cutoff values that were used in previous studies (1, 5, and 10). At baseline, IPS was significantly higher in major responders than in minor responders (p = 0.034, Fig. 5b) and there was an indication that CPS at a cutoff of 10 was predictive of major pathologic response (p = 0.048, Supplementary Table 9). Interestingly, upon one cycle of neo-adjuvant chemotherapy, we observed an increase in PD-L1 expression, resulting in a significantly higher CPS (p = 0.005) and IPS (p = 0.002) in major responders than in minor responders (Fig. 5a, b). After one cycle of neo-adjuvant chemotherapy, CPS at cutoff of 5 and 10 was predictive of major pathologic response (p = 0.036 and p = 0.002, respectively, Supplementary Table 9). In addition, Δ values of TPS showed a significant difference between major responders, in whom TPS increased, and minor responders, in whom TPS remained stable (Fig. 5f).
In patients with non-small cell lung cancer (NSCLC) and gastro-intestinal cancer, the presence of CD8 + PD-1+ TILs in pre-treatment biopsies predicted response to PD-1 blockade and was associated with increased overall survival21,32,33. In our cohort, CD8+PD-1+ T cell density and the ratio of CD8+PD-1+ to total CD8+ T cells at baseline and post-chemotherapy were not associated with pathologic response (Fig. 3g, h). Notably, after one cycle of neo-adjuvant chemotherapy both CD8+PD-1+ T cell density and the CD8+PD-1+/CD8+ ratio decreased significantly in the overall patient population.
At the genomic level, there was no evidence that mutational load was different between major and minor responders (Fig. 4i, two-sided Wilcoxon’s rank-sum test p = 0.075). However, patients whose tumors displayed a homologous recombination deficiency (HRD) signature in their DNA copy number profile at baseline, were more likely to have a major response (Fig. 4j) (Fisher exact test two-sided p = 0.011). All BRCA1 mutation carriers showed HRD signatures, and all three patients with confirmed pathogenic BRCA1 mutations were major responders. Some studies have suggested that HRD is associated with increased PD-L1 expression and mutational burden, resulting in increased neoantigens, leading to an improved response to ICI34,35. In our cohort, HRD was indeed associated with a higher mutational load (two-sided Wilcoxon’s p = 7.3 · 10-4, Supplementary Fig. 1i), although no association between HRD and PD-L1 expression (through MPIF) was found.
Circulating tumor DNA clearance is associated with pathological response and survival
Circulating tumor DNA (ctDNA) was detected with sequencing methods at baseline in 88% (29 of 33) of patients. The comparison of baseline characteristics between patients without available ctDNA and those with ctDNA revealed a significantly lower median baseline CA-125 level in the former group. Specifically, the median CA-125 was 345.5 kU/L (IQR 129.0–566.5) in patients in whom ctDNA was not detected, compared to 1273.0 kU/L (IQR 487.0–4311.0) in patients with ctDNA (p = 0.035). Personalized ddPCR assays were used to analyze ctDNA level and dynamics for response prediction, and could be used in 88% (29 of 33). At baseline, ctDNA level was not associated with response (p = 0.87; Supplementary Fig. 2a), PFS (p = 0.60; Supplementary Fig. 2b), or OS (p = 0.62; Supplementary Fig. 2c). ctDNA dynamics between baseline (t0), after one cycle of neo-adjuvant chemotherapy (t1), and after one subsequent cycle of pembro-chemotherapy (t2) are depicted as relative change (Fig. 7a) and absolute values (Supplementary Fig. 2d). In most patients, ctDNA decreases after one cycle of neo-adjuvant chemotherapy. However, in a few patients, an increase during the first cycle of chemotherapy (t0-t1) is followed by a decrease after pembro-chemotherapy (t1-t2; Fig. 7a). Patients with decrease in ctDNA level of >30% between t0 and t2 had similar PFS (P = 0.17) and OS (P = 0.47) compared to patients without a decrease in ctDNA of >30% (Fig. 7b, c). Preoperative ctDNA clearance was observed more frequently in major responders (100%) than in minor responders (14%) (Fisher’s exact test p < 0.0001; Fig. 7d) and was significantly associated with longer PFS (p < 0.0001; Fig. 7e) and OS (p = 0.0003; Fig. 7f). The association of ctDNA clearance with PFS and OS was irrespective of pathological response (Supplementary Fig. 2e, f).

29 patients were included in the ctDNA analyses. a Relative change in ctDNA level of major responders (green) and minor responders (blue) after one cycle of neo-adjuvant chemotherapy (t1) and after one subsequent cycle of pembro-chemotherapy (t2) compared to baseline (t0). b progression-free survival (PFS) and c overall survival (OS) were similar for patients with a decrease in ctDNA of >30% between t0 and t2 (orange) compared to patients without a >30% decrease (light blue). Both PFS (two-sided log-rank p = 0.08) and OS (two-sided log-rank p = 0.35) were considered not significantly different. d Number of cases that display a clearance (light blue) or no clearance (orange) of ctDNA pre-surgery among the minor responders and major responders. Improved e PFS (two-sided log-rank p < 0.001) and f OS (two-sided log-rank p < 0.01) was observed for patients with a ctDNA clearance (light blue) compared to patients without a clearance (orange). Source data are provided as a Source Data file.
Discussion
The efficacy of immune checkpoint inhibitors in ovarian cancer has been limited to date, primarily due to the cancer’s moderate tumor mutational burden, inadequate anti-tumor immune responses, and the presence of active immunosuppressive mechanisms in the tumor microenvironment36. For the overall patient population, survival outcomes in the present study are consistent with previous ICI trials in the front-line setting12,13 showing no demonstrable benefit of (neo)adjuvant chemotherapy combined with pembrolizumab over standard of care37. However, in patients treated with chemotherapy plus pembro, we identified a subset of patients who achieved a (near) complete response, representing 27% of the patients at the time of surgery. Combining transcriptomic and multiplexed immunofluorescence data on paired tumor samples collected prospectively, we found that these major responders exhibited stronger immune activation and had significantly better OS than minor responders. Homologous recombination deficiency (HRD) and PD-L1 expression were predictive of major pathologic response and may serve as potential biomarkers for selecting HGSOC patients for neo-adjuvant ICI treatment if validated in larger studies.
Previous studies in HGSOC have shown an association between pathologic response to NACT and survival38,39. Our findings, however, indicate that this association may be amplified when neo-adjuvant pembro is added. Specifically, patients who achieved a major response to the pembro-chemotherapy combination had significantly better overall survival compared to those with a major response to chemotherapy alone, based on comparisons with our historical cohort and previous studies39,40,41,42,43,44,45,46,47. While these findings suggests that adding pembro could improve long-term efficacy in this subset of major responders, the lack of randomization and the small sample warrant caution when interpreting these findings. Importantly, patients in the historical cohort were significantly older than those in the interventional cohort (median age 69 vs. 64 years), which may contribute to worse survival outcomes. Additionally, the shorter median interval between neo-adjuvant therapy and surgery in the historical cohort (31 vs. 38 days) may be associated with improved outcomes, potentially favoring the historical cohort in survival analyses. Despite accounting for individual covariates in multivariable analyses, the limited sample size precluded comprehensive adjustment for all factors simultaneously, leaving the potential for residual confounding.
The study design included a single cycle of chemotherapy before adding anti-PD1, allowing us to isolate the effects of carboplatin-paclitaxel and the subsequent combination of chemotherapy plus pembro. After one chemotherapy cycle, we observed reduced proliferation, increased inflammatory signaling, increased MHC class I antigen presentation and PD-L1 expression, and beneficial changes in the effector T cell/regulatory T cell ratio. These patterns may indicate tumor recognition by cytotoxic T cells that produce IFN-γ, which in turn upregulates the expression of genes such as MHC class I and PD-L1, a phenomenon known as adaptive resistance48. After the addition of pembro, significant CD3+ and CD8+ T cell infiltration was observed, predominantly in major pathologic responders. This increased T cell density likely reflects a more effective reactivation and expansion of the anti-tumor immune response, leading to better control and elimination of the tumor. Notably, similar immune activation has been reported after NACT alone, where NACT has been shown to increase T-cell densities, upregulate of MHC class I expression, and modulate the T cell-to-regulatory T cell balance7,26,49,50,51. Additionally, NACT has been found to induce PD-L1 overexpression, which generally inhibits immune responses. However, in the context of immune checkpoint inhibition, elevated PD-L1 levels may sensitize patients to pembro, potentially improving disease control. In the study by How et al., a similar trend was observed among the best responders adjuvant pembro, where an increase in CD3+ and CD8+ T cells was accompanied by elevated PD-L1 expression51. Comparison to a control arm receiving standard of care without the addition of pembro would help clarify the distinct effects of chemotherapy versus pembro. Furthermore, it is important to note that we did not distinguish the locations of T cells in the intraepithelial versus adjacent stromal compartments. Previous studies have shown that changes in T cell subpopulations after NACT ± bevacizumab mostly take place in the stroma, although increases at the intratumoral site have also been described52,53. Whether the ___location of effector immune cells could be predictive of response to ICI warrants further investigation.
