
Abstract
High-entropy alloys (HEAs) are promising catalysts particularly adept for reactions involving multiple intermediates and requiring multifunctional active sites. However, conventional syntheses often result in either (kinetically) random-mixing HEA or (thermodynamically) phase-separated composites-both fail to fine-tune local structures and further optimizing their performances. Here we present finely tailoring the local ensembles in HEA catalysts through rational composition design and sequential pulsed annealing. Employing PdSnFeCoNi HEA as a model, pulsed annealing (e.g., 0.5 s heating at 1300 K for 30 cycles) leverages differences in enthalpic interactions and surface energies to control the formation of ultrafine PdSn clusters within the HEA matrix, yielding the heterostructured HEA/c-PdSn. Compared with random HEAs and commercial Pd/C, HEA/c-PdSn exhibits >5 − 10-fold higher mass activity and good stability (>90.6% retention after 2000 cycles) for ethanol oxidation. This enhancement arises from the synergy between active local ensembles and the multifunctional HEA matrix, which reduces overall limiting potential, mitigates sluggish C-C/C-H breaking, and enhances structural stabilization. Our findings provide a strategy for engineering heterostructured HEAs for broad catalytic applications.
Similar content being viewed by others
Introduction
High entropy alloy (HEA) nanoparticles, characterized by a single-phase solid solution consisting of five or more elements, have recently gained prominence as effective catalysts1,2,3. Their multi-elemental composition and unique high-entropy structures enable a wide range of tunable activities and enhanced stability4,5,6. Particularly in complex reactions involving multiple intermediates, HEA electrocatalysts demonstrate high performance attributed to their multifunctional catalytic surfaces populated with diverse active sites7,8,9,10,11,12. In addition, the broad compositional space in HEAs allows for the exploration of a myriad of different elemental combinations to further optimize the properties13,14,15,16. However, despite being beneficial for multifunctional catalysis, HEAs inevitably result in diluted active sites for each specific intermediate, resulting in limited overall catalysis2,14,17. For example, the ethanol oxidation reaction (EOR) involves multiple intermediates in a 12-electron transfer process where HEA catalysts are deemed beneficial to lower the overall limiting potentials by providing multifunctional active sites. However, the strong C-C and C-H bond-breaking steps often require ensembled adsorption and activation on multiple atom clustering, making the random mixing in HEA ineffective13,14,15,16. Therefore, it would be ideal if one could construct high-density and well-defined active sites within the multi-functional HEAs to achieve activity-multifunctionality-stability harmony.
Local structural modulation and active site design are ubiquitously explored in conventional catalysts, including engineering facets, core-shell structures, and local ordering18,19,20,21. Particularly, structural ensemble sites, which consist of multiple active atoms (referred to as “clusters”) on a catalyst surface22, can operate cooperatively to bind complex or large reactants and effectively activate strong bonds that require ensemble adsorption, e.g., C-C and C-H bond-breaking and hydrocarbon oxidation23,24,25. With the right ensemble configuration (local structure), catalysts can obtain improved activity by allowing for optimal binding, and higher selectivity by promoting specific reaction pathways, while still having improved stability by distributing the catalytic activity across multiple atoms or sites25,26,27,28,29. Hence, there is a compelling need for fine-tuning the nanoscale or local structure of HEAs to further optimize their catalytic properties, going beyond mere compositional design.
Unfortunately, current HEA catalysts are formed mostly under strongly non-equilibrium conditions to ensure multielement random mixing, whose local structures and active sites are often challenging to modulate during the rapid and non-equilibrium synthesis30,31,32,33,34,35,36. Moreover, accounting for the disparate physicochemical properties and small size of HEA catalysts, it is also a significant challenge to tailor their local structure and active site ensembles by post treatments2,15,37. In fact, conventional methods lead to either random mixing HEAs by strongly non-equilibrium synthesis or phase-separated composites using near-equilibrium methods1,30,32,38. At present, there is no widely recognized method that enables dynamic tuning of local order or ensemble structures in the HEA catalysts, thus posing great challenges to further optimize their performance through ensemble activation, multielement synergy, and high entropy stabilization.
In this work, we report a finely controlled pulsed annealing approach combining rational compositional design to synthesize HEA catalysts with dynamically tunable local structures and ensembles. Specifically, in the PdSnFeCoNi HEA, ultrafine PdSn clusters can controllably emerge from the HEA matrix to form heterostructured HEA/c-PdSn (Fig. 1a). The HEA was initially synthesized through rapid high-temperature thermal shock for uniform alloying, followed by a lower-temperature pulsed annealing (~1300 K, 0.5 s, 30 cycles) to facilitate the in-situ formation of PdSn nanoclusters, as guided by their strong interaction and formation energy (−0.532 eV/atom). The pulsed annealing was meticulously controlled to promote the emergence and controllable growth of PdSn clusters from the HEA matrix while avoiding inter-particle sintering, vigorous phase separation, and excessive nanocluster growth, issues commonly associated with traditional annealing. The finely tailored HEA/c-PdSn demonstrates a significantly enhanced EOR activity, benefiting from the multifunctionality of the HEA matrix to lower the overall activation limiting potentials and the site-specific reactivity of PdSn/FeCoNi local clustering to largely mitigate C-C and C-H bond breaking, resulting in a much smoother energy landscape for EOR (Fig. 1b). Therefore, our method provides a viable path for precisely tailoring the local structure and enthalpy-entropy interplay toward a broad distribution of heterostructured HEA catalysts.

a Traditional HEA, PdSnFeCoNi, exhibits a homogeneous solid-solution structure. By applying pulsed annealing (~1300 K, 0.5 s, 30 times), active nanoclusters (e.g., PdSn) can precipitate from the HEA matrix, forming HEA/c-PdSn with well-balanced multifunctionality and site-specific reactivity. Excessively annealing can result in a phase-separated structure that reaches thermodynamic equilibrium and exhibits limited catalytic function and activity. b Schematic EOR process on conventional catalysts (e.g., Pd/C) and HEA/c-PdSn. The overall multifunctionality inherent in the HEA matrix and the site-specific reactivity facilitated by PdSn local clustering enable a much smoother energy landscape and largely enhanced EOR. RDS rate-determining steps.
