
Abstract
Relatlimab and nivolumab combination therapy shows significant efficacy in treating various types of cancer. Current research on the molecular mechanisms of this treatment is abundant, but in-depth investigations into post-treatment resistance remain notably lacking. In this study, we identify significant enrichment of SRY (sex determining region Y)-box 9 (Sox9)+ tumor cells in resistant samples using single cell RNA sequencing (scRNAseq) in a head and neck squamous cell carcinoma (HNSCC) mouse model. In addition, Sox9 directly regulates the expression of annexin A1 (Anxa1), mediating apoptosis of formyl peptide receptor 1 (Fpr1)+ neutrophils through the Anxa1-Fpr1 axis, which promotes mitochondrial fission, inhibits mitophagy by downregulating BCL2/adenovirus E1B interacting protein 3 (Bnip3) expression and ultimately prevents the accumulation of neutrophils in tumor tissues. The reduction of Fpr1+ neutrophils impairs the infiltration and tumor cell-killing ability of cytotoxic Cd8 T and γδT cells within the tumor microenvironment, thereby leading to the development of resistance to the combination therapy. We further validate these findings using various transgenic mouse models. Overall, this study comprehensively explains the mechanisms underlying resistance to the anti-LAG-3 plus anti-PD-1 combination therapy and identifies potential therapeutic targets to overcome this resistance.
Similar content being viewed by others


Introduction
The emergence of immunotherapy targeting programmed cell death protein 1 (PD-1) and its principal ligand PD-L1 signifies a new era in modern oncology1. Monoclonal antibodies inhibiting PD-1 (e.g., pembrolizumab, nivolumab, and cemiplimab) and those targeting PD-L1 (e.g., atezolizumab, durvalumab, and avelumab) have received approval from the U.S. Food and Drug Administration (US FDA) and are widely used in clinical practice, demonstrating promising response rates across various cancer types2,3,4. The molecular mechanisms underlying these therapies are actively being explored through animal models. Researchers have examined genetic alterations, transcriptomic features, immune cell composition, and interspecies genomic conservation between human and mouse tumors at different pathological stages under immunotherapy using a 4-nitroquinoline 1-oxide (4NQO)-induced mouse tumor model5. Furthermore, various multi-omics approaches have been extensively utilized in this model to investigate the effects of immunotherapy6,7,8. While anti-PD-1/PD-L1 therapy has achieved significant success over the past decade, a substantial proportion of patients either fail to exhibit clinical responses or develop resistance following drug administration, preventing them from benefiting from the treatment9. Similarly, studies using anti-PD-1 monotherapy in mice with oral carcinogenesis have shown that although PD-1 blockade effectively delays tumor progression, it does not completely prevent or reverse it10. To overcome this, an increasing number of PD-1 therapies are employing combination strategies with multiple drugs, such as PD-1 with chemotherapy, PD-1 with targeted therapy, and PD-1 with other immunotherapeutic agents11,12,13,14. These approaches often achieve better outcomes than PD-1 monotherapy, and among them, lymphocyte activation gene 3 (LAG-3), a prominent immune molecule, is becoming a significant research focus in combination strategies with PD-115,16,17. LAG-3, a cell surface inhibitory receptor expressed by various lymphoid and non-lymphoid lineage cells, plays a regulatory role in immunity by impacting T cell activation and effector functions, with preclinical studies demonstrating that blocking LAG-3 can reverse T cell exhaustion, enhance T cell activity, and restore T cell functionality through targeted immunotherapies14,18,19.
Combination immunotherapy with anti-PD-1 and anti-LAG-3 antibodies has also demonstrated synergistic anti-tumor effects across various cancers13,20,21,22. In a previous phase 2/3 clinical trial, researchers found that the 12-month progression-free survival rate for previously untreated patients with metastatic or unresectable melanoma was significantly higher with the combination of relatlimab (LAG-3 blocking antibody) and nivolumab (PD-1 blocking antibody) than with nivolumab alone22. Most importantly, based on the results of the phase 2/3 RELATIVITY-047 trials, relatlimab plus nivolumab received approval from the US FDA and the European Medicines Agency (EMA)23. Moreover, in a first-in-human study of previously treated advanced microsatellite-stable colorectal cancer, favezelimab, with or without pembrolizumab, demonstrated a manageable safety profile with no treatment-related deaths and showed promising anti-tumor activity, particularly in participants with PD-L1 CPS ≥ 1 tumors13. Despite recent significant reports on the molecular mechanisms underlying this combination treatment15,16,17, there remains a notable absence of in-depth investigation into post-treatment resistance.
In this work, by utilizing a classic HNSCC mouse model, we demonstrate that anti-PD-1 and anti-LAG-3 combination therapy resulted in better survival outcomes than monotherapy. In addition, we observe a higher expression level of Anxa1, which is regulated by Sox9, in tumor samples displaying higher resistance to combination therapy. Subsequently, our cell interaction analysis revealed that Anxa1+ epithelial tumor cells mediate apoptosis of Fpr1+ neutrophils through the Anxa1-Fpr1 axis, which promotes mitochondrial fission, inhibits mitophagy by suppressing Bnip3 expression, and ultimately prevents neutrophil accumulation in tumor tissues. Our study demonstrates through various transgenic mouse models that elevated Sox9 expression in epithelial cells mediates the apoptosis of Fpr1+ neutrophils by initiating Anxa1 transcription, thereby inhibiting their accumulation, suppressing cytotoxic Cd8 T and γδT cell infiltration into the tumor microenvironment, and leading to resistance in HNSCC cells to combination therapy.
Results
Establishment of mouse cancer model for combination therapy resistance
To evaluate the combination anti-tumor effects of anti-LAG-3 and anti-PD-1 on murine HNSCC, C57BL/6 wild-type (WT) mice were first fed with 4-nitroquinoline 1-oxide (4NQO) water for 16 weeks, followed by normal water for another 8 weeks to induce the formation of HNSCC. Mice with similar sizes of tumor lesions were then randomly divided into 4 groups for control IgG, anti-PD-1, anti-LAG-3, and anti-LAG-3 plus anti-PD-1 combined treatment and assessed every 4 days from the day of initial treatment. Consistent with previous reports13,20, our study found that LAG-3 monotherapy did not improve survival rates compared to the IgG control group, while PD-1 monotherapy significantly enhanced survival compared to the IgG control, and the combination therapy demonstrated a significantly superior effect compared to PD-1 monotherapy (Supplementary Fig. 1A). However, a substantial portion of the samples did not respond to the combination therapy. To comprehensively investigate the principles and mechanisms of resistance to combined anti-PD-1 and anti-LAG-3 antibody therapy in HNSCC, we further treated HNSCC-bearing mice with anti-LAG-3 and anti-PD-1 antibodies. As shown in Fig. 1A, we found mice responded differently to the combined therapy and were then divided into resistant groups and sensitive groups. Basically, 14 days after initial treatment, tumors which grew more than 20% larger in size compared to their original size were considered resistant tumors according to the response evaluation in solid tumor (RECIST) definition (Fig. 1B).

A The tumor growth curves of each mouse receiving combination therapy (n = 14) or control IgG treatment (n = 6) were measured every 4 days. B Representative image of tongue visible lesions in different groups. Scale bar, 2 mm. C Exemplary instances of magnetic resonance imaging (MRI) of the head and neck from various groups. The dashed regions delineate the boundaries of the tumor. D Representative H&E staining of HNSCC and quantification of HNSCC invasion grades in different groups. n = 6 mice in control group, n = 6 mice in resistant group, n = 8 mice in sensitive group. Scale bar, 65 μm. Statistical significance was assessed using the Pearson chi-square test. P value is exact and two-sided. Representative IHC staining and IHC Score of Ki67 (E) and Caspase3 (F) in HNSCC. n = 6 mice in control group, n = 6 mice in resistant group, n = 8 mice in sensitive group. Scale bar, 65 μm. Data are represented as mean ± SD. P values were calculated one-way ANOVA with Tukey’s multiple comparisons test. G The UMAP plot shows the outcomes post-unbiased clustering, delineating subpopulations of epithelial cell, fibroblast, endothelial cell, immune cell, and muscle cell, each color-coded to represent its cell type. Corresponding bar graphs on the right display the quantity of each subpopulation. H The distributions of the three groups, control, resistant, and sensitive group, are illustrated in the UMAP plot (left), along with the proportions of cells between the three groups (center), and the proportions of epithelial cell, fibroblast, endothelial cell, immune cell and muscle cell within these three groups of cells (right). Calculation method for the right figure: First, normalization of the proportion of each cell type was performed within each group, followed by the calculation of the proportion of each group within the cell type. Source data and exact p values are provided as a Source Data file.
There were 57.1% of animals (8 out of 14) which are responsive to the anti-LAG-3 and anti-PD-1 combined therapy and showed a partial reduction in tumor progression or even had almost complete tumor eradication, while 42.9% of animals (6 out of 14) were not sensitive to anti-LAG-3 and anti-PD-1 combined treatment with tumors grew rapidly (Fig. 1A). Simultaneously, we employed magnetic resonance imaging (MRI) to monitor the growth processes of tumors in these three groups of mice (Fig. 1C). Tongue histopathological examination showed resistant samples displayed more advanced tumor phenotype compared to the sensitive ones (Fig. 1D). In addition, we examined the expression of Ki67 in all groups of mouse tongue and noticed a decrease of cell proliferation in the sensitive group compared to control or resistant groups (Fig. 1E). On the contrary, the percentage of apoptotic cells (cleaved-Caspase3) was greatly elevated in the sensitive group as compared to other groups (Fig. 1F).
To gain insight into the potential mechanisms responsible for resistance to combined therapy, we performed single-cell RNA sequencing (scRNA-seq). For each group, tumor tissues dissected from three mice were pooled together and digested into single-cell suspensions, which were then split into two parts for library construction (Supplementary Fig. 1B). After quality control and filtering, we obtained mRNA measurements of more than 33424 single cells across all samples (7210 for control-1, 6542 for control-2, 4726 for resistant-1, 4943 for resistant-2, 5181 for sensitive-1, and 4822 for sensitive-2). Based on the expression of canonical markers, our analysis revealed 5 major cell types with unique transcriptional features, including epithelial cells (Krt14, Krt5 and Krt6a), fibroblasts (Col1a1, Col3a1, and Apod), endothelial cells (Flt1, Pecam1, and Eng), immune cells (Ptprc, Cd74 and Cd3g) and muscle cells (Myl9, Myh11 and Mylk) (Figs. 1G and S1C). All major cell types were consistently detected in all three conditions (Supplementary Fig. 1D). The distribution of cell types captured by scRNA-seq was comparable between all three groups, while epithelial cells were the most predominant cell type in all groups (Fig. 1G, H). However, the proportion of immune cells was dramatically increased in the sensitive group compared to the control or resistant group (Fig. 1H), highlighting the significance of tumor-infiltrating immune cells in response to combined therapy.
Characterization of resistant tumor cells against combination therapy
To investigate how the resistance of tumor was regulated, we extracted epithelial cells and used CopyKAT24 to distinguish the malignant cells from the non-malignant cells. We successfully identified 19917 aneuploid tumor cell subpopulations with distinct gene expression profiles (Supplementary Fig. 2A). Subsequently, we extracted the malignant cells and divided them into five different subclusters, designated E-comm1, E-resi1, E-comm2, E-sens, and E-resi2 (Fig. 2A). The frequencies of the identified epithelial cell subclusters varied among groups (Fig. 2B; Supplementary Fig. 2B). E-comm1 and E-comm2 subclusters were detected in all three groups (Fig. 2B). However, within E-resi1 and E-resi2 populations, resistant cells were the most abundant cells, accounting for 83.55% and 97.67% of E-resi1 and E-resi2 cells, respectively (Fig. 2B). On the contrary, the vast majority cells in E-sens subcluster belongs to cells in sensitive group (Fig. 2B). Of note, we found that the epithelial-mesenchymal transition (EMT) score was significantly increased in the E-resi1 and E-resi2 population (Supplementary Fig. 2C), implying a malignant nature of these cells. Therefore, we regrouped all epithelial cells into three categories, including common cells (E-comm1 and E-comm2), resistant cells (E-resi1 and E-resi2), and sensitive cells (E-sens). The top significant genes in each of the cell populations were demonstrated in a heatmap (Fig. 2C).