In other tumor types, such as lung cancer and urothelial cancer, the number of TLS increased after ICI. However, we did not observe this in our cohort. Although the clinical relevance of TLSs in HGSOC has been previously reported54,55, HGSOC is characterized by a low density of follicular helper T cells. This results in fewer TLS compared to, for example, non-small cell lung cancer (NSCLC) and potentially inadequate preservation of ICI-sensitive T cell phenotypes, such as PD1+CD8+ 56. This disparity may explain the lower efficacy of ICIs in HGSOC than in NSCLC patients.
In our study, PD-L1 expression (CPS > 10) at baseline was predictive of major pathologic response. Additionally, neo-adjuvant chemotherapy was observed to increase PD-L1 expression, particularly in major pathologic responders, which is consistent with previous studies7,49,50,51,57. Similarly, KEYNOTE-100 showed an association between CPS ≥ 10 and improved response rates in patients with advanced recurrent ovarian cancer treated with pembro13. The IMAGYN050 trial highlighted the predictive value of high PD-L1 expression (≥ 5%) on immune cells in newly diagnosed advanced-stage ovarian cancer, while a 1% cutoff did not show significance13. These findings collectively suggest that PD-L1 expression may serve as a valuable biomarker for selecting patients for ICI treatment. However, results from ENGOT-OV41/GEICO 69-O/ANITA and ATALANTE/ENGOT-ov29 did not confirm PD-L1 as a biomarker in the recurrent setting15,58. These trials used a threshold of ≥1% immune cell positivity, which may not be an optimal cutoff for ovarian cancer, and excluded PD-L1 expression on tumor cells, potentially limiting predictive value. Furthermore, variation in assays and scoring methods across studies further complicates interpretation and highlights the need for standardization. Another limitation of PD-L1 as a biomarker is that 5-20% of PD-L1-negative tumors still respond to ICI treatment, emphasizing the need for combining biomarkers to improve predictive accuracy9,59. Advancing assay standardization and biomarker combinations could enhance the clinical utility of PD-L1 in ovarian cancer.
Our findings demonstrate that HRD is associated with major pathologic response. This may be attributed to the favorable prognosis and increased sensitivity to platinum-based chemotherapy and PARPi commonly associated with HRD and BRCA mutations. Another possible explanation is that HRD tumors are related to a higher mutational burden, as also observed in our study, and increased antigen production, which may improve responses to ICI. However, the relatively higher mutational burden does not fully explain the increased immunogenicity, as HRD is a predictor of response to ICI independent of mutational burden60. Furthermore, we found no overall association between mutational burden and pathologic response in our cohort. Beyond mutational burden, HRD tumors can also exhibit altered inflammatory signaling, involving the cGAS/STING pathway61, which may enhance ICI response. If validated in a larger cohort, HRD may help select HGSOC patients for neo-adjuvant ICI treatment.
It is important to note that this study was conducted in a setting where maintenance treatment with bevacizumab is not reimbursed and PARPi use in the frontline setting is largely limited to patients with BRCA mutations, as there is limited evidence supporting an overall survival benefit outside this subgroup62. As a result, none of the patients in our study received front-line bevacizumab and three of the four patients with a BRCA mutation (9.1%) received front-line PARPi therapy, with one patient enrolled before EMA approval of olaparib for frontline use in BRCA-mutated ovarian cancer. Extrapolating our findings to settings with more widespread use of PARPi or first-line bevacizumab therapy requires consideration of the potential interactions between ICIs, PARPi, and bevacizumab, both in terms of efficacy and safety. Given the immunological properties of PARPi, such as its ability to enhance anti-tumor immunity through DNA damage and modulation of the TME63, the potential interaction between PARPi and ICI has led to recent trials investigating the combination. In the recurrent setting, trials such as ENGOT-OV41/GEICO 69-O/ANITA and ATHENA-COMBO reported manageable toxicity profiles, but failed to meet their primary endpoints in improving PFS58,64. Early reports of a study combining neoadjuvant ICI and PARPi in newly diagnosed HRD-positive ovarian cancer have shown promising results65. VEGF blockade through bevacizumab may enhance T cell infiltration and reduce immunosuppression within the tumor microenvironment, offering a rationale for its combination with ICIs. Despite these theoretical benefits, the addition of atezolizumab to chemotherapy and bevacizumab in both frontline and recurrent settings did not improve PFS13,15. The safety profile of ICI plus chemotherapy plus bevacizumab was consistent with safety profiles of the individual medicines and no new safety signals were identified with the combination. Whether the triplet combination offers a benefit for patients with ovarian cancer in the front-line setting is currently being investigated in ongoing trials66,67. In addition to evaluating clinical effects, comprehensive translational research will be important for identifying optimal candidates for ICI in combination with other treatment modalities. The results, once available, may provide further insights into the applicability of our findings to broader clinical contexts.
When designing the trial, we initially selected a dose-dense (weekly) paclitaxel schedule, guided by the results of the Japanese phase 3 RCT (JCOG-3016), which demonstrated significantly improved PFS (28 vs. 17.2 months, HR 0.71; 95% CI 0.58-0.88, p = 0.002) and 3-year OS (72.1% vs. 65.1%; HR 0.75, 0.57-0.98; p = 0.03) compared to the conventional 3-weekly regimen68. As a weekly regimen increases patient and hospital burden and subsequent phase 3 trials did not confirm the efficacy benefits69,70,71, we amended the protocol to include the conventional 3-weekly regimen. Notably, we observed no significant differences in changes of immune infiltrate (i.e. delta scores of CD3 + , CD8 + , or CD8/FOXP3 ratio) between the weekly and 3-weekly paclitaxel cohorts (not reported here).
Patient-specific ctDNA analysis revealed that neither baseline ctDNA levels, considered to represent the tumor load72, nor a > 30% decrease in ctDNA level presurgery were predictive of treatment response and survival. However, ctDNA clearance was observed in all major responders and only in two minor responders. ctDNA clearance was also associated with longer PFS and OS, consistent with previous studies in gastro-intestinal cancer33. These associations with PFS and OS were independent of pathologic response. These data suggest that monitoring ctDNA dynamics may provide valuable information on treatment response and prognosis, with potential to inform clinical decision-making, if further validated. For example, if ctDNA clearance is not achieved after three cycles of neoadjuvant chemotherapy, extending treatment to six cycles may be considered. Alternatively, non-responding patients could benefit from an early switch to a non-cross-resistant regimen. Possibly, in cases where ctDNA clearance is not achieved, the contribution of cytoreductive surgery to improve outcomes becomes uncertain and might be challenged. Such strategies warrant further exploration in larger, randomized studies and may ultimately help to optimize patient management.
Limitations of our study include the relatively small sample size and the single-arm design, which suggests that the findings are more hypothesis-generating than providing definitive conclusions. Although our study offers valuable insights into cell dynamics during treatment, variability in sample ___location requires cautious interpretation due to potential bias from different tissue origins. This variability, along with intra-patient variation in TME composition and immune checkpoint expression across different disease sites, highlights the limitations of single-site biopsies. Future studies could benefit from standardized sampling protocols to minimize these confounding factors. However, serial sampling from matched sites or multiple-site biopsies is particularly challenging in ovarian cancer due to the difficulty of accessing primary and peritoneal metastases. Molecular imaging techniques may offer a non-invasive alternative for monitoring therapeutic response by providing real-time insights into the TME. Another limitation is that immune cell abundance by gene expression signature did not always correspond to the MIF results. This discrepancy could be due to stromal material in the bulk transcriptomic samples, tissue heterogeneity, or the inherent limitations of bulk transcriptomic analysis. Furthermore, HRD status was assessed using the large-scale state transition (LST) score with ploidy-specific cutoffs, which has shown strong correlation with FDA-approved HRD measures, particularly the Myriad myChoice HRD test73,74. However, as the LST score is based on genomic scarring, it may misclassify the HRD status of some tumors, representing a limitation of our study. Lastly, emerging data indicated that PD-1 blockade does not significantly alter the frequency or breadth of circulating tumor-reactive CD8 + T cells but instead exerts more pronounced immune effects within the tumor microenvironment75. Consequently, while the study’s primary objective remained consistent throughout, the primary endpoint shifted to assessing immune activation within the tumor tissue rather than in peripheral blood.