Results and discussion
Compositional design and pulsed-annealing synthesis of HEA/c-PdSn
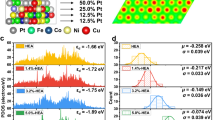
A high-entropy structure with active/ensemble sites firmly embedded in the HEA surface holds promise for achieving superior catalytic performance, yet the method of its construction remains enigmatic. The contribution of entropy to HEA formation is well established, with high entropy stabilizing the single-phase structure, as demonstrated in our previous studies2,28. In this study, the active sites embedded in the HEA surface were deliberately designed based on the binary formation energy between each element (e.g., their enthalpic interactions, Fig. 2a and Table S1 derived from the Open Quantum Materials Database39) and surface energy. Taking the system comprising Pd, Sn, Fe, Co, and Ni elements as an example, the combinations of Fe, Co, and Ni exhibit minimal values (close to 0) for binary formation energy, thus facilitating the formation of a single-phase solid solution. In contrast, the interaction between Pd and Sn demonstrates a more negative formation energy (−0.579 eV/atom, Fig. 2b) compared to the Pd-Fe (−0.080 eV/atom), Pd-Co (~0 eV/atom), and Pd-Ni (~0 eV/atom), indicating a stronger propensity for PdSn bonding and ordering39. This characteristic builds the thermodynamic foundation to enable the construction of PdSn clusters within the HEA matrix.

a The formation energy between each element, guiding the compositional design of heterostructured HEAs. b The formation energy of Pd-Sn, Pd-Fe, Pd-Co, and Pd-Ni, highlighting the ordering and clustering of PdSn. c The surface energy of Pd, Sn, Fe, Co, Ni, FeCoNi, PdSnFeCoNi, Pd2Sn2FeCoNi, and PdSn, indicating easy surface segregation of PdSn. d The temperature profiles used in the synthesis of HEA/c-PdSn, including high-temperature thermal shock and pulsed annealing. e Comparison of the size distribution and (f–h) TEM images of the HEA samples before and after pulsed annealing (PA), and furnace annealing (FA). i, j Elemental mappings of the HEA samples (i) before and (j) after pulsed annealing. The sample before PA shows uniform mixing of each element (i.e., Pd, Sn, Fe, Co, and Ni) throughout the nanoparticle. After PA, the elements of Fe, Co, and Ni remain in uniform mixing, while the Pd and Sn emerge from the HEA matrix, forming nanoclusters embedded in the HEA matrix. Error bars represent standard deviations calculated from nanoparticle size distributions measured by TEM image analysis using ImageJ software. Source data are provided as a Source Data file.
In addition, surface energy also plays a significant role in tailoring the local ensembles in heterostructured HEA. Based on the valence-based surface-energy model, the surface energies of PdSnFeCoNi (HEA) solely depend on the distribution of atoms on the top layers of the surfaces40,41. The surface energies of HEA are negatively linearly correlated with the descriptor Ж, which are determined by the group and period number (Ng and Np), valence-electron number (Sv), and electronegativity (χ) of the surface atoms, which is defined as Ж = \({(\frac{{N}_{{{{\rm{p}}}}}}{\bar{N}_{{{{\rm{p}}}}}})}^{(\sqrt{{N}_{{{{{\rm{g}}}}}^{-}}}\sqrt{{\bar{N}}_{{{{\rm{g}}}}}})}\times \frac{{S}_{{{{\rm{v}}}}}^{2}}{\chi }\) (Note S1). The descriptors of alloys are defined as the geometric mean of the descriptors of the corresponding pure metals. Notably, Pd and Sn have large descriptors (ЖPd = 47.1, ЖSn = 118.0, ЖFe = 35.0, ЖCo = 43.1, and ЖNi = 52.4) and thus easily segregate to the surface to reduce the surface energy (Fig. 2c). Therefore, Pd-Sn bonding is likely thermodynamically favored and easily diffuses to the surface, thus forming PdSn clusters on the surfaces. Note the unique catalytic functions of these metal elements in the EOR were also factored into the compositional design (discussed later).
Guided by the compositional design above, we first employed a high-temperature thermal shock strategy to synthesize a homogeneous PdSnFeCoNi HEA, featuring a strongly non-equilibrium process that enables single-phase alloying. We then applied pulsed annealing (PA) to promote the emergence of PdSn ensemble sites within the HEA matrix. Specifically, we utilized radiative Joule heating with the temperature profiles illustrated in Fig. 2d (for details, see the Experiment section). The synthesis of HEA starts with a rapidly high-temperature synthesis at ~1700 K for 0.5 s to ensure efficient multi-elemental mixing. This is followed by pulsed annealing at ~1300 K for 0.5 s per pulse, repeated 30 times. The pulsed annealing gradually induces the formation of PdSn clusters within the HEA matrix, driven by their lower thermodynamic potential. Both the samples before and after pulsed annealing displayed nanoparticles uniformly distributed on carbon black, with an average nanoparticle size of ~20 nm (Fig. 2e–g and Figure S1), and a single-phase face-centered-cubic structure revealed by X-ray diffraction (XRD) (Figure S2). For comparison, we annealed the HEA sample using a traditional furnace set to 1000 K for 30 minutes in an Ar atmosphere, as the furnace annealing (FA) process. After FA, the HEA sample exhibited significant particle aggregation and growth (with sizes approximating ~33 nm) and notable phase separation (Fig. 2h and Figs. S3–4). This underscores the efficacy of our innovative pulsed annealing method in mitigating particle growth/aggregation and phase separation while enabling finely tailored clustering.
To elucidate the microstructural evolution, we conducted further characterization using a transmission electron microscope (TEM) and elemental mappings. Prior to PA, we observed uniform elemental mixing of Fe, Co, Ni, Sn, and Pd throughout the HEA nanoparticle (Fig. 2i). After PA, while Fe, Co, and Ni maintained homogenous mixing within the HEA matrix, there were noticeable PdSn clusters precipitated from the HEA (Fig. 2j). The distribution density of PdSn clusters in HEA/c-PdSn was roughly ~11.5% ± 3.6% (Figure S5). Pd and Sn are often localized on the nanoparticle surface, likely attributable to their lower surface energy as we calculated above. Notably, their XRD and TEM results revealed no macroscopic phase separation or demixing in HEA/c-PdSn, implying that the pulsed annealing predominantly leads to changes in elemental redistribution and localized clustering without causing serious sintering and phase separation. Our pulsed annealing approach offers a versatile method for forming metal clusters within the HEA matrix, as demonstrated by the PdBiFeCoNi HEA composition (Figure S6). This method also has the potential to produce catalysts at a large scale (Figure S7).