A The UMAP plot shows five epithelial subtypes in distinct colors, with bar graphs indicating their quantities. B The UMAP plot (left) shows the distributions of control, resistant, and sensitive groups, with cell proportions (center) and subtype compositions (right). C Heatmap of top 10 signature genes for three groups. D CellChat circle plot shows differences in intercellular communication number and strength between two groups. E The UMAP plot shows 11 immune cell subtypes, with bar graphs indicating their numbers. F The UMAP plot shows 11 immune subtypes across two groups. G CellChat analysis compares epithelial-immune information flow in Resi and Sens groups, with relative (left) and absolute (right) flow shown. H Scatter plot of ANNEXIN pathway signaling in cell clusters from CellChat analysis. Bubble plots from CellChat (I) and CellPhoneDB (J) show epithelial subtype communication with Neu1 receptors and ligands. P values were calculated using a permutation test, assessing the significance of cell-cell communication by comparing observed mean expression with a null distribution generated by random permutations (I). P values were calculated using a permutation test, which generates a null distribution by randomly shuffling cell labels to determine the specificity of interactions (J). P values are one-sided and exact. K Violin plot (left) and FeaturePlot (right) show Anxa1 expression in epithelial cells across groups. The box plot elements are defined as described in the Statistics and Reproducibility section. Each group includes cells from 3 mice. P values were calculated one-way ANOVA. L Violin plot showing Anxa1 expression across epithelial cell subtypes. The box plot elements are defined as described in the Statistics and Reproducibility section. Each group includes cells from 9 mice. P values were calculated one-way ANOVA. M Representative IHC staining and score of Anxa1 in HNSCC. n = 6 control, n = 6 resistant, n = 8 sensitive mice. Scale bar = 65 μm. Data shown as as mean ± SD. P values were calculated one-way ANOVA with Tukey’s multiple comparisons test. Source data and exact p values are provided as a Source Data file.
To characterize the cell-cell communication patterns in different groups, we performed CellChat analysis25. Our analysis showed that the interaction between epithelial cells and immune cells was more profoundly altered compared to other types (Fig. 2D). In addition, we found a dramatically increased number of interactions between epithelial cells and immune cells in the sensitive group compared to the control or resistant groups, while the strength of interaction was reduced in the resistant group compared to other groups (Fig. 2D). To obtain the detail of epithelial and immune cell interaction, we re-clustered immune cells and regrouped them into Treg, γδT, Neu1, Cd4 T, Macro1, Mono, Neu2, Cd8 T, Macro2, DC and NK cells according to the typical markers (Fig. 2E; Supplementary Table 1). Compared with control and resistant groups, sensitive tumors displayed a higher proportion of Neu1, γδT, Treg, Cd4 T, Cd8 T (Fig. 2F; Supplementary Fig. 2D; Supplementary Table 1).
To identify the differences in signaling pathways between resistant and sensitive tumors, we compared the overall information flow for each signaling pathway and found four signaling pathways (CSF3, CHEMERIN, ANNEXIN, and TNF) enriched in resistant tumor (Fig. 2G). However, we noticed the information flow degrees of CSF3 and CHEMERIN were relatively low, while the difference in the TNF signaling pathway was not dramatic (Fig. 2G). Instead, the information flow of the ANNEXIN signaling pathway was robust and dramatically increased in the resistant compared to the sensitive group (Fig. 2G), suggesting the ANNEXIN signaling pathway might be involved in mediating the resistance to combination therapy. When comparing the outgoing and incoming signals of the ANNEXIN signaling pathway in each cell population, we found that E-resi1 and E-resi2 resistant cells were the major signaling sources and that Neu1 served as the main signaling receiver (Fig. 2H). Simultaneously, a marked suppression of Neu1 cells was observed in Resi samples (Fig. 2F; Supplementary Fig. 2D). Results from CellChat or CellPhoneDB26 analysis confirmed that the communication probability of the Anxa1-Fpr1 signaling pathway was highly elevated between epithelial cells and Neu1 in the resistant group compared to the sensitive group (Fig. 2I, J).
ScRNA-seq analysis showed that Anxa1 expression was highly enriched in the resistant cells (Fig. 2K). Moreover, Anxa1 exhibits significantly elevated expression in the E-resi1 and E-resi2 clusters of epithelial subtypes (Fig. 2L). The IHC data also indicated that the protein level of Anxa1 was significantly upregulated in the resistant samples compared to sensitive ones (Fig. 2M).
Fpr1+ neutrophils are associated with cytotoxic T cells
FeaturePlot analysis reveals a pronounced enrichment of Fpr1 expression in Neu1 (Fig. 3A). Examination of IHC data reveals a marked upregulation in the protein levels of Fpr1 in sensitive specimens, as compared to resistant samples (Fig. 3B). To investigate Fpr1+ neutrophils, we further isolated the entirety of neutrophils and segregated them into two subclusters, denoted as Neu1 and Neu2 (Fig. 3C). Through Velocity and CytoTRACE analyses27, we discerned distinct initial cell populations for each of the two subclusters, suggesting that the relationship between these subclusters might not involve a progenitor-descendant dynamic (Fig. 3D, E). Additionally, our analysis using CellCycleScoring observed no disparity in proliferation between the two neutrophil subclusters28. Cells in both subclusters exhibit a dispersed distribution across the G1, S, and G2M phases without any discernible aggregation (Fig. 3F). The aforementioned characteristics indicate that these two subclusters of neutrophils represent distinct and independently existing populations within the neutrophil group. Subsequently, we selected resistant and sensitive groups after 14 and 21 days of combination therapy, respectively, and conducted immunofluorescence (IF) staining and statistical analysis. We found a significant reduction in Fpr1+ neutrophils in the resistant group compared to the sensitive group at 14 and 21 days (Fig. 3G). Furthermore, consistent results were observed using flow cytometry analysis (Fig. 3H; Supplementary Fig. 3A).

A FeaturePlot shows Fpr1 expression in immune cells. B Representative IHC staining and score of Fpr1 in HNSCC. Scale bar = 65 μm. n = 6 resistant, n = 8 sensitive mice. Data shown as mean ± SD. P value is presented by two-tailed unpaired Student’s t test. C The UMAP plot shows two neutrophil subtypes in different colors, with bar graphs indicating their quantities. D Velocity analysis was conducted on neutrophils from two groups. Arrows in the figure indicate the predicted direction of cellular evolution. E CytoTRACE analysis was performed on neutrophils from two groups. The color intensity in the figure indicates the predicted evolutionary order of cells. F CellCycleScoring analysis showed no proliferative difference between neutrophil subtypes. Left: UMAP plot of overall cell cycle. Middle: Proportions of cell cycle stages in subtypes. Right: UMAP plot of neutrophil cell cycles in each group. G After 14 and 21 days of combination therapy, immunofluorescence staining was performed on HNSCC tissues from both mouse groups. Epithelial tissues were stained with anti-Fpr1 (green), anti-Ly6g (red), and DAPI (blue) for nuclei. Scale bar = 30 μm. On the right, percentages of Ly6g+Fpr1+ cells by area and number are shown. d14: n = 6 resistant, sensitive mice; d21: n = 6 resistant, n = 8 sensitive mice. Data shown as mean ± SD. P values are presented by two-tailed unpaired Student’s t test. H Representative flow cytometry plots (left) and analysis (right) of Ly6g+Fpr1+ neutrophils in different groups. The experiment was independently repeated three times. Data shown as mean ± SD. P value is presented by two-tailed unpaired Student’s t test. I Comparative analysis of Neu1 and T cell information flow in Resi and Sens groups via CellChat. Left: relative flow, right: absolute flow. Source data and exact p values are provided as a Source Data file.
To investigate the link between Neu1 neutrophils and T cell recruitment, we performed ligand-receptor interaction analysis using CellPhoneDB and identified significant interactions between Neu1 (Fpr1+ neutrophils) and γδT and Cd8 T cells (Supplementary Fig. 3B). Subsequently, we conducted GO enrichment analysis on the differentially expressed genes specific to Neu1 and observed its enrichment in pathways such as chemokine production and neutrophil chemotaxis (Supplementary Fig. 3C). Using CellChat analysis, we compared the information flow between Neu1 cells and T cells in the Resi and Sens groups. In the Sens group, we observed that the interaction between Neu1 cells and T cells primarily occurs through CXCL-related ligand-receptor pairs (Fig. 3I). Consequently, we performed ELISA to examine differences in chemokine production between Neu1(Fpr1+ neutrophils) sorted from the Rens and Sens groups, revealing that Cxcl10 exhibited the most significant differential expression (Supplementary Fig. 3D). We sorted Fpr1+ neutrophils and Cd45+Cd3+ T cells from sensitive tumors, followed by co-culture and subsequent flow cytometry analysis of the cells in the lower chamber (Supplementary Fig. 3E). In the presence of Fpr1+ neutrophils, we observed that Cd8 T and γδT cells migrated to the lower chamber (Supplementary Fig. 3F). Double IF staining was then employed to assess the enrichment of Gzma+ γδT cells and Gzma+ Cd8 T cells in Resi and Sens samples. Our analysis revealed a significant enrichment of Gzma+ γδT cells and Gzma+ Cd8 T cells in the epithelial tissue of Sens samples compared to Resi samples (Supplementary Fig. 4A, B). Similarly, we used flow cytometry to demonstrate an increased enrichment of cytotoxic Cd8 T and γδT cells in tumors from the Sens group, with a lower proportion of these cells in an exhausted state (Supplementary Figs. 4C, S4D and S5A). Moreover, we conducted exhaustion assays targeting Cd8 T and γδT cells, demonstrating that in the Sens group mice, the reduction of both cytotoxic T cell populations led to tumor progression (Supplementary Fig. 5B–D). In summary, this suggests that Neu1 neutrophils may be associated with an increase in cytotoxic γδT and cytotoxic Cd8 T cells within target tumors.
To validate our findings in vivo, we engineered conditional knockout Anxa1flox/flox mutant mice (Supplementary Fig. 6A) and crossed them with K14CreER mice to generate K14CreER; Anxa1flox/flox (Anxa1cKO) and K14CreER; Anxa1wt/wt (Anxa1cKO-Con) mice. Anxa1cKO-Con and Anxa1cKO mice were then subjected to 4NQO treatment to induce the formation of HNSCC (Fig. 4A). After 14 days of administering the combination therapy to HNSCC mice, we selected Anxa1cKO and Anxa1cKO-Con mice that developed resistance. Subsequently, a Tamoxifen injection was administered to induce Anxa1 knockout during the continuation of combination therapy, and tongue samples were collected from the mice 3 weeks later. Within the tongue tissues of Anxa1cKO, fewer morphological alterations were observed. Moreover, in comparison to the Anxa1cKO-Con group, there was a notable reduction in both the number and area of oral lesions in Anxa1cKO mice (Fig. 4B, C). Histological analysis of all lesions obtained from the Anxa1cKO-Con and Anxa1cKO groups revealed a significant reduction in the formation of grade 3 HNSCC due to the absence of Anxa1 (Fig. 4D). Additionally, the ablation of Anxa1 resulted in a decreased proliferation within the lesion area of the tongue (Fig. 4E). IHC results reveal the absence of Anxa1 in the tongue mucosa of Anxa1cKO, yet Fpr1 expression is significantly increased (Fig. 4F, G). This suggests that, following the development of resistance in HNSCC mice, the loss of Anxa1 does not inhibit Fpr1+ neutrophils. Simultaneously, an enrichment of cytotoxic γδT cells and cytotoxic Cd8 T cells was observed in the Anxa1cKO group (Fig. 4H, I), indicating that the loss of Anxa1 may lead to the recruitment of Gzma+γδT cells and Gzma+Cd8 T cells by Fpr1+ neutrophils to the HNSCC lesion area.