Our data provide evidence of immune-activation within the TME following treatment with neo-adjuvant chemotherapy plus pembro. Immune-activation was stronger in the subset of major pathologic responders and these patients may derive long-term survival benefits from the addition of pembro. Homologous recombination deficiency and PD-L1 expression were predictive of major pathologic response and may serve as potential biomarkers for selecting HGSOC patients for neo-adjuvant ICI treatment if validated in larger studies. In clinical practice, assessment of pathologic response in surgical resection tissue may provide valuable prognostic information for individual patients. For minor responders, the benefit of continuing adjuvant treatment is questionable, as survival outcomes remain poor despite extensive therapy. For major responders, however, this study provides data to further investigate whether adjuvant treatment provides a clinical benefit and what the optimal number of cycles should be.
Methods
Ethics
The protocol was approved by the local ethics committee at the Netherlands Cancer Institute (NKI). All patients provided written informed consent prior to enrolment. This study was performed in accordance with the Declaration of Helsinki.
Study design
Neo-Pembro is an exploratory, open-label, single-arm, phase 2 study. Patients were recruited at the Netherlands Cancer Institute (NKI), Amsterdam, The Netherlands. This trial was investigator-initiated and conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. The protocol (Supplementary Note 1) was approved by the independent medical ethics committee of the NKI. The first patient was enrolled on December 21, 2017, and the last patient was enrolled on and May 2, 2022. The data were collected by an independent data manager. Final clinical data revisions and analyses were performed by the first author (SLA) in collaboration with the Department of Biometrics of the Netherlands Cancer Institute (ML-Y) according to the Statistical Analysis Plan (Supplementary Note 2). The trial is registered with ClinicalTrials.gov (NCT03126812) and EudraCT (2016-004700-56).
Participants
Patients aged ≥18 years were considered eligible for the study if they had histologically confirmed FIGO stage IV high-grade serous ovarian, fallopian tube, or peritoneal cancer and were willing and able to provide three pre-treatment tumor biopsies from a metastatic site. Other inclusion criteria included a WHO performance status score of 0–1, normal blood count, and adequate organ function. Patients with immunosuppressive treatment, immunodeficiency, human immunodeficiency virus infection, hepatitis B or C infection, active autoimmune disease, or other active malignancies were ineligible. All patients provided written informed consent prior to enrolment.
Procedures
Patients were treated with six cycles of carboplatin (intravenously, area under the curve 6 mg/mL per min, once every 3 weeks) and paclitaxel (intravenously, either 80 mg per m² body-surface area once a week or 175 mg per m² body-surface area, once every 3 weeks). Pembrolizumab was added from the second cycle onwards and was administered at a dose of 200 mg once every 3 weeks during the chemotherapy phase. After completion of the chemotherapy cycles, patients received an additional 7 cycles of pembrolizumab monotherapy at a dose of 400 mg once every six weeks (i.e., maintenance phase). The study flowchart is shown in Fig.1a. Premedication to minimize chemotherapy-related toxicities was administered according to the local practice guidelines. Corticosteroids were omitted for cycles of chemotherapy combined with pembrolizumab. The majority of patients were enrolled prior to the EMA’s approval and reimbursement of PARPi for frontline use in June 2018. Following this approval, the protocol was updated to allow patients to receive PARPi treatment from June 2018 onward. In accordance with national guidelines in the Netherlands, patients did not receive bevacizumab maintenance therapy after frontline treatment.
The response evaluation of neo-adjuvant chemotherapy involved CT scans of the pelvis, abdomen, and thorax after the second cycle. Patients who did not experience disease progression during the neo-adjuvant treatment phase were considered to be eligible for cytoreductive surgery. Ideally, surgery was scheduled within six weeks of the third neo-adjuvant cycle (i.e., within 42 days), and the remaining chemotherapy cycles in combination with pembrolizumab were completed after surgery. If a patient responded but the tumor load was considered too extensive to achieve complete cytoreduction, surgery could be deferred after an additional 2–3 cycles of neo-adjuvant treatment.
Carboplatin or paclitaxel dose reductions were applied following clinically significant toxicity, as determined by the clinician. Dose reductions were not allowed for pembrolizumab, although doses could be omitted in cases of persistent toxicity at the investigator’s discretion.
A safety follow-up visit was performed 30 days after the last dose of the study treatment or prior to the initiation of any new anticancer treatment, whichever occurred first. Adverse events and their relation to study medication were graded and reported throughout the study until the safety follow-up visit using the Common Terminology Criteria for Adverse Events (CTCAE) v4.03. The protocol for further patient follow-up included repeated visits every 12 weeks, without further standardized imaging or laboratory tests. Recurrence status (by investigator assessment) and survival status were obtained from medical records, general practitioner records, and the National Death Registry.
Collection of blood and tumor tissue samples
Blood samples were collected at baseline, during neo-adjuvant chemotherapy, and during the maintenance phase (Fig. 1a). Cell-free plasma and cell pellet were isolated according to the local standard operating procedures [10.1093/clinchem/hvab040]. Tumor tissue samples were obtained at three time points, either from the primary tumor or metastatic sites: (1) prior to treatment, (2) following one cycle of neo-adjuvant chemotherapy, and (3) during surgery. For patients who were considered unfit for cytoreductive surgery, a tumor tissue biopsy was collected in the same window as the pre-planned surgery when possible. The obtained tumor tissue samples were immediately snap-frozen and subsequently formalin-fixed and paraffin-embedded (FFPE).
Endpoints
The primary objective of this clinical trial is to assess the immune-activating capacity of neo-adjuvant pembrolizumab combined with conventional chemotherapy. Initially, we planned to measure this through the expansion of tumor-specific T cells in peripheral blood76,77,78. However, recent studies have shown that PD-1 blockade primarily induces intratumoral immune effects75, and neo-antigen recognition in ovarian cancer significantly differs between circulating T lymphocytes and TILs79. Consequently, we have shifted our focus to evaluating the intratumoral immune-activating dynamics using multiplexed immunofluorescence and immune gene expression profiling.
Secondary endpoints included efficacy as assessed by radiological and pathological response to treatment, PFS, OS, safety, and feasibility.
Exploratory endpoints included the association between (pathologic) response and immunologic, transcriptomic, and genomic findings, including PD-1/PD-L1 expression on cancer and immune cells, tumor mutational burden, immune-related gene signatures and T-cell infiltration. Furthermore, the use of circulating tumor DNA (ctDNA) to monitor treatment response was evaluated.
Pathological assessments and immunohistochemistry
Histopathological assessment of biopsies and resection samples was performed by two experienced gynecology pathologists (HMH and ALC) independently in Slide Score (www.slidescore.com), without knowledge of each patient’s clinical history and treatment results. Hematoxylin and eosin (H&E) stained sections were used to assess tumor cell percentage, tumor-stroma ratio, stromal tumor infiltrating lymphocytes (according to the International Immuno-Oncology Biomarker Working Group80), tertiary lymphoid structures, giant cells, neutrophils, eosinophils, and pathologic response. In case of disagreement, a consensus decision was reached.