Detailed characterization and formation mechanism of HEA/c-PdSn
To further study the chemical state and microstructure of HEA/c-PdSn, we employed X-ray photoelectron spectroscopy (XPS), X-ray adsorption spectrum (XAS), and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). Notably, the XPS feature peaks of Pd and Sn spectra in HEA/c-PdSn shift to a lower binding energy compared to HEA (Fig. 3a and Figure S8), indicating decreased chemical valence for the two metallic elements. The Pd K-edge X-ray absorption near-edge structure (XANES) spectra provided further insight into the electronic structure alterations within the HEA/c-PdSn (Fig. 3b). A conspicuous downshift in the absorption edge energy for the Pd K-edge in HEA/c-PdSn relative to the pristine HEA substantiates the reduced valence state of Pd, aligning with the XPS observations. This valence change may be attributed to enhanced charge-transfer processes resulting from the formation of PdSn clusters42,43.

a XPS of Pd spectra for HEA and HEA/c-PdSn. b XANES and (c) WT of Pd K edge for Pd foil, HEA, HEA/c-PdSn. d Coordination number of HEA and HEA/c-PdSn in terms of Pd-Pd/Sn, Pd-Fe/Co/Ni, and total CN. e HADDF-STEM image of HEA/c-PdSn from the view of [110], including five nanodomains with five-fold symmetry. f Enlarged HADDF-STEM image in the red dash frame of (e), dark atom columns colored blue and bright atom columns colored yellow. g The intensity of the line scan in the white frame of (f). The error bars represent standard deviations derived from the covariance matrix in the least-squares fitting process of the synchrotron radiation data. Source data are provided as a Source Data file.
The detailed local bonding and coordination environments are further elucidated by extended X-ray absorption fine structure spectroscopy (EXAFS). Wavelet transform (WT) analysis is employed, with its high resolution in both reciprocal (K) space and real space, allowing for the discernment of atoms sharing similar coordination environments and proximities44. Interestingly, the WT intensity maximum for the HEA/c-PdSn was observed at elevated positions compared to the pristine HEA (Fig. 3c). This upward shift in K regions is indicative of an increased proportion of heavier elements (Pd and Sn) in the vicinity of Pd atoms, which also demonstrates the aggregation of Pd and Sn after the pulsed annealing process. Further, Fourier transform EXAFS (FT-EXAFS) fitting of the Pd first shell in pristine HEA reveals a Pd-Pd/Sn coordination number (CN) of 8.7 and a Pd-Fe/Co/Ni CN of 3 (Fig. 3d, Figure S9 and Table S2), whereas a Pd-Pd/Sn CN of 6 and a Pd-Fe/Co/Ni CN of 1 were found in HEA/c-PdSn. The relative escalation in the proportion of Pd-Pd/Sn CN within HEA/c-PdSn furnishes further evidence of Pd and Sn aggregation after pulsed annealing. Intriguingly, a reduction in the total CN from 11.7 to 7.3 is observed following pulsed annealing, intimating segregation of PdSn atoms toward the surface45, corroborating TEM findings.
Additionally, HAADF-STEM was employed to examine the atomic structures of HEA/c-PdSn (Fig. 3e). A multi-grain nanoparticle with five-fold symmetry encompasses five domains of varying sizes. From the FFTs of each ___domain (Figure S10), we ascertain that the lattice constants across these five domains vary insignificantly, with differences below 2%. This suggests that PdSn precipitation, induced by pulsed annealing, does not induce noteworthy lattice strain. Elemental mappings (Figure S11) of this nanoparticle display a concentration of PdSn in particular domains, indicating the sporadic emergence of PdSn within the HEA. Line profiling reveals heightened atomic intensity in the PdSn-rich region along the [110] direction (Fig. 3f, g), confirming the formation of PdSn nanoclusters.
The above characterizations prove the presence of PdSn nanoclusters in the HEA/c-PdSn, which can be attributed to the alloy’s composition and pulsed annealing. As illustrated in Fig. 2, Pd-Sn bonding is propelled by robust Pd-Sn pairwise interactions and is likely thermodynamically favored, given it maximizes Pd-Sn bonds with the most substantial formation energy. In contrast, bonds like Pd-Fe, Pd-Co, and Pd-Ni possess lesser formation energies and are not evident in the final configuration. In addition, the PdSn has a lower surface energy compared to other variants such as FeCoNi and PdSnFeCoNi. Therefore, during the pulsed annealing process, the propensity for PdSn clusters to diffuse to the surface can be ascribed to the controllable, thermodynamic-driven transformation of the homogeneous PdSnFeCoNi region into heterostructure HEA/c-PdSn with the outward migration of PdSn atoms from the interior. Notably, configurational entropy plays a critical role in stabilizing the HEA matrix during pulsed annealing. While the enthalpy or strong interactions between Pd and Sn drive local clustering, the entropy among PdSnFeCoNi stabilizes the entire HEA matrix, enabling the formation of the heterostructured HEA/c-PdSn.
Multielement synergy in HEA/c-PdSn for EOR
We now display how such a unique structure is particularly advantageous for catalytic applications in EOR, which is applied for direct ethanol fuel cells46,47,48. Compared to hydrogen, methanol, and formic acid used in fuel cells, ethanol has the advantages of low toxicity, high energy density, and high boiling point, making it safer to store and transport46,47,48. The complete oxidation of ethanol to CO2 and H2O is a complex 12-electron transfer process that involves multiple intermediate reactions such as adsorption dehydrogenation, C-C bond breaking, and adsorption and desorption of intermediates (e.g., carbon monoxide), which particularly require ensemble active sites for effective adsorption and dissociation46,47,48. Although HEA nanocatalysts are suitable for such multi-step catalytic reactions, they are not always the best ones due to the random mixing structure and averaging effect.
To investigate the critical role of HEA/c-PdSn catalysts toward EOR, we tested the catalytic performance of commercial Pd/C, FeCoNi, PdSn, HEA, and HEA/c-PdSn, and compared their mass activities (Fig. 4a, b), where the current was normalized by the noble metal mass of each catalyst. Mass activities were recorded by cyclic voltammetry (CV) curves of these catalysts, tested in 1 M KOH + 1 M EtOH solution with the scan rate of 20 mV s−1 moving from 0.05 to 1.10 V (vs. reversible hydrogen electrode (RHE)). The FeCoNi sample is almost inactive. Remarkably, the HEA/c-PdSn has a lower starting peak potential at ~0.79 V compared to the Pd/C (~0.86 V), PdSn (~0.80 V), and HEA (~0.82 V), denoting good activity. At their respective peak potentials, the mass activity of the HEA/c-PdSn (11.87 A mg−1Pd) significantly surpassed that of the Pd/C (1.14 A mg−1Pd), PdSn (1.90 A mg−1Pd), and HEA (2.11 A mg−1Pd). The performance of the HEA/c-PdSn aligns with that of state-of-the-art catalysts for EOR in terms of their mass activity (Fig. 4c)26,49,50,51,52,53,54.