A The experimental design of the HNSCC tumorigenesis model and treatment strategy in the Resi; Anxa1cKO-Con and Resi; Anxa1cKO groups. B Representative image of tongue visible lesions in Resi; Anxa1cKO-Con and Resi; Anxa1cKO groups. n = 6 mice in each group. Scale bar, 2 mm. C Quantification of HNSCC lesion number and lesion area (mm3) in Resi; Anxa1cKO-Con and Resi; Anxa1cKO groups. n = 6 mice in each group. Data shown as mean ± SD. P values are presented by two-tailed unpaired Student’s t test. D Representative H&E staining of HNSCC and Quantification of HNSCC invasion grades in Resi; Anxa1cKO-Con and Resi; Anxa1cKO groups. Scale bar, 65 μm. n = 6 mice in each group. Statistical significance was assessed using the Pearson chi-square test. P value is exact and two-sided. Representative IHC staining and IHC Score of Ki67 (E), Anxa1 (F), and Fpr1 (G) in HNSCC of Resi; Anxa1cKO-Con and Resi; Anxa1cKO groups. The scale bar is 65 μm. n = 6 mice in each group. Data shown as mean ± SD. P values are presented by two-tailed unpaired Student’s t test. Representative immunofluorescence staining of Gzma+γδT (H) or Gzma+Cd8 T (I) cells in mouse HNSCC tissues from Resi; Anxa1cKO-Con and Resi; Anxa1cKO groups. Epithelial tissues were stained with anti-Gzma antibody (green) and anti-Tcr g/d (H) or anti-Cd8a (I) antibody (red). Cell nuclei were stained with DAPI (blue). Scale bar, 30 μm. On the right, the corresponding percentages of Gzma+γδT (H) or Gzma+Cd8 T (I) cells in terms of area and number are provided. n = 6 mice in each group. Data shown as mean ± SD. P values are presented by two-tailed unpaired Student’s t test. Source data and exact p values are provided as a Source Data file.
Furthermore, we generated conditional knockout mice with the Fpr1flox/flox genotype (Supplementary Fig. 6B). Fpr1flox/flox mice were crossed with Lyz2CreER mice to generate Lyz2CreER; Fpr1flox/flox (Fpr1cKO) and Lyz2CreER; Fpr1wt/wt (Fpr1cKO-Con) mice (Supplementary Fig. 6C). Consistent with the method of treatment applied to Anxa1cKO-Con and Anxa1cKO mice described above, we obtained Fpr1cKO-Con and Fpr1cKO mice resistant to the combination therapy (Supplementary Fig. 6C). Relative to the Fpr1cKO-Con group, the Fpr1cKO group exhibits reduced HNSCC lesion count and area upon Fpr1 deficiency (Supplementary Fig. 6D, E). Histological analysis reveals a marked reduction in the invasiveness of HNSCC in the Fpr1cKO mice group (Supplementary Fig. 6F). Anti-Ki67 immunostaining indicates a significant reduction in cellular proliferation within the tongue lesion region upon the ablation of Fpr1 (Supplementary Fig. 6G). Additionally, IHC staining of anti-Fpr1 and anti-Anxa1 reveals an absence of Fpr1 expression in the Fpr1cKO group, while the expression of Anxa1 in the mucosa of Fpr1cKO tongues remains unaffected (Supplementary Fig. 6H, I). Similarly, an infiltration of Gzma+γδT cells and Gzma+Cd8 T cells is observed in the Fpr1cKO group (Supplementary Fig. 6J, K). Further analysis showed the cell membrane protein Sell was selectively expressed in Neu1, which allowed us to use Sell as a marker for the isolation of Neu1 (Supplementary Fig. 7A; Supplementary Table 3). Subsequently, in normal mice not induced with 4NQO and without combination therapy, we observed no change in neutrophils in the bone marrow of both Fpr1cKO-Con and Fpr1cKO groups (Supplementary Fig. 7B, C). However, after 14 and 21 days of combination therapy, we observed a significant reduction in Sell+ neutrophils in the Fpr1cKO-Con group compared to the Fpr1cKO group at both 14 and 21 days (Supplementary Fig. 7D).
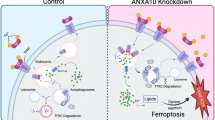
Anxa1-Fpr1 axis regulates the mitochondrial fission of neutrophils
Previous studies have indicated that ANXA1 and its analog peptides induce apoptosis and inhibit the intratissular accumulation of neutrophils by causing rapid and transient changes in intracellular Ca2+ concentration29,30,31. However, the underlying molecular mechanisms remain poorly understood. Therefore, we used flow cytometry to sort Sell+ neutrophils from Resi; Fpr1cKO-Con and Resi; Fpr1cKO groups 3 days after Tamoxifen injection. These isolated cells were then subjected to mRNA-seq for further analysis.
Through gene set enrichment analysis (GSEA), we found that upregulated genes in the Fpr1cKO-Con group were significantly enriched in the “GOBP_ORGANELLE_FISSION” pathway, while downregulated genes were significantly enriched in pathways related to maintaining normal mitochondrial function (Fig. 5A, B). Mitochondria are known as key organelles for regulating apoptosis and maintaining calcium homeostasis32. Gene Ontology (GO) enrichment analysis revealed that downregulated genes were significantly enriched in “regulation of membrane potential” and “response to oxidative stress” pathways, while upregulated genes were significantly enriched in “negative regulation of organelle organization” pathways, consistent with the GSEA results (Fig. 5C). Based on these findings, we hypothesize that mitochondrial fission occurs in the Fpr1cKO-Con group. Electron microscopy and MitoTracker fluorescence results from the HL60 cell line showed that mitochondrial fission mediated by the ANXA1-FPR1 axis results in the fragmentation of mitochondria into smaller units (Fig. 5D; Supplementary Fig. 8A). Moreover, we observed that treatment of sorted Fpr1+ neutrophils with Anxa1 induced mitochondrial fission (Supplementary Fig. 8B). This result was consistently confirmed through electron microscopy (Supplementary Fig. 8C). Additionally, we found a significant downregulation of Bnip3 in the Fpr1cKO-Con group (Fig. 5E–G; Supplementary Fig. 8D). Bnip3 primarily regulates mitophagy33, a cellular self-protection mechanism that eliminates damaged mitochondria caused by fission34. When mitophagy is suppressed, mitochondrial rupture occurs, leading to the release of Cytochrome C and subsequent cell apoptosis35. Figure 5D shows that in the ANXA1+; shCTR group, mitochondria underwent fission but did not undergo mitophagy, and Cytochrome C was detected in the cytoplasm of cells from the ANXA1+; shCTR group (Fig. 5H).

A GSEA was performed in the Resi; Fpr1cKO-Con and Resi-Fpr1cKO groups to compute an enrichment score (ES) for gene set overrepresentation in a ranked mRNA-seq list. A positive ES indicates top enrichment, while a negative ES indicates bottom enrichment. B The ridgeline plot displays GSEA core gene fold changes (positive: upregulated; negative: downregulated). P values were calculated using a permutation test, evaluating the significance of pathway enrichment by comparing the observed enrichment score (ES) with a null distribution generated through random permutations of gene sets. P values are one-sided and adjusted for multiple comparisons using the Benjamini-Hochberg method. C GO-BP enrichment of up/downregulated genes in Resi; Fpr1cKO-Con VS Resi-Fpr1cKO, annotated with p values (yellow numbers). P values were calculated using the hypergeometric test to evaluate the significance of gene set enrichment. The test was one-sided, and P values were adjusted for multiple comparisons using the Benjamini-Hochberg method. D Representative electron microscopy images of mitochondria from four groups. Scale bar, 400 nm. The experiment was independently repeated three times. Knockdown group selected using shFPR1-2. E Volcano plot of Resi; Fpr1cKO-Con VS Resi; Fpr1cKO mRNA-seq data highlights Bnip3(red) among differentially expressed genes (threshold: p < 0.05, |Log2FC| > 1). n = 3 mice in each group. P values were calculated using the Wald test to evaluate the significance. The test was two-sided. P values were adjusted for multiple comparisons using the Benjamini-Hochberg method. F Representative immunofluorescence staining of HL60 cells from four groups. Knockdown group selected using shFPR1-2. Cells were stained with anti-BNIP3 (green) and DAPI (blue) for nuclei. Scale bar = 15 μm. Right: percentages of BNIP3+ cells by area and number. The experiment was independently repeated three times. Data shown as mean ± SD. P values were calculated one-way ANOVA with Tukey’s multiple comparisons test. G BNIP3 protein levels (WB) in four groups, with α-Tubulin as loading control. H WB analysis of Cytochrome c in mitochondria-free cytoplasmic proteins from four groups, with α-Tubulin as loading control. Source data and exact p values are provided as a Source Data file.
To summarize, the Anxa1-Fpr1 axis mediates mitochondrial fission in neutrophils and inhibits mitophagy by downregulating Bnip3 expression. This results in mitochondrial rupture and the release of Cytochrome C, which induces apoptosis in neutrophils. Ultimately, this process may prevent the recruitment of cytotoxic γδT and Cd8 T cells to the HNSCC tumor lesions in resistant tumors.
Sox9 mediates resistance in HNSCC by acting as a transcription factor for Anxa1
To delve further into the specific mechanisms of resistance to combination therapy, we proceeded with an in-depth exploration and analysis of scRNA-Seq data. The transcription factors obtained from SCENIC analysis in epithelial cells were intersected with the specific markers for E-resi1 and E-resi2, resulting in the identification of a unique transcription factor, Sox9 (Fig. 6A). Similar to Anxa1, scRNA-seq analysis reveals significant enrichment of Sox9 expression in both resistant subclusters (E-resi1 and E-resi2) and the resistant group (Fig. 6B, C). IHC results also demonstrated a significantly higher expression level of Anxa1 in the resistant group compared to the sensitive group (Fig. 6D). Analysis from the Cistrome database also revealed significant binding signals of Sox9 on the Anxa1 promoter (Supplementary Fig. 9A). Subsequently, we conducted chromatin immunoprecipitation (ChIP) using a Sox9-specific antibody on genomic DNA isolated from mouse HNSCC tissues of the resistant group and detected the enrichment of Sox9 on the Anxa1 promoter (Fig. 6E). To ascertain whether Sox9 binding to the Anxa1 promoter induces transcription, Anxa1 promoter sequences containing a putative binding motif (the retrieval of binding motif information was sourced from the footprintDB database) were used to generate luciferase reporter constructs and motif-deleted controls (Fig. 6F). As evidenced by luciferase activity, the overexpression of Sox9 in Moc1 cells containing the reporter construct significantly induced the transcription from the Anxa1 gene. The deletion of the Motif abolished Anxa1 promoter induction (Fig. 6G). Furthermore, through double IF staining, we observed colocalization of Sox9 and Anxa1 within the same cells of the resistant group (Fig. 6H). Taken together, these findings suggest that Sox9 binds to the Anxa1 promoter, probably in the Motif region, inducing Anxa1.