Pathologic response was evaluated using omental tissue resected at surgery. If omental tissue was not available, a peritoneal metastasis or primary tumor was used. Evaluation was based on the validated chemotherapy response score (CRS) by Böhm et al.44. Briefly, CRS 1 is characterized by no or minimal tumor response, that is mainly viable cancer cells with no or minimal regression-associated fibroinflammatory changes, limited to a few foci; CRS 2 is characterized by appreciable tumor response amid viable tumor that is readily identifiable, or regression-associated fibroinflammatory changes with multifocal residual tumor, which are easily identifiable; CRS 3 is characterized by complete or near-complete response with no residual tumor or minimal irregularly scattered tumor foci seen as individual cells, cell groups, or nodules up to a maximum size of 2 mm. Mainly regression-associated fibroinflammatory changes are observed. In our study, patients with CRS 3 were categorized as major pathologic responders. Since the study by Böhm et al. did not show a clear prognostic separation of CRS 1 and CRS 2, minor pathologic responders were defined as either CRS 1 or CRS 2. After the consensus meeting, the final score was compared to the initial report for all resected tissues. In cases where the pathology report described a multifocal vital tumor, the HE slides were again reviewed and CRS score was adjusted if appropriate.
For baseline PD-L1 immunohistochemistry, paraffin sections were used for the PD-L1 IHC 22C3 1/40 dilution pharmDx qualitative immunohistochemical assay on a DAKO Autostainer 48 system at the NKI laboratory. PD-L1 expression on cancer and immune cells, as well as the combined positive score (CPS) was assessed. Tumor proportion score (TPS) was defined as the percentage of viable tumor cells showing PD-L1 expression at the membrane. Immune proportion score (IPS) was defined as the percentage of TILs showing PD-L1 expression either at the membrane or cytoplasm. CPS was defined as the number of PD-L1–staining cells (cancer cells, lymphocytes, and macrophages) divided by the total number of viable cancer cells, multiplied by 10081.
For double staining, PD1 (yellow) was detected first using clone CAL20 (1:250, 60 min at 37 °C), visualized with Anti-Mouse NP and Anti-NP AP (both 12 min at 37 °C), followed by the Discovery Yellow detection kit (Ventana Medical Systems). Next, CD8 (purple) was detected using clone C8/144B (1:200, 32 min at 37 °C), visualized with Anti-Mouse HQ and Anti-HQ HRP (both 12 min at 37 °C), followed by the Discovery Purple Detection Kit (Ventana Medical Systems). Slides were counterstained with Hematoxylin and Bluing Reagent.
Radiological response evaluation
Radiological assessment of the response was performed using CT scans after two cycles of neo-adjuvant treatment according to RECIST v1.1. by an experienced gynecology radiologist (ML) who was blinded to the outcome. Objective response rate (ORR) was defined as the proportion of patients with partial (reduction of at least 30% in the size of the tumor) or complete response (the disappearance of all detectable signs of the tumor) to treatment with chemotherapy plus pembrolizumab after two neoadjuvant cycles of treatment.
Survival
Progression-free survival (PFS) was defined as the time from initiation of treatment to disease recurrence, progression, or death from any cause, whichever occurred first. Disease recurrence or progression was confirmed by imaging (according to RECIST v1.1) or elevated CA125 concentrations, according to the Gynecologic Cancer InterGroup criteria82. Overall survival (OS) was defined as the time from initiation of treatment to death from any cause. PFS was censored at the date of last contact (i.e., physical or telephone consultation) and OS was censored at the date last known to be alive for patients who had no evidence of disease and remained alive, respectively, with a cutoff date of April 1, 2024.
Safety and feasibility
Safety was assessed according to the Common Terminology Criteria for Adverse Events (CTCAE) v.4.03. Grades 2 and higher were reported from the onset of treatment until 30 days after treatment cessation. A safety follow-up visit was conducted approximately 30 days after the last dose of trial treatment or before the initiation of a new anticancer treatment, whichever came first. Serious AEs were reported until 90 days after the last trial treatment or until the initiation of a new anticancer treatment, whichever came first.
Feasibility was assessed based on any treatment-related complications leading to delays in surgery beyond 6 weeks after the last neo-adjuvant cycle, or unexpected post-surgery complications. Additionally we considered the relative dose intensity (RDI, %), a composite measure that accounts for dose reductions and treatment delays. To calculate the RDI for each individual study drug we used the following formula: (Delivered total dose, in mg/m2)/(actual time to complete chemotherapy with imputation for missed cycles, in days) divided by (Standard total dose, in mg/m2)/(standard time to complete chemotherapy, in days) * 100. In cases where a cycle was not administered, a dose of 0 mg was imputed for ‘delivered total dose’, the dose of the last cycle administered was imputed for ‘standard total dose’, and the standard cycle duration according to protocol was imputed for both ‘actual time to complete chemotherapy’ and ‘standard time to complete chemotherapy’.
Multiplexed immunofluorescence
Analysis of tumor immune cell infiltrates was performed by multiplexed immunofluorescence technology on a Ventana Discovery Ultra automated stainer, using the Opal 6-Plex Detection Kit (50 slides kit, Akoya Biosciences, cat NEL871001KT). 3-μm FFPE sections were cut and heated at 75 °C for 28 min and subsequently deparaffinized with Discovery Wash. Pretreatment was performed with Discovery CC1 buffer for 64 minutes at 95 °C, after which Discovery Inhibitor was applied for 8 minutes to block endogenous peroxidase activity. Specific markers were detected consecutively on the same slide with the following antibodies, Anti-CD68(Clone KP1, Cat M0814, DAKO, 1/300 dilution 1 h at RT), anti-CD8 (Clone C8/144B, Cat M7103, DAKO, 1/100 dilution, 1 h at RT), anti-FoxP3 (clone 236 A/E7, Cat ab20034, Abcam, 1/100 dilution, 2 h at RT), anti-CD3 (Clone SP7, Cat RM-9107-S, Thermo Scientific, 1/400 dilution 1 h at RT), Anti-PanCK (Clone AE1/AE3, cat MS343P, Thermo Scientific, 1/100 dilution, 2 h at RT) and Anti-CD20 (Clone L26, Cat M0755, DAKO, 1/500 dilution, 1 h at RT). Each staining cycle was composed of four steps: Primary Antibody incubation, Opal polymer HRP Ms+Rb secondary antibody incubated for 1 h at RT, OPAL dye incubation (OPAL480, OPAL520, OPAL570, OPAL620, OPAL690, OPAL780, 1/40 or 1/50 dilution as appropriate for 1 hour at RT) and an antibody denaturation step using CC2 buffer for 20 minutes at 95 °C. Cycles were repeated for each new antibody to be stained. At the end of the protocol slides were incubated with DAPI (1/25 dilution in Reaction Buffer) for 12 minutes. After the run was finished slides were washed with demi water and mounted with Fluoromount-G (Southern Biotech, cat 0100-01) mounting medium.
Multispectral imaging and quantitative image analysis
After staining, slides were imaged using the PhenoImager HT automated imaging system (Akoya). Scans were made with the MOTiF protocol. Using the InForm software version 2.5.0 the MOTIF images were unmixed into 8 channels: DAPI, OPAL480, OPAL520, OPAL570, OPAL620, OPAL690, OPAL780 and Auto Fluorescence and exported to a multilayered TIFF file. The multilayered TIFF’s were fused with HALO software (version 3.2, Indica Labs) to create one file for each sample. The HALO software (V3.5.3577.285, Indica Labs) was used for image-analysis. A trained biopathologist manually annotated the regions of interest (ROI) to be analyzed. The random forest classifier was used to distinguish between tumor and stroma within the ROI. Fat tissue areas and artifacts (necrosis, tissue folding, massive blood cell areas, non-specific auto-fluorescent structures) were manually excluded. The Indica Labs Highplex FL v4.0.2 analysis algorithm was used for analysis using AI nuclei segmentation. All annotation layers were analyzed and both the summary data and cell object data were exported. Several rules were employed to determine the cell type of each segmented cell: All cells positive for PanCK were classified as cancer cells, regardless of other positive markers. Cells positive for both CD3 and CD20 were classified as CD3 if the CD3 intensity was higher than the CD20 intensity. If CD20 intensity was higher than the CD3 intensity, cells were classified as B cells. Cells positive for CD68 were classified as macrophages. Cells without positive markers were classified as unknown. Cytotoxic and regulatory CD3 positive cells were distinguished based on their positivity for CD8 or FOXP3, respectively. The number of immune cells within 150 micron of a cancer cell, within either the stromal or intratumoral compartments, was quantified and normalized to the total surface area within 150 micron of cancer cells. Visual verification of the results was done in QuPath83. Additional information on antibodies used is provided in the Life Sciences Reporting Summary.