a, b CV curves and mass activities at different potentials of HEA/c-PdSn, HEA, PdSn, FeCoNi, and commercial Pd/C (tested in the Ar-saturated aqueous solution containing 1.0 M KOH and 1.0 M EtOH at a scan rate of 20 mV s−1). c Performance comparison of HEA/c-PdSn with other reported catalysts in terms of mass activity at their corresponding peaks. d Free energy profiles of FeCoNi, PdSn, and HEA/c-PdSn in the EOR process including dehydrogenation, C-C bond breaking, and oxidation. PDS potential determined steps in DFT calculation. The optimized computational models are provided in Supplementary Data 1. e The maximal reaction energy of FeCoNi, PdSn, and HEA/c-PdSn in the EOR process based on DFT calculations. f The reaction energies of specific-intermediate (C-C bond, *CH oxidation) on FeCoNi, PdSn, and HEA/c-PdSn surface in the EOR process based on DFT calculations. g CV curves of HEA/c-PdSn at the initial and 2000th cycles. h CO stripping curves of HEA/c-PdSn, PdSn alloy, and commercial Pd/C. i Temperature programmed reduction spectra of HEA and HEA/c-PdSn. Source data are provided as a Source Data file.
To understand the origin of the better performance of HEA/c-PdSn, we performed density functional theory (DFT) calculations to analyze the EOR processes on a single-layer PdSn cluster embedded in the FeCoNi surface (corresponding to HEA/c-PdSn), PdSn alloy surface, and FeCoNi alloy surface, by comparing all potential reaction pathways with respect to their adsorption and reaction energies. The atomic coordinates of the optimized computational models are provided in Supplementary Data 1. The EOR process from ethanol to carbon dioxide involves several intricate processes, including dehydrogenation, C-C bond breaking, and oxidation (Fig. 4d). In particular, the maximum reaction energy is 0.69 eV (*CH2CHO → *CHCHO) on the HEA/c-PdSn surface compared with 0.93 eV (*CHCO → *CH + *CO) on the PdSn alloy, and 1.77 eV (*CH + *OH → *CHOH) on the FeCoNi alloy surface (Fig. 4e). This indicates that EOR on the heterostructure HEA/c-PdSn catalysts has a much lower overall limiting potential and importantly, the traditional potential-determined steps (PDS) of C-C and C-H bond breaking are no longer the PDSs for HEA/c-PdSn, indicating significant changes in the reaction pathways.
We then analyzed the site-specific reactivity for C-C and C-H bond breaking (Fig. 4f). We identified the relatively stable configurations of the intermediates during the EOR process (Figure S12). It has been found that most adsorption and reactions occur at the edge of FeCoNi and PdSn, which exhibit multiple adsorption sites. This leads to stronger adsorption energies for intermediates and lower dehydrogenation reaction energies. For example, the cleavage of the C-H bond from *CH3CH2OH to *CHCHO consistently takes place at the edges of PdSn clusters since PdSn provides lower potential reaction energies for C-H bond breaking compared with the FeCoNi matrix (Fig. 4d). The intermediates *CHCHO and *CHCO tend to be adsorbed at the boundary between PdSn clusters and FeCoNi, causing C-C bond breaking to occur at the FeCoNi matrix with the regulation of the PdSn clusters. Whereas the CO oxidation returns to take place at the top site of the edge of the PdSn clusters. In other words, the synergistic effect of both FeCoNi and PdSn promotes the EOR process. In comparison, the individual FeCoNi or PdSn provides only a single type of active site.
FeCoNi can effectively break C-C bonds but performs poorly in *CH oxidation (C-H breaking), while PdSn facilitates the oxidation of *CH intermediates but is inefficient at C-C breaking. This corroborates the fact that conventional catalysts have limited active sites and can only facilitate specific steps in a multistep reaction, resulting in constrained catalytic activity. In contrast, the HEA/c-PdSn catalyst can significantly accelerate these PDSs through site-specific reactivity derived from the synergy between the PdSn clusters and the FeCoNi-dominated matrix. The combination of multifunctionality and site-specific reactivity in HEA/c-PdSn renders an overall smoother energy landscape for the EOR, leading to significantly improved catalytic activity. In addition, we found that the reaction energy for *CO oxidation on the HEA/c-PdSn surface slightly decreases compared to FeCoNi and PdSn, demonstrating improved resistance to CO poisoning. These results prove the significance of heterostructured HEA/c-PdSn for overall multifunctionality and site-specific reactivity, thus synergistically benefiting complex catalysis.
Furthermore, we investigated the stability of the HEA/c-PdSn sample using continuous CV scans (Fig. 4g) and chronoamperometry (Figure S13). After 2000 cycles, the mass density of HEA/c-PdSn at the peak had decreased by ~9.4%, as evidenced by the CV curves. The stability performance of HEA/c-PdSn was superior to both the HEA (PdSnFeCoNi) and commercial Pd/C catalysts24. In chronoamperometry tests (at 0.45 V vs. RHE, 7200 s), the steady current density of HEA/c-PdSn (~0.09 A mg−1Pd) was notably higher than that of Pd/C (~0.01 A mg−1Pd) and PdSn (~0.03 A mg−1Pd). After the EOR, the HEA/c-PdSn nanoparticles remain uniformly dispersed on the carbon black substrate, while retaining the PdSn clusters (Figure S14). The excellent stability of HEA/c-PdSn can be attributed to its high-entropy structure and the stable interface between the PdSn clusters and the FeCoNi matrix. The surface stability of alloys under the electrochemical conditions can be measured by the gradient electronic descriptor ψ‘ of surface atoms according to the reported surface-ejection-energy model (ψ‘ = ψ1/ψ02, here ψ1 is the geometric mean of descriptor ψ of the atoms at the active sites and ψ0 is the electronic descriptor of the ejected atom)55. Compared with Fe, Co, and Ni, Pd and Sn exhibit the large descriptor ψ0 (ψPd = 45.4, ψSn = 100, ψFe = 35.0, ψCo = 43.1, and ψNi = 52.4), leading to the small gradient descriptor ψ‘ and high stability on surfaces, in terms of the positively linear relationship between ψ‘ and ejection energies. Fe, Co, and Ni thus are preferentially ejected followed by Pd and Sn. Therefore, the emergence of PdSn clusters on HEA/c-PdSn surfaces maintains the high activity of the surfaces during the CV cycles, contributing to the high activity and stability in HEA/c-PdSn.