A Venn diagram of genes identified by FindMarkers and SCENIC analyses. B Violin plot (left) shows Sox9 expression across groups, and FeaturePlot (right) depicts Sox9 expression in epithelial cells. The box plot elements are defined as described in the Statistics and Reproducibility section. Each group includes cells from 3 mice. P values were calculated one-way ANOVA. C Violin plot showing Sox9 expression across epithelial cell subtypes. The box plot elements are defined as described in the Statistics and Reproducibility section. Each group includes cells from 9 mice. P values were calculated one-way ANOVA. D Representative IHC staining and score of Sox9 in HNSCC. n = 6 control, n = 6 resistant, n = 8 sensitive mice. Scale bar = 65 μm. Data shown as mean ± SD. P values were calculated one-way ANOVA with Tukey’s multiple comparisons test. E ChIP of genomic DNA from resistant mouse HNSCC tissues was performed with a Sox9-specific antibody. Left: gel image, right: qPCR analysis of Anxa1 promoter ChIP signal relative to IgG group. Two primer pairs were used. The experiment was independently repeated three times. Data shown as mean ± SD. P values are presented by two-tailed unpaired Student’s t test. F Luciferase reporter assay assessed Anxa1 promoter activity in Moc1 cells. A schematic diagram shows cloning the Anxa1 promoter into the pZX001 vector to generate pGL3 luciferase plasmid. Binding motif information was retrieved from footprintDB database. G The relative activity of the Anxa1 promoter was detected by luciferase assay across the three groups. The experiment was independently repeated three times. Data shown as mean ± SD. P values were calculated one-way ANOVA with Tukey’s multiple comparisons test. H Representative immunofluorescence colocalization of Anxa1 (red) and Sox9 (green) in Resi mouse HNSCC tissues. Nuclei counterstained with DAPI (blue). Scale bar = 20 μm. Source data and exact p values are provided as a Source Data file.
To further explore the role of Sox9+ cells in drug resistance, we employed a multi-generational crossbreeding strategy involving K14DreER, Sox9CreER, Ai66-Tomato, and DTR tool mice. This yielded K14DreER; Sox9CreER; Ai66-Tomato; DTR+/− (DTR+) and K14DreER; Sox9CreER; Ai66-Tomato (DTR−) mice (Fig. 7A and Supplementary Fig. 9B). Firstly, using K14DreER; Sox9CreER; Ai66-Tomato mice, we observed continuous development of Sox9+ epithelial tumor cells in resistant samples (Supplementary Fig. 9C). Employing the aforementioned treatment regimen, we obtained DTR− and DTR+ mice resistant to combined therapy (Fig. 7A). Compared to the DTR− group, the loss of Sox9+ cells resulted in a significant reduction in both lesion count and area (Fig. 7B, C). Histological analysis revealed that the loss of Sox9+ cells reversed the resistance of HNSCC to combination therapy (Fig. 7D, E). IHC staining reveals that the absence of Sox9+ cells in the DTR+ group leads to the concomitant loss of Anxa1 compared to the DTR− group (Fig. 7F, G). Concurrently, the depletion of Sox9+ cells reversed the accumulation of Fpr1+ neutrophils in the lesion area (Fig. 7H). Gzma+γδT cells and Gzma+Cd8 T cells became enriched in the lesion area of the DTR+ group (Fig. 7I, J).

A The experimental design of the HNSCC tumorigenesis model and treatment strategy in the Resi; DTR+ and Resi; DTR− groups. B Representative image of tongue visible lesions in Resi; DTR+ and Resi; DTR− groups. n = 6 mice in each group. Scale bar, 2 mm. C Quantification of HNSCC lesion number and lesion area (mm3) in Resi; DTR+ and Resi; DTR− groups. n = 6 mice in each group. Data shown as mean ± SD. P values are presented by two-tailed unpaired Student’s t test. D Representative H&E staining of HNSCC and Quantification of HNSCC invasion grades in Resi; DTR+ and Resi; DTR− groups. Scale bar, 65 μm. n = 6 mice in each group. Statistical significance was assessed using the Pearson chi-square test. P value is exact and two-sided. Representative IHC staining and IHC Score of Ki67 (E), Sox9 (F), Anxa1 (G), and Fpr1 (H) in HNSCC of Resi; DTR+ and Resi; DTR− groups. The scale bar is 65 μm. n = 6 mice in each group. Data shown as mean ± SD. P values are presented by two-tailed unpaired Student’s t test. Representative immunofluorescence staining of Gzma+γδT (I) or Gzma+Cd8 T (J) cells in mouse HNSCC tissues from Resi; DTR+ and Resi; DTR− groups. Epithelial tissues were stained with anti-Gzma antibody (green) and anti-Tcr g/d (I) or anti-Cd8a (J) antibody (red). Cell nuclei were stained with DAPI (blue). Scale bar, 30 μm. On the right, the corresponding percentages of Gzma+γδT (I) or Gzma+Cd8 T (J) cells in terms of area and number are provided. n = 6 mice in each group. Data shown as mean ± SD. P values are presented by two-tailed unpaired Student’s t test. Source data and exact p values are provided as a Source Data file.
Specific ablation of Anxa1 in Sox9+ cells will facilitate the in vivo validation of our findings. Therefore, we proceeded to crossbreed Sox9CreER tool mice with Anxa1flox/flox mutant mice, resulting in Sox9CreER; Anxa1flox/flox (Sox9-Anxa1cKO) and Sox9CreER; Anxa1wt/wt (Sox9-Anxa1cKO-Con) mice (Supplementary Fig. 10A). Following the previously mentioned protocol, we acquired Sox9-Anxa1cKO-Con and Sox9-Anxa1cKO mice resistant to combined therapy (Supplementary Fig. 10A). Consistently, in resistant samples of HNSCC, the elimination of Anxa1 from Sox9+ cells resulted in a reduction in the number, area, and malignancy of the lesion surface (Supplementary Fig. 10B–D). Moreover, in comparison to the Sox9-Anxa1cKO-Con group, there was a reduction in cell proliferation within the HNSCC lesion area in the Sox9-Anxa1cKO group (Supplementary Fig. 10E). IHC with anti-Sox9 and anti-Anxa1 revealed that, in resistant samples, the expression of Sox9 in the control and knockdown groups remained unaffected, while the expression of Anxa1 was significantly suppressed in the Sox9-Anxa1cKO group (Supplementary Fig. 10F, G). Consistently, the absence of Anxa1 results in the failure to induce apoptosis in Fpr+ neutrophils, leading to their accumulation in the lesion area (Supplementary Fig. 10H). This, in turn, may recruit Gzma+γδT cells and Gzma+Cd8 T cells for the eradication of HNSCC cells (Supplementary Fig. 10I, J).
Discussion
PD-1, an immune checkpoint inhibitor (ICI), is crucial in local immune suppression within the tumor microenvironment (TME). Unlike traditional approaches such as surgery, radiotherapy, and chemotherapy, which aim to target and kill tumor cells, the core principle of immunotherapy is to mobilize and activate the patient’s immune system to control and eliminate tumor cells, thereby achieving cancer inhibition or cure. This has made immunotherapy a new trend in controlling tumor initiation, growth, and progression36,37,38,39. With the widespread use of anti-PD-1/PD-L1 therapy, resistance issues have emerged across various types of tumors9,40. Consequently, this led to the development of a combination of anti-PD-1 and anti-LAG-3 antibody therapy, with LAG-3 being referred to as “the third checkpoint inhibitor”41. Nonetheless, our research and that of others indicate the presence of resistant individuals even with the use of this combination therapy5. We discovered through techniques such as transgenic mice and single-cell RNA sequencing (scRNA-seq) that in HNSCC, epithelial cells with high Sox9 expression initiate the transcription of Anxa1. This process mediates mitochondrial fission and inhibits mitophagy in Fpr1+ neutrophils, leading to apoptosis and preventing their accumulation in tumor tissue. Consequently, this hinders the migration of cytotoxic Cd8 T and γδT cells into the tumor tissue, leading to therapy resistance in HNSCC cells.
Research has indicated that SOX9 is overexpressed in some cases of HNSCC, and its increased expression has been associated with more aggressive tumor behavior and poorer prognosis42. It has been linked to promoting cell proliferation, invasion, and metastasis in HNSCC cells43. Additionally, in a study on oral squamous cell carcinomas (OSCC), researchers found that resistance to cisplatin and drug-induced adaptation was associated with increased expression of SOX944. In our study, we identified Sox9 as a transcription factor for Anxa1, initiating its expression and thereby mediating HNSCC resistance to combination therapy.
ANXA1 belongs to the annexin superfamily of membrane proteins, binding to acidic phospholipids in a calcium-dependent manner. While ANXA1 is normally located in the cytoplasm, it can also be secreted into the extracellular environment, where it modulates cellular behavior through activation of FPR1 via autocrine, paracrine, or exocrine pathways45,46. ANXA1 is known to mediate neutrophil apoptosis during inflammatory responses, thereby inhibiting the persistence of inflammation47. While neutrophils play a crucial role in effective host defense, their excessive accumulation and prolonged activity can cause tissue damage and inflammatory stimuli48,49. Persistent inflammatory stimuli can lead to chronic inflammation, ultimately harming the host50. While ANXA1 plays a positive role in suppressing chronic inflammation during inflammatory stimuli, it exhibits pro-carcinogenic effects in tumor tissues. Recent studies increasingly suggest that ANXA1 promotes tumor progression51,52. Compared to adjacent non-cancerous tissues, elevated expression of ANXA1 is observed in cancerous tissues, which is often associated with poorer prognosis and lower overall survival rates in patients53,54, such as in lung cancer (LC)55, colorectal cancer (CRC)56, and triple-negative breast cancer (TNBC)57,58. Particularly, high levels of ANXA1 expression are consistently observed in TNBC57,58. ANXA1 induces a favorable tumor microenvironment, promoting invasion and tumor progression across various cancers51. In this study, we found that in samples resistant to combination therapy, tumor cells activated mitochondrial fission in neutrophils within the tumor tissue via the Anxa1-Fpr1 axis and inhibited the expression of Bnip3, which mediates mitophagy through the same axis.
Neutrophils, circulating in the bloodstream, serve as the first line of defense against microbial infections and can be rapidly recruited to sites of injury to combat infections59. There are numerous interactions and regulatory mechanisms between epithelial cells and neutrophils. During intestinal inflammation, neutrophils are rapidly recruited by epithelial cells through a series of chemotactic gradients from the microcirculation60. In inflammatory bowel disease (IBD), neutrophil activity is elevated, leading to the release of inflammatory mediators such as cytokines and chemokines, which promote the infiltration of inflammatory cells, tissue damage, and ulcer formation, resulting in mucosal inflammation and injury61. Studies have also shown that in IBD, PAD4 derived from neutrophils can enter intestinal epithelial cells via extracellular vesicles (EVs), inducing epithelial cell apoptosis62. Additionally, alveolar epithelial cells can mitigate neutrophil-induced inflammation in lung injury by regulating mitochondrial fatty acid oxidation63. In mice, an inducible long non-coding RNA (lncRNA) called lnc-Cxcl2, located at the Cxcl2 gene locus in lung epithelial cells; feedback inhibits neutrophil-mediated pulmonary inflammation during influenza infection64. However, increasing attention has been paid to the role of neutrophils in cancer65. Recent studies have reported that, in transplant tumor models, a subset of neutrophils expressing the receptor tyrosine-protein kinase MET (also known as HGFR) can directly kill cancer cells through the release of nitric oxide upon activation66. Neutrophils can also indirectly combat cancer by creating an anti-tumor microenvironment. In human lung cancer, researchers have identified neutrophils with an antigen-presenting cell-like phenotype that triggers anti-tumor T cell responses, thereby supporting anti-cancer activity67.