DNA and RNA isolation
DNA and RNA were isolated from fresh frozen tumor samples, if available, or from FFPE samples. The pathologist scored tumor percentage and indicated tumor region, relevant for isolation, on a consecutive H&E stained cryostat frozen section (5 µm) slide (Using SlideScore). DNA and RNA was isolated simultaneously from frozen sections (10 µm) with the AllPrep DNA/RNA/miRNA Universal isolation kit (Qiagen, 80224) by using the QIAcube, according to manufacturer’s protocol and from FFPE sections with the AllPrep DNA/RNA FFPE isolation kit (Qiagen, #80234) by using the QIAcube, according to manufacturer’s protocol. To evaluate germline variants and variants associated with clonal hematopoiesis of indeterminate potential (CHIP), DNA was isolated from patient-matched blood cell pellets using the DNeasy Blood & Tissue Kit (Qiagen).
Whole-transcriptome sequencing
The quality and quantity of total RNA isolated from FF and FFPE samples were assessed on a 2100 Bioanalyzer using a NanoChip (Agilent, Santa Clara, CA, USA). The percentage of RNA fragments >200 nt fragment distribution values (DV200) was determined using the region analysis method according to the manufacturer’s instructions (Illumina, technical-note-470-2014-001).
Strand-specific libraries were generated using the TruSeq RNA Exome Library Prep Kit (Illumina Inc., San Diego, CA, USA) according to the manufacturer’s instructions (Illumina, # 1000000039582v01). Briefly, intact total RNA was fragmented (94 °C for 8 min), randomly primed, and reverse transcribed using SuperScript II Reverse Transcriptase (Invitrogen, part # 18064-014) with the addition of actinomycin D (fragmentation was omitted for FFPE total RNA). Second-strand synthesis was performed using Polymerase I and RNaseH with dTTP substituted for dUTP. Generated cDNA fragments were 3’-end adenylated and ligated to IDT xGen UDI(10 bp)-UMI(9 bp) paired-end sequencing adapters (Integrated DNA Technologies, Inc., Coralville) followed by 15 cycles of PCR. Libraries were validated on a 2100 Bioanalyzer using a 7500 chip (Agilent, Santa Clara, CA, USA) followed by 1-4 plex library pooling containing up to 200 ng of each sample.
The pooled libraries were enriched for target regions using the Probe Coding Exome Oligos set (CEX, 45MB) according to the manufacturer’s instructions (Illumina, # 1000000039582v01). Briefly, cDNA libraries and biotin-labeled capture probes were combined and hybridized using a denaturation step at 95 °C for 10 minutes and an incubation step of 94 °C to 58 °C with a ramp of 18 cycles of 1 minute incubation and 2 °C per cycle. Hybridized target regions were captured using streptavidin magnetic beads and subjected to two stringency washes, an elution step, and a second round of enrichment, followed by purification using AMPure XP beads (Beckman, A63881) and PCR amplification for 10 cycles. The target-enriched pools were analyzed on a 2100 Bioanalyzer using a 7500 chip (Agilent, Santa Clara, CA, USA), diluted, and then pooled equimolar into a multiplex sequencing pool.
Libraries were sequenced with 54 paired-end reads on a NovaSeq6000 using a Reagent Kit v1.5 (100 cycles) (Illumina Inc., San Diego, CA, USA). Transcript abundance was estimated using Salmon (v1.10.0)84, with hg38.p13 and Gencode v42 as reference, removing unexpressed pseudogenes and keeping only transcripts with transcript support level of 1-3. Salmon was run with GC-bias correction to reduce differences between snap-frozen and FFPE samples. Transcripts were summarized to gene level before downstream analysis. Resulting read count were normalized using DESeq2 and log2-transformed. Gene set scores were calculated using Singscore85. Hallmark gene sets were obtained from MSigDB86. The interferon-γ signature was obtained from the publication of Ayers et al.87. The MHC class-I signature consisted of the expressed MHC genes HLA-A,B,C,E and F, and B2M. Immune cell subtypes abundances were estimated by taking the average expression of marker genes88. Transcriptome sequencing samples where the corresponding exome sequencing sample had a FACETS purity estimates below 0.1 were excluded from the analysis.
Whole-exome sequencing
For each tumor tissue and cell pellet sample the amount of double stranded DNA was quantified by using the Qubit dsDNA HS Assay Kit (Invitrogen, cat no Q32851). A maximum amount of 2 μg of double stranded genomic DNA was fragmented by covaris AFA technology to obtain fragment sizes of 200-300 bp. Samples were purified using Agencourt AMPure XP Reagent (Beckman Coulter, cat no A63881) in a 2x reaction volume settings according to manufacturer’s instructions. The fragmented DNA was quantified and qualified on a BioAnalyzer system using the DNA7500 assay kit (Agilent Technologies cat no. 5067- 1506). With a maximum input amount of 1 μg fragmented DNA, NGS library preparation for Illumina sequencing was performed using the KAPA Hyper Prep Kit (KAPA Biosystems, KK8504) in combination with xGen UDI-UMI Adapters of IDT 10 bp (Integrated DNA Technologies). During the library amplification step 4 cycles of PCR were performed to obtain enough yield for the exome enrichment assay. All DNA libraries were quantified on a BioAnalyzer system using the DNA7500 assay kit. Exome enrichment was performed on library pools of 9 unique dual indexed libraries, 500 ng each, using the xGen™ Exome Hyb Panel v2 (IDT, cat no 10005152) and xGen™ Hybridization Capture Core Reagents according to manufacturer’s protocol were hybridization time was adjusted to 20 hours and 10 cycles of PCR were performed during post-capture PCR. All exome-enriched library pools were quantified on a BioAnalyzer system using the DNA7500 assay kit, pooled equimolar to a final concentration of 10 nM and subjected to paired-end 100 bp sequencing on an Illumina Novaseq 6000 instrument using a NovaSeq 6000 S4 Reagent Kit v1.5 (Illumina, 20028313) according to the manufacturer’s instructions.
After seqpurge89 adapter trimming, reads were aligned with bwa (v0.7.17)90 mem algorithm to GRCh38, duplicates were marked UMI-aware using rumidup (https://github.com/NKI-GCF/rumidup), and base call qualities were re-calibrated. Somatic variants were called for paired tumor and blood sample alignments using Mutect2 from the genome analysis toolkit (GATK, v4.3)91. For paired samples, tumor purity and ploidy were estimated using FACETS92. Samples with a FACETS purity estimate of less than 0.1 were excluded from analysis. Mutation load was estimated by dividing the number of filtered Mutect2 somatic variant calls by the total length of the targeted exome sequence, multiplied by one million. BRCA1/2 germline variants were called with Freebayes (v1.3.6). Homologous recombination deficiency signatures were calculated as described by Popova et al.93, based on the genomic copy number profiles estimated from the whole-exome sequencing reads. The copy number profiles were estimated using CNVkit (v0.9.10)94 using default settings with the addition of the “—drop-low-coverage” option as recommended for whole exome sequencing data. From these copy number profiles, the number of large-scale state transitions were calculated; that is, the number of breakpoints between segments longer than 3 Mb, after filtering out smaller variations as described by Popova et al. Ploidy estimates were obtained from FACETS as mentioned above. Samples were then classified as homologous recombination deficient based on the thresholds described by Popova et al.; that is, near-diploid samples with more than 15 large-scale state transitions, and near-tetraploid samples with more than 20 large-scale state transitions were classified as homologous recombination deficient. We used only the baseline DNA samples to estimate homologous recombination deficiency.
ctDNA analysis
Circulating cell-free DNA (ccfDNA) was extracted from blood plasma using QIAsymphony DSP Circulating DNA Kit (Qiagen). To identify tumor-derived mutations detectable in plasma, 6 to 50 ng of ccfDNA was sequenced using the AVENIO ctDNA Expanded Kit V2 in accordance with the manufacturer’s instructions (Roche). True circulating tumor DNA (ctDNA) mutations were defined as either 1) present in tumor tissue and plasma DNA and absent in cell pellet DNA, or 2) present in plasma DNA with a variant allele frequency >1% and absent in tumor tissue and cell pellet DNA. Based on these criteria, individual droplet digital PCR (ddPCR) assays were designed for 39 tumor-derived mutations identified in 29 patients through plasma sequencing (Supplementary Table 10) using the Bio-Rad PrimePCR Mutation Detection Design tool (Bio-Rad) and ordered at Integrated DNA Technologies (IDT). ddPCR analysis was performed on the QX600 Droplet Digital PCR System (Bio-Rad) following the manufacturer’s recommendations. ddPCR results were interpreted based on experimentally determined Limit of Blank (LoB) following a CLSI EP17 protocol95. A difference in ctDNA levels was defined as a relative change of more than 30% in accordance with the technical variance of ddPCR analyses96.