In addition, the DFT results show that the *CO oxidation to *COOH is the essential process in the EOR path and CO easily poisons the catalysts. We thus conducted CO stripping experiments to evaluate the CO tolerance for the Pd/C, PdSn, and HEA/c-PdSn (Figure S15). CO oxidation peaks manifested in the initial forward scan, whereas in the subsequent scan, these peaks were absent, attributed to the total removal of CO from the catalyst surface. All three catalysts demonstrated CO anti-poisoning capability. However, the HEA/c-PdSn with its lower CO oxidation peak at ~0.81 V, outperformed the the Pd/C (~0.87 V) and PdSn (~0.86 V) (Fig. 4h). The CO temperature-programmed desorption (CO-TPD) profiles for HEA and HEA/c-PdSn (Fig. 4i) showed that the HEA/c-PdSn, exhibiting a peak at ~560 °C, binds CO more weakly compared to the HEA (~590 °C). This characteristic is advantageous for EOR, as rapid oxidative removal of CO enhances both catalytic activity and stability. Our experimental and theoretical results further demonstrate that the HEA-coordinated PdSn plays a critical role in affecting the activity and stability of the PdSn active sites, through the multielement synergy and ensemble effect.
Finely tailoring and ensemble effect of HEA/c-PdSn in EOR
Our pulsed annealing method, which offers flexible control over both the annealing temperature and duration, enables precise modulation of the number or size of active sites on the HEA matrix. To demonstrate the structural modulation and ensemble active sites of HEA/c-PdSn (Fig. 5a), we employed three distinct pulsed annealing (PA) profiles: a pulse temperature of 1300 K, a pulse duration of 0.5 s, and varied pulse counts (5, 30, and 80 times). These profiles were used to anneal HEAs and assess their subsequent EOR performance. The increase in pulsed annealing times results in a slight increase in particle size (~1–3 nm, Figure S16). From the EDS characterization, a lower pulse number (e.g., 5 times) is not enough to induce obvious migration of PdSn, while a higher pulse number (e.g., 80 times) leads to the formation of phase-separated nanoparticles because of over-annealing, resulting in composite PdSn/FeCoNi. Thus, an intermediate pulse number (e.g., 30 times) is necessary to promote the precipitation of PdSn nanoclusters strongly embedded in the HEA matrix and keep a high-entropy environment.

a Schematic and elemental mappings of various HEAs annealed at different temperatures (i.e., 900 K, 1300 K, and 1700 K) for 0.5 s with 30 heating cycles. b CV curves and (c) mass activities and peak potential of various catalysts annealed at different temperatures for EOR (Ar-saturated solution containing 1.0 M KOH and 1.0 M EtOH at a scan rate of 20 mV s−1). d Energy schematic of widely distributed, dynamically tunable heterostructured HEAs achieved by using pulsed annealing. Source data are provided as a Source Data file.
We tested the catalytic performance of these HEA catalysts annealed at different conditions and compared their mass activities (Fig. 5b, c). Compared to traditional HEA (i.e., before pulsed annealing, 2.11 A mg−1Pd at peak potential), all HEA catalysts after pulsed annealing showed enhanced mass activity (6.09 A mg−1Pd, 11.87 A mg−1Pd, and 2.40 A mg−1Pd at peak potentials for HEA-PA-1300-5, HEA-PA-1300-30, and HEA-PA-1300-80, respectively). Notably, samples after pulsed annealing with fewer (5 times, i.e., HEA-PA-1300-5) or more (80 times, i.e., HEA-PA-1300-80) pulses showed lower mass activities compared to HEA-PA-1300-30, indicating an obvious ensemble effect that requires just-right size clusters. Moreover, conventional furnace-annealed HEA shows a poor mass activity of only 0.56 A mg−1Pd due to sintered particles as well as phase-separated structures that have very limited multielement synergy and highly active interfaces (Figure S17). In addition, the corresponding Tafel slope (Figure S18) of HEA-PA-1300-30 was 183.7 mV dec−1, which is lower than that of HEA-PA-1300-5 (197.4 mV dec−1), HEA-PA−1300-80 (199.6 mV dec−1), and commercial Pd/C (227.0 mV dec−1). This indicates that the ethanol oxidation rate and reaction kinetics of HEA-PA-1300-30 are significantly enhanced compared to the other catalysts.
We also conducted nuclear magnetic resonance (NMR) to investigate the product selectivity of HEA-PA-1300-5, HEA-PA-1300-30, and commercial Pd/C. HEA-PA-1300-30 exhibited a significantly higher yield of C1 products, achieving a high Faradaic efficiency of 64.1% (Figure S19). This efficiency surpasses those of HEA-PA-1300-5 (9.6%) and commercial Pd/C (3.5%), indicating that the formation of PdSn clusters within the HEA matrix effectively promotes the C1 pathway. Therefore, the HEA-PA-1300-30 demonstrates the highest catalytic activity, suggesting the critical role of finely tailoring the local structures with the right-size active PdSn nanoclusters (cluster density: ~11.5% ± 3.6%, Figure S5) in achieving high performance: either too small/faint PdSn (i.e., no distinct clusters) or over-annealed PdSn phase separation (>50% cluster density) all leads to inferior performances.
The experimental results were further analyzed based on DFT calculations with varied sizes: the 5-atom-width PdSn, 9-atom-width PdSn, and 9-atom-width double-layer PdSn on HEAs (Figure S20). With the increase of the size of PdSn nanoclusters, the C-H bond breaking can be easier activated: the reaction energies of *CHCH2O → *CHCHO step are 0.98 eV, 0.88 eV, and 0.84 eV, respectively. In contrast, the smaller size of the PdSn nanoclusters can promote the C-C bond breaking thermodynamically, while this process turns to endothermic on the double-layer PdSn clusters. Additionally, the smaller PdSn clusters on the HEA surface exhibit better CO oxidation activity, with the reaction energies of *CO + *OH → *COOH step 0.51 eV, 0.57 eV, and 0.6 eV for 5-atom-width, 9-atom-width, and the double-layer PdSn nanoclusters, respectively. The above calculation revealed that the optimal or appropriate size of the PdSn clusters balances the reaction potential of C-H and C-C bond breaking and CO oxidation on the HEA surface, leading to a minimum potential of the overall EOR performance.