Tumor-infiltrating immune cells are crucial for the efficacy of immunotherapy, with CD8 T cells being key effectors of anti-tumor immunity68. CD8 T cells mediate their functions through various mechanisms, including the release of granules containing perforin and granzymes at target cells. Granzymes comprise a family of cytotoxic proteases, each targeting unique substrates within target cells, typically inducing cell death69. Recent studies have indicated that CD8+ TILs are considered pivotal in the treatment of HNSCC with neoadjuvant chemoimmunotherapy70. γδT cells, a non-traditional T cell subset, are emerging as a promising component in immunotherapy, demonstrating remarkable anti-tumor potential and therapeutic prospects71. γδT cells are a major component of intraepithelial lymphocytes in mucosal tissues72. Numerous experimental and clinical data suggest that γδT cells possess anti-tumor properties73,74. High levels of γδT cells are positively correlated with lower clinical stages and improved overall survival75. γδT cells can exert direct cytotoxic effects on cancer cells by releasing cytotoxic molecules, such as perforin and granzymes76. Studies have shown that γδT cells are present in the tumor microenvironment of some HNSCC cases, suggesting their involvement in the immune response against cancer77. However, HNSCC can create an immunosuppressive microenvironment, which may hinder the activity of immune cells, including CD8 T, γδT, and CD4 T cells77. PD-1 inhibits γδT cell activation by dampening TCR signaling and interfering with co-stimulatory signals, resulting in γδT cell dysfunction78. Elevated expression of PD-1, TIM-3, and LAG-3 may even drive γδT cells into a “super anergy” state79.
This study primarily employed the 4NQO-induced HNSCC mouse model. The advantages of using mouse models include shorter breeding cycles, higher tumor growth rates, and ease of genetic manipulation. In addition, a comprehensive and widely available transgenic mouse repository, such as the Jackson Laboratory, has been well-established80. In immunotherapy research, mouse models facilitate the design of more complex treatment regimens, particularly for novel immune inhibitors. Furthermore, they allow for the collection of a broader range of experimental samples and data during subsequent observations6,7,8. Cancer induced by carcinogen treatment in mice more closely resembles sporadic human cancers and allows for the tracking of the entire precancerous progression81,82. Mice expressing fluorescent protein (FP)-tagged genes provide intuitive visualization and enhanced cellular and molecular resolution during signal pathway studies83. These features offer significant advantages and open up more possibilities for investigating the molecular signaling pathways of immunotherapy. However, the translation of promising immunotherapy results or resistance mechanisms from preclinical trials to clinical settings remains a challenge, as high failure rates have been observed in human clinical trials despite promising outcomes in mouse models84,85,86. This may be due to limitations inherent in transgenic technologies, which result in discrepancies between the resistance mechanisms validated in mouse models and their applicability to clinical settings. For example, inserting reporter genes or recombinant enzyme genes into a framework containing the endogenous ATG gene results in the loss of gene function in heterozygotes, leading to a 50% reduction in gene expression, which significantly impacts cellular biology87,88. Besides, differences in the immune systems of humans and mice (60% homology) may contribute to the divergence between preclinical and clinical findings. For instance, inhibitory receptors on NK cells that recognize MHC class I molecules differ and do not exhibit cross-reactivity (human KIR versus mouse Ly49s)84,89. In mouse models, the genome is homogeneous, which makes it challenging to fully replicate the comprehensive landscape of immune therapy and resistance mechanisms observed in clinical settings due to the individual tumor characteristics84. These limitations hinder the translation of resistance mechanisms identified in immune therapy mouse models into clinical practice.
In sum, our findings offer a comprehensive molecular mechanism explanation for the combined anti-PD-1 and anti-LAG-3 antibody therapy in cancer.
Methods
Ethics statement
All animal studies described in this research were approved by the Institutional Animal Care and Use Committee, Sun Yat-Sen University (IACUC, SYSU). The animals were housed under specific pathogen-free conditions and managed at the Experimental Animal Center of Sun Yat-sen University. Approval numbers are SYSU-IACUC-2020-000437, SYSU-IACUC-2021-000121, and SYSU-IACUC-2023-000861. The Committee limits tumor growth to no more than 10% of the animal’s original body weight and the average tumor diameter to no more than 20 mm.
Animal studies
K14CreER (Strain ID: 005107) and Sox9CreER (Strain ID: 018829) mouse was obtained from the Jackson Laboratory (Bar Harbor, ME, USA). K14DreER (Strain ID: NM-KI-190125) and R26-Ai66-DTR (Strain ID: NM-KI-190086) mice were purchased from Shanghai Model Organisms (Shanghai, China). R26-Ai66-tdTomato mice were generated as previously described90. WT C57BL/6, LyzCreER (Strain ID: T052789), Anxa1flox (Strain ID: T016067), and Fpr1flox (Strain ID: T052005) mice were obtained from GemPharmatech (Nanjing, China). All animal subjects were accommodated under specific pathogen-free conditions, maintained on a 12 h light/dark circadian cycle, and granted ad libitum access to sustenance and hydration. All animal protocols received ethical clearance from the IACUC, SYSU, under the protocol numbers SYSU-IACUC-2020-000437, SYSU-IACUC-2021-000121, and SYSU-IACUC-2023-000861.
In our in vivo experiments, we ensured no bias in the selection of the sex of the mice. Each experimental group was randomly assigned an approximately equal distribution of male and female mice to maintain gender balance. For HNSCC induction in mice, 6-week-old mice were provided with water containing 4NQO (Sigma, N8141, diluted to 100 µg/mL) for 16 weeks. After that period, mice were given normal drinking water without 4NQO for another 8 weeks.
For the single or dual antibody blockade experiments, following a previously published dosing strategy20, mice were injected i.p. at a dosage of 10 mg/kg for anti-PD-1 (BioXcell, BE0146) and/or anti-LAG-3 (BioXcell, BE0174) after 8 weeks of normal water intake and the development of palpable tumors. Control rat IgG (BioXCell; BE0089, BE0088) was dosed at 20 mg/kg or added to individual anti-PD-1 or anti-LAG-3 antibody treatments at 10 mg/kg20. The injections were administered every 3 days. As previously described91, tumor volume was measured using calipers or MRI, with maximum diameters recorded in three orthogonal directions (length, width, height). The tumor volume was then calculated using the cuboid formula V = (π/6) × (L × W × H)/1000. For tumors that were not solitary, individual volumes were summed after calculation. The invasiveness of HNSCC was assessed using previously published methods92: showing signs of normal or epithelial dysplasia appearance (grade 1); distinct invasion, unclearness of the basement membrane, drop and diffuse infiltration into the superficial portion of the muscle layer (grade 2); loss of the basement membrane, extensive invasion into deep muscle layer (grade 3). As previously described93,94,95, CD8 T and γδT cell depletion in mice was performed. After identifying the Sens mice, they were injected i.p. with 200 µg of anti-CD8α (BioXcell, BE0061) and anti-TCR γ/δ (BioXcell, BE0070) or the appropriate isotype-matched control antibodies (BioXcell, BE0090, BE0091) every 4 days. This treatment was carried out over a 2-week period.
Cell culture and generation of mutant cell lines
The mouse Moc1 cell line was purchased from KERAFAST, INC. (Boston, MA, USA, EWL001-FP). HL60 cell line was purchased from the National Collection of Authenticated Cell Cultures (NCACC). 293 T cell line was obtained from the American Type Culture Collection (ATCC). Moc1 and 293T cell lines were in a 1:1 mixture of Dulbecco’s modified Eagle’s medium and Ham’s F12 medium (DMEM/F12, Gibco, C11330500BT) media supplemented with 10% fetal bovine serum (FBS, Gibco, 10270-106) and 1% penicillin/streptomycin (Gibco, 15140-122). Using previously published co-culture methods96,97,98, sorted Fpr1+ neutrophils were co-cultured with (or) Cd45+Cd3+ T cells in RPMI 1640 (Gibco, 11875500BT) supplemented with 10% FBS and 1% penicillin/streptomycin. To maintain neutrophil activity, Lipopolysaccharides (LPS, MCE, HY-D1056) were added at a concentration of 100 ng/mL97. HL60 was cultured in Iscove’s Modified Dulbecco’s Medium (IMDM, Gibco, 12440053) supplemented with 20% fetal bovine serum (FBS, Gibco, 10270-106) and 1% penicillin/streptomycin (Gibco, 15140-122) at a 37 °C incubator containing 5% CO2. Following previous research methods, 1.25% dimethyl sulfoxide (DMSO, TargetMol, T0341) was used to induce HL60 cells to differentiate along the granulocytic pathway99.
For stable knockdown of FPR1 in HL60 cells, we utilized the experimental protocol from our previous study100. Briefly, shRNAs (Supplementary Table 2) targeting FPR1 were inserted into the pLKO.1 plasmid and then co-transfected into 293T cells along with the packaging vector psPAX2 and the envelope vector pMD2.G using Lipofectamine 2000 reagent (Invitrogen, 11668019). The supernatant containing lentivirus was collected and added to the culture medium of HL60 cells in the presence of 10 μg/mL Polybrene (YEASEN, 40804ES76). After 48 h, positive clones were selected using 2.5 μg/mL puromycin (Beyotime, ST551-250 mg). Following a previously published method, HL60 cells were incubated with 500 nM ANXA1 (MCE, HY-P7512) for 6 h, or sorted Fpr1+ neutrophils were incubated with 500 nM Anxa1 (MCE, HY-P72078) for 6 h31. Following the manufacturer’s instructions, Fpr1+ neutrophils treated with or without Anxa1 were stained with MitoTracker Red (Thermo Fisher, A66443) and Hoechst 33342 (Beyotime, R0305S-6) under light-protected conditions at 37 °C for 20 min. After staining, images were captured using a super-resolution microscope (Nikon, Tokyo, Japan) in SIM mode. For luciferase reporter assay, pZX001-Anxa1-promoter-WT or pZX001-Anxa1-promoter- MUT was transfected into Moc1 cell lines with Sox9 overexpression. After incubation at 37 °C for 48 h, the luciferase activity was measured and analyzed using Renilla-Firefly Luciferase Dual Assay Kit (MCE, HY-K1013) according to the manufacturer’s instructions. Further details on reagents can be found in Supplementary Table 2.
MRI acquisition and interpretation
For MRI imaging, mice were anesthetized with an intraperitoneal administration of ketamine/xylazine. All mice were scanned with a 3.0T MR scanner (MAGNETOM Vida, Siemens Healthcare, Erlangen, Germany) using a 16-channel mouse coil (CG-MUC48-H300-AS, Shanghai Chenguang Medical Technology Co., Ltd., Shanghai, China). The imaging protocol comprised axial T2-weighted imaging (T2WI) with turbo spin echo (TSE), coronal T2WI with TSE, and axial T1-weighted imaging (T1WI) with StarVIBE.
ELISA
Following the instructions provided by the kit (Supplementary Table 2), briefly, the cell culture supernatant was centrifuged at 1000 × g for 10 min to remove impurities and cell debris, and the supernatant was set aside. Standard, blank, and sample wells were prepared. In the standard wells, 100 μL of diluted standard solution was added, while 100 μL of standard and sample dilution buffer was added to the blank wells. In the remaining wells, 100 μL of the test samples were added. The plate was then sealed and incubated at 37 °C for 90 min. After incubation, the liquid was discarded from each well, and 100 μL of biotinylated antibody working solution was added to each well, followed by incubation at 37 °C for 1 h. Subsequent steps included washing, HRP enzyme binding, addition of substrate solution, and termination solution. The optical density (OD) at 450 nm was measured using a microplate reader.
Histologic evaluation and immunohistochemical staining
For hematoxylin and eosin (H&E) staining, tumor tissues collected from mice were immediately fixed in formalin post-dissection. The fixed tissues underwent dehydration in ethanol, followed by soaking in histoclear, and were then embedded in paraffin. The processed tissues were sectioned (5 μm) with a microtome (Leica Histocore Biocut). The sections were deparaffinized and stained using the H&E staining kit (Solarbio, G1120-3). For immunohistochemistry (IHC) staining, sections were deparaffinized and treated with 3% hydrogen peroxide (H2O2) in water for 10 min. Antigen retrieval was conducted by immersing sections in 10 mM citrate buffer (pH 6.0) for 10 min. Subsequently, the sections were treated with 5% BSA Blocking Buffer (Solarbio, SW3015) for 10 min and incubated overnight at 4 °C with primary antibodies: anti-Caspase3 (Cell Signaling Technology, 9661S, 1:200), anti-Ki67 (Novus, NB500-170, 1:200), anti-Annexin I (Santa Cruz, sc-12740, 1:200), anti-Sox9 (Merck millipore, AB5535, 1:200), and anti-Fpr1 (Immunoway, YT1768, 1:100). The slides were then incubated with goat anti-rabbit or goat anti-mouse horseradish peroxidase-conjugated secondary antibodies for 30 min at room temperature, followed by treatment with 3,3′-diaminobenzidine (DAB) and counterstaining with hematoxylin. Further details on antibodies and reagents can be found in Supplementary Table 2. The intensity of staining was scored, and the percentage of positively stained areas in tumor cells relative to the entire tumor area was calculated for analysis.