Historical cohort formation
We identified all patients diagnosed with stage IV high-grade serous ovarian cancer who received neo-adjuvant chemotherapy at the Netherlands Cancer Institute between November 1, 2017 and May 2, 2022. After approval by the NKI Institutional Review Board (file number IRBd23-332), data were extracted from the medical records of patients who met the inclusion and exclusion criteria of the Neo-Pembro study. Pathologic response was assessed by two experienced gynecology pathologists (HMH and ALC) utilizing the CRS classification on omental tissue obtained during surgery, in accordance with the approach employed in the study cohort. Patients who experienced disease progression during neo-adjuvant treatment were categorized as minor responders. The comparison of pathologic response and survival between the historical and the study cohorts was a post-hoc analysis.
Statistical considerations
This exploratory study had no formal sample size calculation as the data were descriptive in nature.
Descriptive statistics for continuous variables included total counts, mean, median, standard deviation (SD), range and interquartile range (IQR). Continuous variables between groups or strata were compared using parametric (t-test/ANOVA) or non-parametric (Mann Whitney U-test/Kruskall-Wallis test) tests depending on the distribution. Categorical variables were presented as frequencies and percentages, and comparisons were done using Fisher’s exact test.
Differences between major and minor pathologic responders were analyzed using the nonparametric Wilcoxon’s rank-sum test (Mann–Whitney U-test), whereas differences between time points within the subgroups were analyzed using paired Wilcoxon’s signed-rank test.
The Kaplan-Meier method was used to generate time-to-event curves, from which medians, 95% CIs, and survival probabilities were calculated. Survival curves were compared using the log-rank test. Hazard ratios (HRs) and their corresponding 95% CIs were estimated using Cox proportional hazards models. Due to the limited sample size, univariable analyses were adjusted for prognostic factors individually to assess their individual effects on the time-to-event outcomes. Specifically, age, Charlson comorbidity index, BRCA status, interval between neo-adjuvant therapy and surgery, interval from surgery to adjuvant therapy, and result of cytoreduction were examined separately owing to their clinical relevance. The assumption of proportionality of hazards was assessed using tests for independence between scaled Schoenfeld residuals and time, as well as by visual inspection of scaled Schoenfeld residuals versus time with a smoothed curve and 95% CI. Subgroup analyses and comparisons between radiologic and pathologic response were performed post hoc. All reported p values are two sided and a threshold of p < 0.05 was used for statistical significance. Only significant p values are displayed. No adjustments were made for multiple comparisons. All statistical analyses were performed using SAS (v9.4) and R software (v4.2.2).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The study protocol and statistical analysis plan are available in Supplementary Note 1 and 2. The RNA and DNA data generated in this study have been deposited in the European Genome-Phenome Archive under accession code EGAS50000000781. Both genomic data and deidentified clinical data are available under restricted access for academic use, within the limitations of the provided informed consent and under General Data Protection Regulation law. Requests for access to genomic data can be submitted via https://ega.nki.nl/, while requests for clinical data should be sent to [email protected]. All data requests will be reviewed by the NKI Institutional Review Board (IRB) and must be supported by the Principal Investigator of the study. The researcher will need to sign a data access agreement with the NKI after approval. Estimated time to response is 2–4 weeks. All remaining data can be found in the Article, Supplementary and Source data files. Source data are provided with this paper.
References
Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 11, 3801 (2020).
Varga, A. et al. Pembrolizumab in patients with programmed death ligand 1-positive advanced ovarian cancer: Analysis of KEYNOTE-028. Gynecol. Oncol. 152, 243–250 (2019).
Hamanishi, J. et al. Safety and antitumor activity of anti-pd-1 antibody, nivolumab, in patients with platinum-resistant ovarian cancer. J. Clin. Oncol. 33, 4015–4022 (2015).
Lawrence, M. S. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013).
Hwang, W. T., Adams, S. F., Tahirovic, E., Hagemann, I. S. & Coukos, G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: a meta-analysis. Gynecol. Oncol. 124, 192–198 (2012).
Webb, J. R., Milne, K., Kroeger, D. R. & Nelson, B. H. PD-L1 expression is associated with tumor-infiltrating T cells and favorable prognosis in high-grade serous ovarian cancer. Gynecol. Oncol. 141, 293–302 (2016).
Böhm, S. et al. Neoadjuvant chemotherapy modulates the immune microenvironment in metastases of tubo-ovarian high-grade serous carcinoma. Clin. Cancer Res 22, 3025–3036 (2016).
Webb, J. R., Milne, K. & Nelson, B. H. PD-1 and CD103 are widely coexpressed on prognostically favorable intraepithelial cd8 t cells in human ovarian cancer. Cancer Immunol. Res. 3, 926–935 (2015).
Disis, M. L. et al. Efficacy and safety of avelumab for patients with recurrent or refractory ovarian cancer: phase 1b results from the javelin solid tumor trial. JAMA Oncol. 5, 393–401 (2019).
Varga, A. et al. Antitumor activity and safety of pembrolizumab in patients (pts) with PD-L1 positive advanced ovarian cancer: Interim results from a phase Ib study. J. Clin. Oncol. 33, 5510–5510 (2015).
Matulonis, U. A. et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: results from the phase II KEYNOTE-100 study. Ann. Oncol. 30, 1080–1087 (2019).
Monk, B. J. et al. Chemotherapy with or without avelumab followed by avelumab maintenance versus chemotherapy alone in patients with previously untreated epithelial ovarian cancer (JAVELIN Ovarian 100): an open-label, randomised, phase 3 trial. Lancet Oncol. 22, 1275–1289 (2021).
Moore, K. N. et al. Atezolizumab, bevacizumab, and chemotherapy for newly diagnosed stage iii or iv ovarian cancer: placebo-controlled randomized Phase III Trial (IMagyn050/GOG 3015/ENGOT-OV39). J. Clin. Oncol. 39, 1842–1855 (2021).
Pujade-Lauraine, E. et al. Avelumab alone or in combination with chemotherapy versus chemotherapy alone in platinum-resistant or platinum-refractory ovarian cancer (JAVELIN Ovarian 200): an open-label, three-arm, randomised, phase 3 study. Lancet Oncol. 22, 1034–1046 (2021).
Kurtz, J. E. et al. Atezolizumab combined with bevacizumab and platinum-based therapy for platinum-sensitive ovarian cancer: placebo-controlled randomized Phase III ATALANTE/ENGOT-ov29 trial. J. Clin. Oncol. 41, 4768–4778 (2023).
Zamarin, D. et al. Randomized phase II trial of nivolumab versus nivolumab and ipilimumab for recurrent or persistent ovarian cancer: an nrg oncology study. J. Clin. Oncol. 38, 1814–1823 (2020).
Liu, J. F. et al. Assessment of combined nivolumab and bevacizumab in relapsed ovarian cancer: a phase 2 clinical trial. JAMA Oncol. 5, 1731–1738 (2019).
Blank, C. U. et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat. Med. 24, 1655–1661 (2018).
Cloughesy, T. F. et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 25, 477–486 (2019).
Vos, J. L. et al. Neoadjuvant immunotherapy with nivolumab and ipilimumab induces major pathological responses in patients with head and neck squamous cell carcinoma. Nat. Commun. 12, 7348 (2021).
Chalabi, M. et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat. Med 26, 566–576 (2020).
Huang, A. C. et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat. Med 25, 454–461 (2019).
Powles, T. et al. Clinical efficacy and biomarker analysis of neoadjuvant atezolizumab in operable urothelial carcinoma in the ABACUS trial. Nat. Med. 25, 1706–1714 (2019).
Forde, P. M. et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N. Engl. J. Med. 378, 1976–1986 (2018).
Liu, J. et al. Improved efficacy of neoadjuvant compared to adjuvant immunotherapy to eradicate metastatic disease. Cancer Discov. 6, 1382–1399 (2016).