Notably, many reactions require a careful balance between multifunctionality and site-specific reactivity, particularly for complex multistep reactions, corroborating the necessity and significance of heterostructured HEAs. Importantly, different reactions often demand varying local clusters and ensemble sizes, further highlighting the versatility and adaptability of heterostructured HEAs. Benefited by our rational composition design and flexible pulsed annealing method in adjusting the local structures, we are able to achieve a broad distribution of dynamically tunable heterostructured HEAs that span between non-equilibrium and equilibrium states (Fig. 5d). Such finely tailored catalysts can be suitable for a range of complex multistep catalysis reactions.
In conclusion, we developed a pulsed annealing method to synthesize heterostructured HEA/c-PdSn catalysts with controllable PdSn active nanoclusters embedded in the HEA matrix. The formation of such a heterostructured HEA is based on the composition design considering their formation energy and surface energy, whose structure can be further finely tailored by adjusting the pulsed annealing temperatures and durations to control the emerging and growth of clustering. As a proof-of-concept, the HEA/c-PdSn was used for EOR catalysis and demonstrated a high mass activity of 11.87 A mg−1Pd and good stability (>90.6% retention after 2000 cycles), substantially outperforming traditional HEA, PdSn, and commercial Pd/C catalysts. Theoretical and experimental results demonstrate that such high performance is attributed to the appropriate size and dispersion of PdSn ensemble active sites as well as their intimate synergy with the HEA matrix to largely lower overall limiting potentials, mitigate C-C/C-H bond breaking, and promote structural stability. Our work provides a viable way for local structural control and property optimization of HEA catalysts, which can be potentially applied to a series of multistep complex reactions necessitating ensemble activation.
Methods
Chemicals
The metal salts such as palladium chloride (CAS: 7647-10-1, 99.99%), tin chloride (CAS: 7772-99-8, 99%), iron (III) chloride (CAS: 7647-10-1, 99.99%), cobalt chloride hexahydrate (CAS: 7791-13-1, 98%), nickel chloride hexahydrate (CAS: 13478-00-7, 99.99%), and commercial 10.0 wt% Pd/C was purchased from were purchased from Aladdin. Commercial carbon black (Vulcan XC72R, Cabot) was utilized as the substrate.
Material preparation
The precursor solution (0.05 mol L−1) was prepared by dissolving various metal salts with an equimolar concentration in ethanol. The mixed non-noble metal solution was added into carbon black with a targeted loading concentration and then sonicated for 2.0 h. The mixture was dried in a vacuum drying oven to achieve precursor-loaded carbon black. A pulsed heating procedure was used to synthesize our catalysts. The obtained powders were subjected to high-temperature shock by a commercial carbon cloth heating element driven by a DC power supply (MR50040, BK PRECISION). The corresponding temperature was captured by an Endurance IR camera (E1RH-R59-V-0-0) with a temperature resolution of ±2 K and a response time of 10 ms.
Materials characterization
The morphology of the catalysts was examined by a JEOL 2100 TEM at the acceleration voltages of 200 kV. HRTEM and HAADF-STEM images were performed on a Talos F200X with a probe spherical aberration corrector at an operation voltage of 300 kV. The chemical state and electron structure of catalysts were probed using X-ray absorption spectroscopy (XAS) at the Australian Synchrotron (ANSTO). Measurements at both Pd K-edge and Sn K-edge were conducted utilizing a Si (311) monochromator crystal at the Hutch B experimental station. All samples were tested using fluorescence mode at room temperature. Energy calibration was performed based on Pd and Sn metal foils. Subsequent data processing via the Athena and Artemis in the Demeter package according to the standard data analysis procedures. Morlet wavelet transformation (WT) is employed to map the EXAFS data to (k, R) space56.
Electrocatalytic tests
A three-electrode system connected with CHI 760 electrochemical workstation (and electrochemical station (CX310X, Corrtest, China) was used for the test. A glassy carbon (5 mm in diameter) was used as the working electrode. The counter electrode was the Pt sheet and the reference electrode was the calomel (saturated KCl) or Ag/AgCl: (saturated KCl). The electrolytes of 0.5 M H2SO4, 1.0 M KOH with 1.0 M ethanol or without ethanol were used. In terms of electrode preparation, 5 mg catalysts were dispersed in 1.0 mL solution composed of water (200.0 μL), isopropanol (800.0 μL) and 5.0 wt% Nafion (20.0 μL) and then sonicated for 30.0 min. The as-prepared ink 10.0 μL was dropped onto the glassy carbon for the working electrode, and further dried at room temperature. The potentials were converted to that versus reversible hydrogen electrode (RHE) according to the Nernst equation (ERHE = Ecalomel + 0.0591 pH + 0.242) for calomel. All potentials reported in three-electrode half-cell in this work are versus RHE without iR-compensation, and all electrolytes were purged with Ar for 30 min.
For the EOR, the CVs were recorded in a 1.0 M KOH solution with 1.0 M ethanol at the potential from 0.01 to 1.10 V (vs. RHE). The sweep rate was fixed at 20 mV s−1. For the CO stripping, first, the catalysts underwent electrochemical pre-treatment by potential cycling between 0.01 and 1.20 V for multiple cycles at a scan rate of 200 mV s−1 under an N2-saturated 1.0 M KOH electrolyte. Then, two CVs cycling between 0.05 and 1.10 V were recorded at 20 mV s−1. Next, the working electrode was maintained a CO-saturated solution for 20 min. Afterward, two additional CVs were measured between 0.01 and 1.1 V with a scan rate of 20 mV s−1 in an N2-saturated solution.
For product quantification, the catalyst was deposited onto a carbon paper electrode to achieve a loading of 1 mg Pd/cm2. The electrolyte consisted of 1 M KOH and 1 M ethanol. Following a brief activation process, chronoamperometric measurements were performed on the carbon paper working electrode at 0.79 V vs. RHE, accumulating a total charge of 40 C. Acetate concentrations in the electrolyte post-electrolysis were quantitatively determined by analyzing the area of the characteristic peak in the 1H NMR spectrum, recorded on a VARIAN 400 MHz spectrometer. Maleic acid was employed as an internal standard for quantifying reaction species, while D2O was used to provide the field frequency lock, ensuring field stability. The Faraday efficiency (FE) of the products was calculated using the equation: \({FE}=eF\times {nQ}\), where e represents the number of electrons transferred for the respective products, n is the total amount of products (in moles), Q is the total charge, and F is the Faraday constant.