Immunofluorescence staining
Mouse HNSCC tumors were dissected and fixed overnight in 4% paraformaldehyde in PBS. The samples were then rinsed with cold PBS, equilibrated in 30% sucrose in PBS overnight, and embedded in OCT (Tissue Tek, 25608-930). Tissue sections or cell climbing sheets were permeabilized with 1% Triton™ X-100 (Sigma-Aldrich, X-100) and stained with primary antibodies: anti-BNIP3 (Cell Signaling Technology, 44060, 1:100), anti-Sell (Santa Cruz, sc-390756, 1:200), anti-Ly6g (Abcam, ab25377; 1:100), anti-Fpr1 (Immunoway, YT1768; 1:100), anti-Granzyme A (ABclonal, A6231; 1:100), anti-Cd8a (Abcam, ab93278; 1:100), anti-Tcr g/d (Invitrogen, 13-5711-82; 1:100), anti-Annexin I (Santa Cruz, sc-12740, 1:200) and anti-Sox9 (Merck millipore, AB5535, 1:200). The antigens were then visualized using corresponding secondary antibodies conjugated with DyLight 488 and Fluor 594. Nuclei were counterstained with DAPI (Solarbio, C0065) at a dilution of 1:1000 for 1 min. Further details on antibodies and reagents can be found in Supplementary Table 2. Images were captured using an upright fluorescence microscope (ZEISS, Germany).
Protein extraction and western blot
Total proteins from the cells were extracted using RIPA lysis buffer (TIANYA BIO, P1024) supplemented with protease inhibitors. To obtain cytoplasmic proteins free of mitochondria, we followed the instructions of the Cell Mitochondria Isolation Kit (Beyotime Biotechnology, C3601) to isolate mitochondria-free cytoplasmic proteins from the cells. Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE, EpiZyme, PG11X) and transferred onto polyvinylidene fluoride (PVDF) membranes (Merck Millipore, IPVH00010). The membranes were incubated with primary antibodies against BNIP3 (Cell Signaling Technology, 44060, 1:2000), Cytochrome C (Cell Signaling Technology, 11940, 1:2000) and α-Tublin (Proteintech, 66031-1-Ig, 1:2000). Visualization of the blots was performed using the enhanced chemiluminescence (ECL) method with species-specific horseradish peroxidase-conjugated secondary antibodies from Proteintech (anti-rabbit, SA00001-2, 1:2000; anti-mouse, SA00001-1, 1:2000).
Sample preparation and imaging for transmission electron microscopy
Cell lysates were fixed at 4 °C for over 2 h using 2.5% neutral glutaraldehyde (Alfa Aesar, A17876). Following fixation, the samples were washed six times with PBS, each for 30 min. Subsequently, they were fixed again using 1% osmium acid (Ted Pella, 18456) at 4 °C for 1.5–2 h, followed by three PBS washes, each for 10 min. After performing a series of dehydration and resin infiltration steps, the samples were embedded in resin (Ted Pella, GP18010). The embedded samples were then cured in an oven at 38 °C for 6 h, 60 °C for 6 h, and finally at 80 °C for 12 h. The cured samples were sectioned into 50–70 nm slices using a Leica ultramicrotome (UC-7, Germany). The slices were stained with 2% uranyl acetate (EMS, 22400) and 3% lead citrate (Ted Pella, 19314). Finally, the stained slices were imaged and measured using a transmission electron microscope (JEM-1400, Japan Electron Optics Laboratory Co., Ltd., Tokyo, Japan).
Single-cell RNA sequencing
Fresh oral tumor tissues from mice were carefully minced and dissociated at 37 °C using the Mouse Tumor Dissociation Kit (130-096-730, Miltenyi) on a gentle MACS Octo Dissociator. The dissociated tissues were then filtered to produce single-cell suspensions. These cells were subsequently washed with ACK lysing buffer (Thermo Fisher Scientific, A1049201), and their viability and count were evaluated using trypan blue staining. Prepared single-cell suspensions were processed according to the 10x Genomics protocol using the Chromium Controller, which rapidly generated single-cell GEMs (Gel Beads in Emulsions) and synthesized cDNA through reverse transcription. In each microwell, a magnetic bead and a single cell were present, with each cell uniquely labeled by multiple molecular markers known as Unique Molecular Identifiers (UMIs). These UMIs enabled efficient RNA capture once the cells were lysed, allowing the RNA to hybridize with the beads. Subsequently, a GEMs-breaking reaction system was prepared to disrupt the GEMs, followed by purification and recovery of the resulting product. The purified product was then subjected to cDNA amplification and another round of purification. The amplified cDNAs are subjected to fragmentation, end repair, the addition of “A” base at the 3′-end of each strand and purification. The product was then ligated with adapters, followed by another round of purification. A PCR reaction system was prepared, and PCR amplification was performed to generate the library product. After stringent quality control of the library, the PCR product was denatured into single strands, and a cyclization reaction was conducted to obtain single-stranded circular products, with linear DNA molecules that were not circularized being digested. The single-stranded circular DNA molecules underwent rolling circle replication to form DNA nanoballs (DNBs) containing multiple copies. These DNBs were then loaded into the mesh-like wells on a high-density DNA nanofabricated chip and sequenced using combinatorial Probe-Anchor Synthesis (cPAS) technology.
Processing and clustering of scRNA-seq data
After sequencing, raw reads were processed using the mkfastq command from Cell Ranger (Version 4.0.0, 10x Genomics), which assigns unique barcodes to reads, enabling the identification of sequences corresponding to individual cells. The resulting FASTQ files were then aligned to the mm10 reference genome (GRCm38.91) using Cell Ranger’s count command, generating cell-gene-barcode matrices that capture gene expression profiles for each cell. These matrices were subsequently merged for downstream analysis using the Seurat R package (version 4.0.0).
Before proceeding with downstream analysis, several quality control measures were implemented to exclude low-quality cells and genes. Genes appearing in fewer than three cells per sample were removed. Cells were filtered out if they expressed fewer than 200 genes or more than 5000 genes, to eliminate empty droplets or doublets. Additionally, cells exhibiting over 10% mitochondrial or hemoglobin gene expression were also excluded, as these are indicators of low-quality or dying cells. Potential doublets were further identified and removed using the DoubletFinder R package (version 2.0.3) with default settings. These quality assurance steps were conducted individually for each sample before merging the count matrices.
Post quality control, we employed the “harmony” function in Seurat to integrate and normalize the data across different samples, thereby minimizing batch effects. Following data integration and normalization, principal component analysis (PCA) was performed on a selection of highly variable genes using the RunPCA function in Seurat. We selected a resolution of 0.2 for clustering the cells, which were subsequently visualized using the UMAP (Uniform Manifold Approximation and Projection) technique with the RunUMAP function. Cell clusters were delineated using the FindClusters function.
Each cell cluster was annotated based on the expression of canonical marker genes, which was accomplished using the FindAllMarkers function with a likelihood-ratio test for single-cell gene expression analysis. This method facilitates the identification of significant marker genes within each cluster, enabling precise cell type classification. To evaluate the cell cycle state of each cluster, the CellCycleScoring function in Seurat was utilized, calculating scores based on established markers of the S and G2/M phases.
Cell type annotation
Marker genes were identified using the FindAllMarkers function, and cell types were annotated based on the most prominent marker genes along with well-known classical markers. This analysis uncovered five primary cell types within the studied population, including various immune cells and non-immune cells such as epithelial cells, endothelial cells, muscle cells, and fibroblasts. To further refine the analysis, a second round of clustering was performed using the same parameters to detect subclusters within the major cell types identified earlier.
Identification of malignant cells
To identify malignant cells within the epithelial populations, the CopyKAT algorithm was applied to estimate copy number variations (CNVs). A systematic sampling approach was used, where 10 to 100 malignant cells were selectively sampled in increments of 10 cells. Cells classified as aneuploid by CopyKAT were then specifically targeted for further analysis.
Cell-cell communication analysis
To explore intercellular communication within the tumor microenvironment, we utilized both CellPhoneDB (https://www.cellphonedb.org) and the CellChat (version 1.1.3; http://www.cellchat.org/) R package. scRNA-seq data from various cell types, including epithelial cells, endothelial cells, muscle cells, immune cells, and fibroblasts, were input into CellPhoneDB or CellChat for analysis. The kpy.plot_cpdb function was then used to identify upregulated and downregulated signaling ligand pairs across various pathways. For additional insights, the CellChat R package (version 1.1.3) was employed to construct a cellchat object with the normalized expression matrix. The cell type annotations were assigned based on the results from the Harmony integration, which combined all single-cell datasets. Cellular interaction strength and number were visualized using the netVisual_circle function. Based on the differences in overall information flow inferred from the networks of epithelial and immune cell subgroups, the rankNet function was employed to rank significant signaling pathways. Subsequently, the netAnalysis_signalingRole_scatter function was used to visualize the signal incoming and outgoing within the ANNEXIN pathway between these cell subgroups. The netVisual_bubble function was then used to identify upregulated and downregulated signaling ligand pairs within various pathways. Default parameters were applied for all steps in the analysis. CellPhoneDB was employed to investigate potential ligand-receptor interactions between Neu1 neutrophils and T cells, following the default pipeline of the package. To facilitate cross-species comparisons, mouse genes were mapped to human homologs using Biomart. For the interaction analysis, non-log-transformed UMI counts were used to calculate the expression levels of receptors and ligands, as described by the methodology in the original reference26.
Trajectory analysis of neutrophils
For velocity analysis, we began with FASTQ files, aligned them to the genome to obtain sam files, converted sam files to bam files, and then extracted spliced, unspliced, and ambiguous information from the bam files to generate.loom files. The plotting process was conducted in R using the velocyto.R package. Cell trajectory UMAP plots were generated with the show.velocity.on.embedding.cor function. CytoTRACE assesses cellular differentiation states based on gene expression diversity: undifferentiated stem cells typically exhibit high gene expression diversity, while differentiated cells show lower diversity. By analyzing gene expression data, CytoTRACE infers cell differentiation trajectories and assigns a “differentiation potential score” to each cell. This process was conducted in R using the CytoTRACE function to calculate the scores and the plotCytoTRACE function for visualization.