Peng, J. et al. Chemotherapy induces programmed cell death-ligand 1 overexpression via the nuclear factor-κb to foster an immunosuppressive tumor microenvironment in ovarian cancer. Cancer Res 75, 5034–5045 (2015).
Apetoh, L., Ladoire, S., Coukos, G. & Ghiringhelli, F. Combining immunotherapy and anticancer agents: the right path to achieve cancer cure? Ann. Oncol. 26, 1813–1823 (2015).
Galluzzi, L., Humeau, J., Buqué, A., Zitvogel, L. & Kroemer, G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 17, 725–741 (2020).
Hanna, R. K. et al. Predictors of reduced relative dose intensity and its relationship to mortality in women receiving multi-agent chemotherapy for epithelial ovarian cancer. Gynecologic Oncol. 129, 74–80 (2013).
Denduluri, N. et al. Dose delays, dose reductions, and relative dose intensity in patients with cancer who received adjuvant or neoadjuvant chemotherapy in community oncology practices. J. Natl Compr. Cancer Netw. J. Natl Compr. Canc Netw. 13, 1383–1393 (2015).
Wonders, K. Y., Schmitz, K. & Harness, J. Dose delays, dose reductions, and relative total dose intensity in patients with advanced cancer who exercised during neoadjuvant chemotherapy treatment. Integr. Cancer Ther. 22, 15347354231168368 (2023).
Thommen, D. S. et al. A transcriptionally and functionally distinct PD-1(+) CD8(+) T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med. 24, 994–1004 (2018).
Verschoor, Y. L. et al. Neoadjuvant atezolizumab plus chemotherapy in gastric and gastroesophageal junction adenocarcinoma: the phase 2 PANDA trial. Nat. Med. 30, 519–530 (2024).
Wieser, V. et al. BRCA1/2 and TP53 mutation status associates with PD-1 and PD-L1 expression in ovarian cancer. Oncotarget 9, 17501–17511 (2018).
Strickland, K. C. et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 7, 13587–13598 (2016).
Kandalaft, L. E., Dangaj Laniti, D. & Coukos, G. Immunobiology of high-grade serous ovarian cancer: lessons for clinical translation. Nat. Rev. Cancer 22, 640–656 (2022).
Timmermans, M., Sonke, G. S., Van de Vijver, K. K., van der Aa, M. A. & Kruitwagen, R. No improvement in long-term survival for epithelial ovarian cancer patients: A population-based study between 1989 and 2014 in the Netherlands. Eur. J. Cancer 88, 31–37 (2018).
Cohen, P. A. et al. Pathological chemotherapy response score is prognostic in tubo-ovarian high-grade serous carcinoma: A systematic review and meta-analysis of individual patient data. Gynecologic Oncol. 154, 441–448 (2019).
Santoro, A. et al. Pathological chemotherapy response score in patients affected by high grade serous ovarian carcinoma: the prognostic role of omental and ovarian residual disease. Front Oncol. 9, 778 (2019).
Michaan, N., Chong, W. Y., Han, N. Y., Lim, M. C. & Park, S. Y. Prognostic value of pathologic chemotherapy response score in patients with ovarian cancer after neoadjuvant chemotherapy. Int. J. Gynecologic Cancer 28, 1676 (2018).
Lawson, B. C. et al. A 3-Tier Chemotherapy Response Score for Ovarian/Fallopian Tube/Peritoneal High-grade Serous Carcinoma: Is it Clinically Relevant? The American J. Surgic. Pathol. 44, 206–213 (2020).
Lee, J. Y. et al. External validation of chemotherapy response score system for histopathological assessment of tumor regression after neoadjuvant chemotherapy in tubo-ovarian high-grade serous carcinoma. J. Gynecol. Oncol. 28, e73 (2017).
Rajkumar, S. et al. Prognostic implications of histological tumor regression (Böhm’s score) in patients receiving neoadjuvant chemotherapy for high grade serous tubal & ovarian carcinoma. Gynecologic Oncol. 151, 264–268 (2018).
Böhm, S. et al. Chemotherapy response score: development and validation of a system to quantify histopathologic response to neoadjuvant chemotherapy in tubo-ovarian high-grade serous carcinoma. J. Clin. Oncol. 33, 2457–2463 (2015).
Ditzel, H. M. et al. Assessment of a chemotherapy response score (CRS) system for tubo-ovarian high-grade serous carcinoma (HGSC). Int J. Gynecol. Pathol. 38, 230–240 (2019).
Singh, P. et al. The chemotherapy response score is a useful histological predictor of prognosis in high-grade serous carcinoma. Histopathology 72, 619–625 (2018).
Coghlan, E. et al. Prognostic role of histological tumor regression in patients receiving neoadjuvant chemotherapy for high-grade serous tubo-ovarian carcinoma. Int J. Gynecol. Cancer 27, 708–713 (2017).
Sharma, P., Hu-Lieskovan, S., Wargo, J. A. & Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723 (2017).
Lo, C. S. et al. Neoadjuvant chemotherapy of ovarian cancer results in three patterns of tumor-infiltrating lymphocyte response with distinct implications for immunotherapy. Clin. Cancer Res 23, 925–934 (2017).
Lee, Y. J. et al. Dynamics of the tumor immune microenvironment during neoadjuvant chemotherapy of high-grade serous ovarian cancer. Cancers (Basel) 14, 2308 (2022).
How, J. A. et al. Pembrolizumab plus chemotherapy in frontline treatment of advanced ovarian cancer: Clinical and translational results from a phase 2 trial. Med. (2024).
Leary, A. et al. Neoadjuvant chemotherapy alters the balance of effector to suppressor immune cells in advanced ovarian cancer. Cancer Immunol. Immunother. 70, 519–531 (2021).
Tavira, B. et al. Analysis of tumor microenvironment changes after neoadjuvant chemotherapy with or without bevacizumab in advanced ovarian cancer (GEICO-89T/MINOVA Study). Clin. Cancer Res 30, 176–186 (2024).
Kroeger, D. R., Milne, K. & Nelson, B. H. Tumor-infiltrating plasma cells are associated with tertiary lymphoid structures, cytolytic t-cell responses, and superior prognosis in ovarian cancer. Clin. Cancer Res 22, 3005–3015 (2016).
Chaurio, R. A. et al. TGF-β-mediated silencing of genomic organizer SATB1 promotes Tfh cell differentiation and formation of intra-tumoral tertiary lymphoid structures. Immunity 55, 115–128.e119 (2022).
Kasikova, L. et al. Tertiary lymphoid structures and B cells determine clinically relevant T cell phenotypes in ovarian cancer. Nat. Commun. 15, 2528 (2024).
Mesnage, S. J. L. et al. Neoadjuvant chemotherapy (NACT) increases immune infiltration and programmed death-ligand 1 (PD-L1) expression in epithelial ovarian cancer (EOC). Ann. Oncol. 28, 651–657 (2017).
González-Martín, A. et al. Atezolizumab Combined With Platinum and Maintenance Niraparib for Recurrent Ovarian Cancer With a Platinum-Free Interval >6 Months: ENGOT-OV41/GEICO 69-O/ANITA Phase III Trial. J Clin Oncol, 42, 4294–4304 (2024).
Fusi, A. et al. PD-L1 expression as a potential predictive biomarker. Lancet Oncol. 16, 1285–1287 (2015).
Hsiehchen, D. et al. DNA Repair Gene Mutations as Predictors of Immune Checkpoint Inhibitor Response beyond Tumor Mutation Burden. Cell Rep. Med. 1, 100034 (2020).
van Vugt, M. A. T. M. & Parkes, E. E. When breaks get hot: inflammatory signaling in BRCA1/2-mutant cancers. Trends Cancer 8, 174–189 (2022).
Monk, B. J. et al. Niraparib first-line maintenance therapy in patients with newly diagnosed advanced ovarian cancer: final overall survival results from the PRIMA/ENGOT-OV26/GOG-3012 trial. Ann Oncol 35, 981–992 (2024).
Musacchio, L. et al. Combining PARP inhibition and immune checkpoint blockade in ovarian cancer patients: a new perspective on the horizon? ESMO Open 7, 100536 (2022).
Monk, B. J. et al. LBA30 ATHENA-COMBO, a phase III, randomized trial comparing rucaparib (RUCA) + nivolumab (NIVO) combination therapy vs RUCA monotherapy as maintenance treatment in patients (pts) with newly diagnosed ovarian cancer (OC). Ann. Oncol. 35, S1223–S1224 (2024).