DFT calculations
DFT calculations are conducted with the software package Vienna Ab-initio Simulation Package (VASP)57 by using the projector augmented wave (PAW) potential method58 and Perdew-Burke-Ernzerhof (PBE) exchange functional59. All the calculations are spin-polarized with the plane wave cutoff energy of 500 eV. The k-point mesh is 4 × 2 × 1 for HEA/c-PdSn and FeCoNi(111) and 4 × 4 × 1 for PdSn(010). A vacuum of at least 15 Å is adopted to separate the adjacent slabs. The space group of PdSn binary alloys is Pnma and (010) facet is used with a 4-layer (2 × 2 × 1) supercell. The FeCoNi(111) surface with a 3-layer and 96-atom supercell is generated with the special quasi-random structure (SQS) method in the Alloy Theoretic Automated Toolkit (ATAT) code60. The bottom 2-layer atoms are fixed and other atoms are relaxed until the force on each of them is less than 0.01 eV/atom in our calculations.
Data availability
All the data generated in this study are provided in the Source Data file. Data are also available from the corresponding author upon request. Source data are provided with this paper.
References
Cao, G. et al. Liquid metal for high-entropy alloy nanoparticles synthesis. Nature 619, 73–77 (2023).
Yao, Y. et al. High-entropy nanoparticles: Synthesis-structure-property relationships and data-driven discovery. Science 376, 151 (2022).
Sun, Y. & Dai, S. High-entropy materials for catalysis: A new frontier. Sci. Adv. 7, eabg1600 (2021).
Hao, J. et al. Unraveling the electronegativity-dominated intermediate adsorption on high-entropy alloy electrocatalysts. Nat. Commun. 13, 2662 (2022).
Wu, D. et al. Noble-metal high-entropy-alloy nanoparticles: atomic-level insight into the electronic structure. J. Am. Chem. Soc. 144, 3365–3369 (2022).
Löffler, T., Ludwig, A., Rossmeisl, J. & Schuhmann, W. What makes high-entropy alloys exceptional electrocatalysts? Angew. Chemie - Int. Ed. 60, 26894–26903 (2021).
Feng, G. et al. Sub-2 nm ultrasmall high-entropy alloy nanoparticles for extremely superior electrocatalytic hydrogen evolution. J. Am. Chem. Soc. 143, 17117–17127 (2021).
Li, T. et al. Interface engineering between multi-elemental alloy nanoparticles and a carbon support toward stable catalysts. Adv. Mater. 34, 2106436 (2022).
Li, H. et al. Fast site-to-site electron transfer of high-entropy alloy nanocatalyst driving redox electrocatalysis. Nat. Commun. 11, 5437 (2020).
Wu, D. et al. Platinum-group-metal high-entropy-alloy nanoparticles. J. Am. Chem. Soc. 142, 13833–13838 (2020).
Yao, Y. et al. High-throughput, combinatorial synthesis of multimetallic nanoclusters. Proc. Natl. Acad. Sci. USA. 117, 6316–6322 (2020).
Tao, L. et al. A general synthetic method for high-entropy alloy subnanometer ribbons. J. Am. Chem. Soc. 144, 10582–10590 (2022).
Hu, X. et al. Ultrafast materials synthesis and manufacturing techniques for emerging energy and environmental applications. Chem. Soc. Rev. 52, 1103–1128 (2023).
Kusada, K., Mukoyoshi, M., Wu, D. & Kitagawa, H. Chemical synthesis, characterization, and properties of multi-element nanoparticles. Angew. Chemie - Int. Ed. 61, e202209616 (2022).
Ren, J. T., Chen, L., Wang, H. Y. & Yuan, Z. Y. High-entropy alloys in electrocatalysis: from fundamentals to applications. Chem. Soc. Rev. 52, 8319–8373 (2023).
Batchelor, T. A. A. et al. High-entropy alloys as a discovery platform for electrocatalysis. Joule 3, 834–845 (2019).
Mei, Y. et al. MoZn-based high entropy alloy catalysts enabled dual activation and stabilization in alkaline oxygen evolution. Sci. Adv. 10, 1–13 (2024).
He, T. et al. Mastering the surface strain of platinum catalysts for efficient electrocatalysis. Nature 598, 76–81 (2021).
Xie, C., Niu, Z., Kim, D., Li, M. & Yang, P. Surface and interface control in nanoparticle catalysis. Chem. Rev. 120, 1184–1249 (2020).
Shen, B. et al. Crystal structure engineering in multimetallic high-index facet nanocatalysts. Proc. Natl. Acad. Sci. USA. 118, e2105722118 (2021).
Koolen, C. D. et al. Low-temperature non-equilibrium synthesis of anisotropic multimetallic nanosurface alloys for electrochemical CO2 reduction. Nat. Synth. https://doi.org/10.1038/s44160-023-00387-3 (2023)
Shi, W., Chen, J. & Tan, F. Heterostructure Engineering in High-Entropy Alloy Catalysts. SusMat 0, e261 (2024).
Liu, P. & Nørskov, J. K. Ligand and ensemble effects in adsorption on alloy surfaces. Phys. Chem. Chem. Phys. 3, 3814–3818 (2001).
Cuesta, A. Atomic ensemble effects in electrocatalysis: The site-knockout strategy. ChemPhysChem 12, 2375–2385 (2011).
Guo, Y., Wang, M., Zhu, Q., Xiao, D. & Ma, D. Ensemble effect for single-atom, small cluster and nanoparticle catalysts. Nat. Catal. 5, 766–776 (2022).
Zeng, K. et al. Surface-decorated high-entropy alloy catalysts with significantly boosted activity and stability. Adv. Funct. Mater. 32, 2204643 (2022).
Lao, X. et al. Pd‐enriched‐core/Pt‐enriched‐shell high‐entropy alloy with face‐centred cubic structure for C1 and C2 alcohol oxidation. Angew. Chemie 62, e202304510 (2023).
Liu, Y. H. et al. Toward controllable and predictable synthesis of highentropy alloy nanocrystals. Sci. Adv. 9, eadf9931 (2023).
Shi, W. et al. High‐entropy alloy stabilized and activated Pt clusters for highly efficient electrocatalysis. Sus. Mat. 2, 186–196 (2022).
Yao, Y. et al. Carbothermal shock synthesis of high-entropy-alloy nanoparticles. Science (80-.) 359, 1489–1494 (2018).
Han, Y. C., Cao, P. Y. & Tian, Z. Q. Controllable synthesis of solid catalysts by high-temperature pulse. Accounts Mater. Res. 4, 648–654 (2023).
Wang, B. et al. General synthesis of high-entropy alloy and ceramic nanoparticles in nanoseconds. Nat. Synth. 1, 138–146 (2022).