Flow cytometry
As previously mentioned in the “Single-cell RNA sequencing” section of the Methods, a single-cell suspension of mouse oral tumor cells was prepared. Before staining, the cells were washed with staining buffer (2% bovine growth serum in PBS) and adjusted to a concentration of 1 × 107 cells/mL. The cells were then stained with antibodies and incubated in a staining buffer for 2 h at 4 °C to complete the extracellular staining process. For intracellular marker staining, cells underwent surface staining followed by fixation and permeabilization using a fixation/permeabilization solution (BD Biosciences, 554714) according to the manufacturer’s instructions. Intracellular cytokine staining was then performed using antibodies against Prf1 and Gzmb. The antibodies and dyes listed below were utilized for flow cytometry: Ghost Dye™ Red 780 (Tonbo Biosciences, 13-0865), PE-Cyanine7 Anti-Mouse CD45 (Tonbo Biosciences, 60-0451), APC Anti-Mouse CD3 (Tonbo Biosciences, 20-0032), PE Anti-Mouse Ly-6G (Tonbo Biosciences, 50-1276-U025), anti-Sell (Santa Cruz, sc-390756), anti-Fpr1 (Novus, NLS1878), Brilliant Violet 510™ Donkey anti-rabbit (BioLegend, 406419) and Brilliant Violet 510™ Goat anti-mouse (BioLegend, 405331). Further details on antibodies and reagents can be found in Supplementary Table 2. Samples were analyzed on a flow cytometer (CytoFLEX, Beckman Coulter, Brea, USA), and the resulting data were processed using CytExpert software (version 2.5.0.77). Sorting of live Fpr1+, Sell+ neutrophils or Cd45+Cd3+ T cells were performed on a flow cytometer (FACSAria Fusion, BD, Franklin Lakes, USA).
mRNA sequencing
Cell pellets obtained from flow cytometry sorting were collected, and total RNA (three mice in each group) was extracted using the AG RNAex Pro Reagent (Accurate Biotechnology, AG21102) according to the manufacturer’s instructions. Subsequently, mRNA transcriptome libraries were constructed using the Hieff NGS® Ultima Dual-mode mRNA Library Prep Kit for Illumina® (Yeasen, 12308ES96), following the manufacturer’s instructions. In brief, mRNA Capture Beads were added to 1 μg of total RNA to bind the mRNA to the beads. The beads were then washed and resuspended with Beads Wash Buffer and Tris Buffer. Fragmentation of the mRNA was performed by adding Frag/Prime Buffer and incubating at 94 °C for 7 min. The fragmented mRNA was reverse-transcribed to synthesize the first-strand cDNA, followed by the synthesis of the second-strand cDNA. Adapters were then ligated to the synthesized cDNA, and the ligated products were purified twice using DNA Selection Beads. The purified ligated products were subjected to PCR amplification, and the amplified products were further purified with DNA Selection Beads to obtain the final library. The libraries were subsequently examined using an Agilent 2100 Bioanalyzer and a Qubit 3.0 Fluorometer (Invitrogen). Finally, the libraries were sequenced using the high-throughput sequencing platform (DNBSEQ-T7, MGI, Shenzhen, China).
Statistics and reproducibility
Statistical analyses were conducted using GraphPad Prism 8 software. Data are expressed as mean ± SD unless otherwise noted. Statistical significance was defined as p < 0.05. For comparisons, unpaired Student’s T test (including nonparametric tests for two groups), one-way ANOVA (for three or more groups), Log-rank test (for overall survival curve), and Pearson chi-square test (for analyzing tumor invasion in transgenic mice) were employed. Significance levels were set at *p < 0.05, **p < 0.01, ***p < 0.001. Source data and exact p values are provided as a Source Data file. The violin plot is a combination of a box plot and a kernel density plot. In the box plot, the upper side of the box represents the third quartile (Q3), the lower side represents the first quartile (Q1), and the length of the box represents the interquartile range (IQR). The second quartile (Q2), also known as the median (Md), is depicted as a horizontal line inside the box. The endpoints of the two vertical lines outside the box represent the minimum and maximum values, respectively, and the points outside the box represent outliers. Kernel density estimation is displayed on each side of the box plot to illustrate the distribution shape of the data. Wider sections of the violin plot indicate a higher probability of the population members taking on the given value, while narrower sections indicate a lower probability. The definition of the box plots applies to Fig. 2K, L, as well as Fig. 6B, C. The experiments in Fig. 5G, H were independently repeated twice, and the experiment in Fig. 6H was independently repeated three times.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The scRNA-seq data generated in this study have been deposited in the Genome Sequence Archive in the National Genomics Data Center under accession number code CRA018502. The remaining data are available within the Article, Supplementary Information, or Source Data file. For additional details or requests for resources and reagents, please contact the corresponding author, Demeng Chen, at [email protected]. Source data are provided with this paper.
Change history
30 May 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41467-025-60473-2
References
Topalian, S. L., Drake, C. G. & Pardoll, D. M. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27, 450–461 (2015).
Yarchoan, M., Hopkins, A. & Jaffee, E. M. Tumor mutational burden and response rate to PD-1 inhibition. N. Engl. J. Med. 377, 2500–2501 (2017).
Nghiem, P. T. et al. PD-1 blockade with pembrolizumab in advanced Merkel-cell carcinoma. N. Engl. J. Med. 374, 2542–2552 (2016).
Topalian, S. L., Taube, J. M., Anders, R. A. & Pardoll, D. M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 16, 275–287 (2016).
Gutierrez, M. et al. Biomarker-directed, pembrolizumab-based combination therapy in non-small cell lung cancer: phase 2 KEYNOTE-495/KeyImPaCT trial interim results. Nat. Med. 29, 1718–1727 (2023).
Fu, Y., Tian, G., Li, J., Zhang, Z. & Xu, K. An HNSCC syngeneic mouse model for tumor immunology research and preclinical evaluation. Int. J. Mol. Med. 46, 1501–1513 (2020).
Xiong, H. et al. Near-infrared-II Ag(2)S quantum dot probes targeting podoplanin enhance immunotherapy in oral squamous cell carcinoma. Biomed. Pharmacother. 170, 116011 (2024).
Elmusrati, A. & Wang, C. Y. The expression of immune checkpoint proteins PD-L1 and TIM3 in mouse and human head and neck squamous cell carcinoma. Eur. J. Oral Sci. 132, e13010 (2024).
Lei, Q., Wang, D., Sun, K., Wang, L. & Zhang, Y. Resistance mechanisms of anti-PD1/PDL1 therapy in solid tumors. Front. Cell Dev. Biol. 8, 672 (2020).
Dong, Y. et al. Mass cytometry and transcriptomic profiling reveal PD1 blockade induced alterations in oral carcinogenesis. Mol. Carcinog. 63, 563–576 (2024).
Zhang, Y. et al. Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell 39, 1578–1593.e1578 (2021).
Zhou, L. et al. Anlotinib plus camrelizumab achieved long-term survival in a patient with metastatic esophageal neuroendocrine carcinoma. Cancer Rep. 6, e1855 (2023).
Garralda, E. et al. A first-in-human study of the anti-LAG-3 antibody favezelimab plus pembrolizumab in previously treated, advanced microsatellite stable colorectal cancer. ESMO Open 7, 100639 (2022).
Harris-Bookman, S. et al. Expression of LAG-3 and efficacy of combination treatment with anti-LAG-3 and anti-PD-1 monoclonal antibodies in glioblastoma. Int. J. Cancer 143, 3201–3208 (2018).
Cillo, A. R. et al. Blockade of LAG-3 and PD-1 leads to co-expression of cytotoxic and exhaustion gene modules in CD8(+) T cells to promote antitumor immunity. Cell 187, 4373–4388.e4315 (2024).
Andrews, L. P. et al. LAG-3 and PD-1 synergize on CD8(+) T cells to drive T cell exhaustion and hinder autocrine IFN-gamma-dependent anti-tumor immunity. Cell 187, 4355–4372.e4322 (2024).
Ngiow, S. F. et al. LAG-3 sustains TOX expression and regulates the CD94/NKG2-Qa-1b axis to govern exhausted CD8 T cell NK receptor expression and cytotoxicity. Cell 187, 4336–4354.e4319 (2024).
Kisielow, M., Kisielow, J., Capoferri-Sollami, G. & Karjalainen, K. Expression of lymphocyte activation gene 3 (LAG-3) on B cells is induced by T cells. Eur. J. Immunol. 35, 2081–2088 (2005).
Workman, C. J., Dugger, K. J. & Vignali, D. A. Cutting edge: molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J. Immunol. 169, 5392–5395 (2002).
Woo, S. R. et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 72, 917–927 (2012).
Deutsch, J. S. et al. Immune microenvironment of basal cell carcinoma and tumor regression following combined PD-1/LAG-3 blockade. J. Immunother. Cancer 11, https://doi.org/10.1136/jitc-2023-007463 (2023).
Tawbi, H. A. et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 386, 24–34 (2022).
Albrecht, L. J., Livingstone, E., Zimmer, L. & Schadendorf, D. The latest option: nivolumab and relatlimab in advanced melanoma. Curr. Oncol. Rep. 25, 647–657 (2023).
Gao, R. et al. Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat. Biotechnol. 39, 599–608 (2021).
Jin, S. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Efremova, M., Vento-Tormo, M., Teichmann, S. A. & Vento-Tormo, R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 15, 1484–1506 (2020).
Gulati, G. S. et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science 367, 405–411 (2020).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e1821 (2019).
Dalli, J. et al. Proresolving and tissue-protective actions of annexin A1-based cleavage-resistant peptides are mediated by formyl peptide receptor 2/lipoxin A4 receptor. J. Immunol. 190, 6478–6487 (2013).
Cooray, S. N. et al. Ligand-specific conformational change of the G-protein-coupled receptor ALX/FPR2 determines proresolving functional responses. Proc. Natl. Acad. Sci. USA 110, 18232–18237 (2013).
Solito, E. et al. A novel calcium-dependent proapoptotic effect of annexin 1 on human neutrophils. FASEB J. 17, 1544–1546 (2003).
Srinivasan, S., Guha, M., Kashina, A. & Avadhani, N. G. Mitochondrial dysfunction and mitochondrial dynamics—the cancer connection. Biochim. Biophys. Acta Bioenerg. 1858, 602–614 (2017).
Lin, Q. et al. Inhibiting NLRP3 inflammasome attenuates apoptosis in contrast-induced acute kidney injury through the upregulation of HIF1A and BNIP3-mediated mitophagy. Autophagy 17, 2975–2990 (2021).
Yao, J. et al. CDK9 inhibition blocks the initiation of PINK1-PRKN-mediated mitophagy by regulating the SIRT1-FOXO3-BNIP3 axis and enhances the therapeutic effects involving mitochondrial dysfunction in hepatocellular carcinoma. Autophagy 18, 1879–1897 (2022).
Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 11, 872–884 (2010).
Zhang, Y. & Zhang, Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 17, 807–821 (2020).
Hegde, P. S. & Chen, D. S. Top 10 challenges in cancer immunotherapy. Immunity 52, 17–35 (2020).
Luo, B. et al. Anlotinib benefits the alphaPDL1 immunotherapy by activating ROS/JNK/AP-1 pathway to upregulate PDL1 expression in colorectal cancer. Oxid. Med. Cell. Longev. 2022, 8965903 (2022).
Wang, D. et al. A comprehensive profile of TCF1(+) progenitor and TCF1(-) terminally exhausted PD-1(+)CD8(+) T cells in head and neck squamous cell carcinoma: implications for prognosis and immunotherapy. Int. J. Oral Sci. 14, 8 (2022).
Tichet, M. et al. Bispecific PD1-IL2v and anti-PD-L1 break tumor immunity resistance by enhancing stem-like tumor-reactive CD8(+) T cells and reprogramming macrophages. Immunity 56, 162–179.e166 (2023).
Aggarwal, V., Workman, C. J. & Vignali, D. A. A. LAG-3 as the third checkpoint inhibitor. Nat. Immunol. 24, 1415–1422 (2023).
Barbosa, S. et al. The role of SOX2 and SOX9 in radioresistance and tumor recurrence. Cancers 16, https://doi.org/10.3390/cancers16020439 (2024).
Khorani, K. et al. Establishment of a plasticity-associated risk model based on a SOX2- and SOX9-related gene set in head and neck squamous cell carcinoma. Mol. Cancer Res. 19, 1676–1687 (2021).
Sharma, A. et al. Longitudinal single-cell RNA sequencing of patient-derived primary cells reveals drug-induced infidelity in stem cell hierarchy. Nat. Commun. 9, 4931 (2018).
Al-Ali, H. N. et al. A therapeutic antibody targeting annexin-A1 inhibits cancer cell growth in vitro and in vivo. Oncogene 43, 608–614 (2024).
Perretti, M. & D’Acquisto, F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat. Rev. Immunol. 9, 62–70 (2009).