Harano, K. et al. Neoadjuvant combination treatment of olaparib and pembrolizumab for patients with HRD-positive advanced ovarian cancer. J. Clin. Oncol. 42, 5545–5545 (2024).
Harter, P. et al. Durvalumab with paclitaxel/carboplatin (PC) and bevacizumab (bev), followed by maintenance durvalumab, bev, and olaparib in patients (pts) with newly diagnosed advanced ovarian cancer (AOC) without a tumor BRCA1/2 mutation (non-tBRCAm): Results from the randomized, placebo (pbo)-controlled phase III DUO-O trial. J. Clin. Oncol. 41, LBA5506–LBA5506 (2023).
Vergote, I. et al. ENGOT-OV43/KEYLYNK-001: A phase III, randomized, double-blind, active- and placebo-controlled study of pembrolizumab plus chemotherapy with olaparib maintenance for first-line treatment of BRCA-nonmutated advanced epithelial ovarian cancer. J. Clin. Oncol. 37, TPS5603–TPS5603 (2019).
Katsumata, N. et al. Dose-dense paclitaxel once a week in combination with carboplatin every 3 weeks for advanced ovarian cancer: a phase 3, open-label, randomised controlled trial. Lancet 374, 1331–1338 (2009).
Chan, J. K. et al. Weekly vs. Every-3-Week Paclitaxel and Carboplatin for Ovarian Cancer. N. Engl. J. Med 374, 738–748 (2016).
Clamp, A. R. et al. Weekly dose-dense chemotherapy in first-line epithelial ovarian, fallopian tube, or primary peritoneal cancer treatment (ICON8): overall survival results from an open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 23, 919–930 (2022).
Pignata, S. et al. Carboplatin plus paclitaxel once a week versus every 3 weeks in patients with advanced ovarian cancer (MITO-7): a randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 15, 396–405 (2014).
van der Leest, P. & Schuuring, E. Critical Factors in the Analytical Work Flow of Circulating Tumor DNA-Based Molecular Profiling. Clin. Chem. 70, 220–233 (2024).
Christinat, Y. et al. Normalized LST Is an Efficient Biomarker for Homologous Recombination Deficiency and Olaparib Response in Ovarian Carcinoma. JCO Precision Oncology 7, e2200555 (2023).
Sztupinszki, Z. et al. Comparative Assessment of Diagnostic Homologous Recombination Deficiency–Associated Mutational Signatures in Ovarian Cancer. Clin. Cancer Res. 27, 5681–5687 (2021).
Gangaev, A. et al. Differential effects of PD-1 and CTLA-4 blockade on the melanoma-reactive CD8 T cell response. Proc. Natl Acad. Sci. USA 118, e2102849118 (2021).
Andersen, R. S. et al. Parallel detection of antigen-specific T cell responses by combinatorial encoding of MHC multimers. Nat. Protoc. 7, 891–902 (2012).
Hadrup, S. R. et al. High-throughput T-cell epitope discovery through MHC peptide exchange. Methods Mol. Biol. 524, 383–405 (2009).
Hadrup, S. R. et al. Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers. Nat. Methods 6, 520–526 (2009).
Bobisse, S. et al. Sensitive and frequent identification of high avidity neo-epitope specific CD8+T cells in immunotherapy-naive ovarian cancer. Nat. Commun. 9, 1092 (2018).
Salgado, R. et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: recommendations by an International TILs Working Group 2014. Ann. Oncol. 26, 259–271 (2015).
Burtness, B. et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet 394, 1915–1928 (2019).
Rustin, G. J. et al. Definitions for response and progression in ovarian cancer clinical trials incorporating RECIST 1.1 and CA 125 agreed by the Gynecological Cancer Intergroup (GCIG). Int J. Gynecol. Cancer 21, 419–423 (2011).
Bankhead, P. et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 7, 16878 (2017).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Foroutan, M. et al. Single sample scoring of molecular phenotypes. BMC Bioinforma. 19, 404 (2018).
Liberzon, A. et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Ayers, M. et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest 127, 2930–2940 (2017).
Danaher, P. et al. Gene expression markers of Tumor Infiltrating Leukocytes. J. Immunother. Cancer 5, 18 (2017).
Sturm, M., Schroeder, C. & Bauer, P. SeqPurge: highly-sensitive adapter trimming for paired-end NGS data. BMC Bioinforma. 17, 208 (2016).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv: Genomics 1303.3997 (2013).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Shen, R. & Seshan, V. E. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res 44, e131 (2016).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. (Austin) 6, 80–92 (2012).
Talevich, E., Shain, A. H., Botton, T. & Bastian, B. C. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol. 12, e1004873 (2016).
Vessies, D. C. L. et al. An automated correction algorithm (ALPACA) for ddPCR data using adaptive limit of blank and correction of false positive events improves specificity of mutation detection. Clin. Chem. 67, 959–967 (2021).
van der Leest, P. et al. Circulating tumor DNA as a biomarker for monitoring early treatment responses of patients with advanced lung adenocarcinoma receiving immune checkpoint inhibitors. Mol. Oncol. 15, 2910–2922 (2021).
Popova, T. et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 72, 5454–5462 (2012).
Bouwman, P. et al. Functional categorization of BRCA1 variants of uncertain clinical significance in homologous recombination repair complementation assays. Clin. Cancer Res. 26, 4559–4568 (2020).
Acknowledgements
We thank all the patients and their families and caregivers for participating in the present study. We thank medical oncologists C.H. Smorenburg and J. Stouthard, gynecological oncologists C. Lok, H. Trum, F. Amant, and clinical trial nurses L. Boog and S. Veldstra for patient care; K. Kersten and K. Hoefakker for processing of samples; D. Peters, W. Kievit, S. Cornelissen and the Core Facility of Molecular Pathology and Biobanking for their support in processing samples and performing immunohistochemistry; M. Nieuwland, W. Brugman, C. van Steenis, R.J.C. Kluin and the Genomics Core Facility for their support in sequencing and data analysis; L. Valkenet and Y. Wijnands from the scientific administration department for data management; T. Kroon and K. van den Berg for trial monitoring; N. Brandhorst and K. Hoefakker for sample processing; T. Korse, M. Lucas and E. Platte for PBMC acquisition and processing; P. Kvistborg and A. Gangaev for providing scientific input during design of the study. The study was funded by MSD and the funder was involved in study design. The funder had no role in data collection, data analysis, data interpretation, or writing of the report. The manuscript was reviewed by MSD before submission.
Author information
Authors and Affiliations
Contributions
S.L.A. coordinated trial procedures, analyzed and interpreted clinical and translational data and drafted the manuscript. G.S.S. was the coordinating principal investigator and was responsible for funding acquisition. B.H. performed and interpreted bioinformatics analyses, under supervision of L.F.A.W. S.N.K. coordinated trial procedures and wrote the trial protocol. A.L.-C., H.M.H., and J.S. performed the histo- and immunopathological scoring and annotated tumor areas for IHC quantification analysis. M.L.-Y. performed statistical analyses of clinical data. M.A.R., S.L.A., S.N.K., G.S.S., and W.J.v.D. were responsible for patient enrollment and patient care. I.K. and M.J.H. were responsible for patient care. M.J.L. revised CT scans. M.M.A. performed processing of samples and staining, under supervision of A.B. I.S. and S.L.A. performed digital imaging analysis using HALO software. R.H. performed quantification of cells using QuPath. P.v.d.L. and D.v.d.B. performed ctDNA analysis and interpreted the data together with S.L.A., W.J.v.D., and G.S.S. I.A.M.M. supported in trial conduct and management. All authors reviewed and approved the manuscript. All authors were involved in critically revising the report. All authors approved the final version and took responsibility for the decision to submit the manuscript for publication.
Corresponding author
Ethics declarations
Competing interests
G.S.S. has received institutional research support from MSD, Agendia, AstraZeneca, Roche, and Novartis, and consulting fees from Biovica and Seagen. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Aronson, S.L., Thijssen, B., Lopez-Yurda, M. et al. Neo-adjuvant pembrolizumab in stage IV high-grade serous ovarian cancer: the phase II Neo-Pembro trial. Nat Commun 16, 3520 (2025). https://doi.org/10.1038/s41467-025-58440-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58440-y