Song, J. Y. et al. Generation of high-density nanoparticles in the carbothermal shock method. Sci. Adv. 7, eabk2984 (2021).
Mori, K. et al. Hydrogen spillover-driven synthesis of high-entropy alloy nanoparticles as a robust catalyst for CO2 hydrogenation. Nat. Commun. 12, 3884 (2021).
Cha, J. H. et al. Flash-thermal shock synthesis of high-entropy alloys toward high-performance water splitting. Adv. Mater. 35, 2305222 (2023).
Gao, S. et al. Synthesis of high-entropy alloy nanoparticles on supports by the fast moving bed pyrolysis. Nat. Commun. 11, 2016 (2020).
Chang, X., Zeng, M., Liu, K. & Fu, L. Phase engineering of high-entropy alloys. Adv. Mater. 32, 1907226 (2020).
Chen, P.-C. et al. Polyelemental nanoparticle libraries. Science. 352, 1565–1569 (2016).
Formation energy. Available at: https://www.oqmd.org/materials/composition/.
Li, B., Li, X., Gao, W. & Jiang, Q. An effective scheme to determine surface energy and its relation with adsorption energy. Acta Mater 212, 116895 (2021).
Yang, Z., Li, X. & Gao, W. Quantitative prediction of surface energy of high-entropy-alloys based on intrinsic descriptors. Surfaces and Interfaces 42, 103442 (2023).
Fernández-García, M. et al. Study of the heterometallic bond nature in PdCu(111) surfaces. J. Phys. Chem. B 102, 141–147 (1998).
Xu, C. Q. et al. Enhanced electron correlations in the binary stannide PdSn4: A homologue of the dirac nodal arc semimetal PtSn4. Phys. Rev. Mater. 1, 064201 (2017).
Funke, H., Scheinost, A. C. & Chukalina, M. Wavelet analysis of extended x-ray absorption fine structure data. Phys. Rev. B - Condens. Matter Mater. Phys 71, 094110 (2005).
Marcella, N. et al. Decoding reactive structures in dilute alloy catalysts. Nat. Commun. 13, 832 (2022).
Li, M. et al. Single-atom tailoring of platinum nanocatalysts for high-performance multifunctional electrocatalysis. Nat. Catal. 2, 495–503 (2019).
Yin, P. F. et al. Synthesis of palladium-based crystalline@amorphous core–shell nanoplates for highly efficient ethanol oxidation. Adv. Mater. 32, 2000482 (2020).
Qiu, Y. et al. Construction of Pd-Zn dual sites to enhance the performance for ethanol electro-oxidation reaction. Nat. Commun. 12, 5273 (2021).
Kim, H. C., Kim, Y., Bando, Y., Yamauchi, Y. & Hong, J. W. Shape-controlled Pd nanocrystal-polyaniline heteronanostructures with modulated polyaniline thickness for efficient electrochemical ethanol oxidation. J. Mater. Chem. A 7, 22029–22035 (2019).
Fan, X. et al. Promoting the electrocatalytic performance of noble metal aerogels by ligand‐directed modulation. Angew. Chemie 132, 5755–5760 (2020).
Li, S. et al. Engineering three-dimensional nitrogen-doped carbon black embedding nitrogen-doped graphene anchoring ultrafine surface-clean Pd nanoparticles as efficient ethanol oxidation electrocatalyst. Appl. Catal. B Environ. 280, 119464 (2021).
Liu, C. et al. Multiple twin boundary-regulated metastable Pd for ethanol oxidation reaction. Adv. Energy Mater. 12, 2103505 (2022).
Pei, A. et al. Nickel hydroxide-supported Ru single atoms and Pd nanoclusters for enhanced electrocatalytic hydrogen evolution and ethanol oxidation. Adv. Funct. Mater. 32, 2208587 (2022).
Wang, H. et al. Pd metallene aerogels with single-atom W doping for selective ethanol oxidation. ACS Nano 16, 21266–21274 (2022).
Li, B., Gao, W. & Jiang, Q. A universal picture for ejecting atoms on metallics. Acta Mater 228, 117792 (2022).
Timoshenko, J. & Kuzmin, A. Wavelet data analysis of EXAFS spectra. Comput. Phys. Commun. 180, 920–925 (2009).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B - Condens. Matter Mater. Phys 54, 11169–11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Van De Walle, A. et al. Efficient stochastic generation of special quasirandom structures. Calphad Comput. Coupling Phase Diagrams Thermochem. 42, 13–18 (2013).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 52371223 (Y. Yao), 52101255 (Y. Yao), 22173034 (W. Gao)), Beijing National Laboratory for Molecular Sciences (BNLMS202405 (Y. Yao)). Shenzhen International Cooperation Program (GJHZ20220913143406013 (Y. Yao)), the Interdisciplinary Research Program of HUST (2023JCYJ004(Y. Yao)), and Collaborative Innovation Center of Suzhou Nano Science and Technology (Z. Liang). The XAFS tests were supported by grants from NAP-SUG (NTU, Singapore (D. Wu)), AcRF Tier 1 grants (RG81/22 (D. Wu)), AcRF Tier 2 grants (MOE-T2EP10123-0003) (Ministry of Education, Singapore (D. Wu)), AS251/XAS/22987 and AS232/XAS/20067 (National Research Foundations, Singapore (D. Wu)). The authors thank test support from the Analytical and Testing Center of Huazhong University of Science & Technology and the State Key Laboratory of Materials Processing and Die & Mould Technology.
Author information
Authors and Affiliations
Contributions
Y. Yao and K. Zeng conceived the concept of HEA/c-PdSn catalysts and designed the pulsed heating approach. K. Zeng, R. Hu, S. Liu, H. Liu, and J. Chen synthesized the samples and collected the electrochemical experimental data. J. Zhang and X. Mu conducted the TEM characterization under the supervision of Z. Liang and J. Zhou. X. Li, H. Wu, and W. Gao performed DFT calculations. Y. Xu and D. Wu performed XAS and analyzed the electronic structure results. Z. Wang developed and optimized the pulsing instrument for preparing the material. Y. Yao and K. Zeng collectively wrote the paper with input from all authors. Y. Yao supervised the project. All authors discussed the results and contributed to the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ziyun Wang, Yafei Li, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zeng, K., Hu, R., Zhang, J. et al. Finely tailoring the local ensembles in heterostructured high entropy alloy catalysts through pulsed annealing. Nat Commun 16, 3403 (2025). https://doi.org/10.1038/s41467-025-58495-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58495-x