Sugimoto, M. A., Vago, J. P., Teixeira, M. M. & Sousa, L. P. Annexin A1 and the resolution of inflammation: modulation of neutrophil recruitment, apoptosis, and clearance. J. Immunol. Res. 2016, 8239258 (2016).
Hallett, J. M. et al. Novel pharmacological strategies for driving inflammatory cell apoptosis and enhancing the resolution of inflammation. Trends Pharmacol. Sci. 29, 250–257 (2008).
Headland, S. E. & Norling, L. V. The resolution of inflammation: principles and challenges. Semin. Immunol. 27, 149–160 (2015).
Nathan, C. & Ding, A. Nonresolving inflammation. Cell 140, 871–882 (2010).
Araujo, T. G. et al. Annexin A1 as a regulator of immune response in cancer. Cells 10, https://doi.org/10.3390/cells10092245 (2021).
Boudhraa, Z., Bouchon, B., Viallard, C., D’Incan, M. & Degoul, F. Annexin A1 localization and its relevance to cancer. Clin. Sci. 130, 205–220 (2016).
Bai, X. F. et al. Overexpression of annexin 1 in pancreatic cancer and its clinical significance. World J. Gastroenterol. 10, 1466–1470 (2004).
Cheng, T. Y. et al. Annexin A1 is associated with gastric cancer survival and promotes gastric cancer cell invasiveness through the formyl peptide receptor/extracellular signal-regulated kinase/integrin beta-1-binding protein 1 pathway. Cancer 118, 5757–5767 (2012).
Rong, B. et al. Elevated serum annexin A1 as potential diagnostic marker for lung cancer: a retrospective case-control study. Am. J. Transl. Res. 6, 558–569 (2014).
Ydy, L. R. et al. Study of the annexin A1 and its associations with carcinoembryonic antigen and mismatch repair proteins in colorectal cancer. J. Gastrointest. Cancer 47, 61–68 (2016).
Yom, C. K. et al. Clinical significance of annexin A1 expression in breast cancer. J. Breast Cancer 14, 262–268 (2011).
Gibbs, L. D. & Vishwanatha, J. K. Prognostic impact of AnxA1 and AnxA2 gene expression in triple-negative breast cancer. Oncotarget 9, 2697–2704 (2018).
Liew, P. X. & Kubes, P. The neutrophil’s role during health and disease. Physiol. Rev. 99, 1223–1248 (2019).
Kang, L. et al. Neutrophil-epithelial crosstalk during intestinal inflammation. Cell. Mol. Gastroenterol. Hepatol. 14, 1257–1267 (2022).
Karmakar, M. et al. N-GSDMD trafficking to neutrophil organelles facilitates IL-1beta release independently of plasma membrane pores and pyroptosis. Nat. Commun. 11, 2212 (2020).
Wang, S. et al. Neutrophil-derived PAD4 induces citrullination of CKMT1 exacerbates mucosal inflammation in inflammatory bowel disease. Cell. Mol. Immunol. 21, 620–633 (2024).
Chung, K. P. et al. Alveolar epithelial cells mitigate neutrophilic inflammation in lung injury through regulating mitochondrial fatty acid oxidation. Nat. Commun. 15, 7241 (2024).
Liu, S. et al. Cis-acting lnc-Cxcl2 restrains neutrophil-mediated lung inflammation by inhibiting epithelial cell CXCL2 expression in virus infection. Proc. Natl. Acad. Sci. USA 118, https://doi.org/10.1073/pnas.2108276118 (2021).
Xiong, S., Dong, L. & Cheng, L. Neutrophils in cancer carcinogenesis and metastasis. J. Hematol. Oncol. 14, 173 (2021).
Finisguerra, V. et al. MET is required for the recruitment of anti-tumoural neutrophils. Nature 522, 349–353 (2015).
Singhal, S. et al. Origin and role of a subset of tumor-associated neutrophils with antigen-presenting cell features in early-stage human lung cancer. Cancer Cell 30, 120–135 (2016).
Li, C. et al. Spatial and single-cell transcriptomics reveal a cancer-associated fibroblast subset in HNSCC That restricts infiltration and antitumor activity of CD8+ T cells. Cancer Res. 84, 258–275 (2024).
Hay, Z. L. Z. & Slansky, J. E. Granzymes: the molecular executors of immune-mediated cytotoxicity. Int. J. Mol. Sci. 23, https://doi.org/10.3390/ijms23031833 (2022).
Ren, S. et al. Intratumoral CD103(+) CD8(+) T cells predict response to neoadjuvant chemoimmunotherapy in advanced head and neck squamous cell carcinoma. Cancer Commun. 43, 1143–1163 (2023).
Gao, Z. et al. Gamma delta T-cell-based immune checkpoint therapy: attractive candidate for antitumor treatment. Mol. Cancer 22, 31 (2023).
Haas, W., Pereira, P. & Tonegawa, S. Gamma/delta cells. Annu. Rev. Immunol. 11, 637–685 (1993).
Lo Presti, E., Dieli, F., Fournie, J. J. & Meraviglia, S. Deciphering human gammadelta T cell response in cancer: lessons from tumor-infiltrating gammadelta T cells. Immunol. Rev. 298, 153–164 (2020).
Wu, Y. et al. An innate-like Vdelta1(+) gammadelta T cell compartment in the human breast is associated with remission in triple-negative breast cancer. Sci. Transl. Med. 11, https://doi.org/10.1126/scitranslmed.aax9364 (2019).
Lu, H. et al. High abundance of intratumoral gammadelta T cells favors a better prognosis in head and neck squamous cell carcinoma: a bioinformatic analysis. Front. Immunol. 11, 573920 (2020).
Song, Y., Liu, Y., Teo, H. Y. & Liu, H. Targeting cytokine signals to enhance gammadeltaT cell-based cancer immunotherapy. Front. Immunol. 13, 914839 (2022).
Chaudhary, S. et al. Differential mutation spectrum and immune landscape in African Americans versus Whites: a possible determinant to health disparity in head and neck cancer. Cancer Lett. 492, 44–53 (2020).
Willcox, C. R. et al. Butyrophilin-like 3 directly binds a human Vgamma4(+) T cell receptor using a modality distinct from clonally-restricted antigen. Immunity 51, 813–825.e814 (2019).
Castella, B., Melaccio, A., Foglietta, M., Riganti, C. & Massaia, M. Vgamma9Vdelta2 T cells as strategic weapons to improve the potency of immune checkpoint blockade and immune interventions in human myeloma. Front. Oncol. 8, 508 (2018).
Young, L. The Jackson Laboratory. Mol. Med. 4, 201–204 (1998).
Ngiow, S. F., Loi, S., Thomas, D. & Smyth, M. J. Mouse models of tumor immunotherapy. Adv. Immunol. 130, 1–24 (2016).
Vitale-Cross, L. et al. Chemical carcinogenesis models for evaluating molecular-targeted prevention and treatment of oral cancer. Cancer Prev. Res. 2, 419–422 (2009).
Garcia-Gonzalez, I., Muhleder, S., Fernandez-Chacon, M. & Benedito, R. Genetic tools to study cardiovascular biology. Front. Physiol. 11, 1084 (2020).
Chiu, W. C., Ou, D. L. & Tan, C. T. Mouse models for immune checkpoint blockade therapeutic research in oral cancer. Int. J. Mol. Sci. 23, https://doi.org/10.3390/ijms23169195 (2022).
Li, E., Lin, L., Chen, C. W. & Ou, D. L. Mouse models for immunotherapy in hepatocellular carcinoma. Cancers 11, https://doi.org/10.3390/cancers11111800 (2019).
Chulpanova, D. S., Kitaeva, K. V., Rutland, C. S., Rizvanov, A. A. & Solovyeva, V. V. Mouse tumor models for advanced cancer immunotherapy. Int. J. Mol. Sci. 21, https://doi.org/10.3390/ijms21114118 (2020).
Oladipupo, S. S., Kabir, A. U., Smith, C., Choi, K. & Ornitz, D. M. Impaired tumor growth and angiogenesis in mice heterozygous for Vegfr2 (Flk1). Sci. Rep. 8, 14724 (2018).
Gale, N. W. et al. Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc. Natl. Acad. Sci. USA 101, 15949–15954 (2004).
Sternberg-Simon, M. et al. Natural killer cell inhibitory receptor expression in humans and mice: a closer look. Front. Immunol. 4, 65 (2013).
Han, X. et al. Lineage tracing reveals the bipotency of SOX9(+) hepatocytes during liver regeneration. Stem Cell Rep. 12, 624–638 (2019).
Zhang, C. et al. ITGB6 modulates resistance to anti-CD276 therapy in head and neck cancer by promoting PF4(+) macrophage infiltration. Nat. Commun. 15, 7077 (2024).
Jia, L., Zhang, W. & Wang, C. Y. BMI1 inhibition eliminates residual cancer stem cells after PD1 Blockade and activates antitumor immunity to prevent metastasis and relapse. Cell Stem Cell 27, 238–253.e236 (2020).
Kak, G. et al. IL-10 production by granulocytes promotes Staphylococcus aureus craniotomy infection. J. Neuroinflamm. 20, 114 (2023).
Abou-El-Hassan, H. et al. Vgamma1 and Vgamma4 gamma-delta T cells play opposing roles in the immunopathology of traumatic brain injury in males. Nat. Commun. 14, 4286 (2023).
Burrack, A. L. et al. Distinct myeloid antigen-presenting cells dictate differential fates of tumor-specific CD8+ T cells in pancreatic cancer. JCI Insight 7, https://doi.org/10.1172/jci.insight.151593 (2022).
Swamydas, M. & Lionakis, M. S. Isolation, purification and labeling of mouse bone marrow neutrophils for functional studies and adoptive transfer experiments. J. Vis. Exp. e50586 https://doi.org/10.3791/50586 (2013).
Wu, Y. et al. Neutrophil profiling illuminates anti-tumor antigen-presenting potency. Cell 187, 1422–1439.e1424 (2024).
Wishart, A. L., Swamydas, M. & Lionakis, M. S. Isolation of mouse neutrophils. Curr. Protoc. 3, e879 (2023).
Hallows, K. R. & Frank, R. S. Changes in mechanical properties with DMSO-induced differentiation of HL-60 cells. Biorheology 29, 295–309 (1992).
Wang, X., Ling, R., Peng, Y., Qiu, W. & Chen, D. RNPS1 stabilizes NAT10 protein to facilitate translation in cancer via tRNA ac(4)C modification. Int. J. Oral Sci. 16, 6 (2024).
Acknowledgements
We thank the members of the Lin and Zhu labs for reagents and helpful discussions. This work is supported by the National Natural Science Foundation of China (82173362, 81872409, 82273109, and 82304069), Guangzhou Municipal Science and Technology Bureau (2024B01J1172 and 2024B03J1384), Guangdong Natural Science Foundation (2022A1515012234), and China Postdoctoral Science Foundation (2023M734003). The schematic diagrams in Supplementary Figs. 3E and 5B were created with MedPeer (medpeer.cn).
Author information
Authors and Affiliations
Contributions
Conceptualization: X.W. and D.C.; methodology: X.W. and M.C.; data analysis and curation: X.W., C.Z., R.L., S.Q., K.C. and D.C.; investigation and validation: X.W., M.C., S.Q. and K.C.; resources: X.W., S.C., B.Z., W.L., and D.C.; writing—original draft: X.W. and D.C.; writing—review and editing: X.W. and D.C.; supervision and funding acquisition: S.C., Q.L., and D.C.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Taiwen Li, Theresa Whiteside, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, X., Cheng, M., Chen, S. et al. Resistance to anti-LAG-3 plus anti-PD-1 therapy in head and neck cancer is mediated by Sox9+ tumor cells interaction with Fpr1+ neutrophils. Nat Commun 16, 3975 (2025). https://doi.org/10.1038/s41467-025-59050-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59050-4
