
Abstract
The rational design of robust nanocatalysts containing the suitable active sites for building relevant organic compounds, such as lactams, is a desired approximation towards the development of a sustainable fine chemistry field. In that sense, the design of a proper nanomaterial able to mediate the selective hydrodeoxygenation of cyclic imides to lactams with high tolerance to the preservation of aromatic rings remains rather unexplored. Here, we show the design of a bimetallic AgRe nanomaterial with notable activity and selectivity to mediate this transformation affording more than 60 lactams from the corresponding imides. Interestingly, in this work we disclose that the optimal AgRe nanocatalyst is constituted by AgReO4 nanoaggregates that undergo an in situ hydrogenative dispersion to form the active centers composed by Ag0 nanoparticles and ReOx species. Deep characterization, together with kinetic and mechanistic studies, have revealed that the intimate Ag-Re contact intrinsic to AgReO4 species is key for the formation of the most active catalytic sites and the proper bimetallic cooperation required for mediating the desired process.
Similar content being viewed by others
Introduction
Catalysis is one of the main driving forces of current organic synthesis as it allows not only the development of new transformations, but also paves the way for a more sustainable fine chemical industry. In this direction, the rational design of multifunctional, solid and stable nanomaterials with the specific structural and catalytic features for promoting challenging chemical transformations with a total selectivity control is a very interesting approach1,2,3. Furthermore, this strategy combined with the use of clean reagents, such as hydrogen, and atom-economical processes follows the desired direction for meeting the requirements of green chemistry4,5.
Hydrogenation and hydrogenative functionalization of carboxylic acid derivatives are atom-economical protocols employing stable and readily available substrates6,7,8,9,10,11,12 (Fig. 1a). However, the activation of these compounds is a challenge due to the low susceptibility towards hydride attack of their poorly electrophilic carbonyl group7,8,10,13. Moreover, another difficulty associated with the hydrogenation of carboxylic acid derivatives is related with the selectivity control. In general, esters, amides and imides hydrogenation can follow either a C-X hydrogenolysis pathway or a C-O hydrogenation via, also known as hydrodeoxygenation (HDO), being the occurrence of one of these pathways strongly influenced by the reaction media pH (Fig. 1a). C-O hydrogenation of esters or amides, favoured in acidic media, is a highly sought transformation as it can afford ethers and amines, respectively, in a straightforward manner10,11. In the last years, a successful strategy to activate carboxylic acid derivatives and drive their hydrogenation through a C-O pathway has been developed based on the design of bimetallic nanomaterials that combine a transition metal presenting oxophilic character, and hence able to activate the carbonyl group, (commonly V, Mo, Sn, W or Re), with another metal with capacity to activate H2 (usually Pd or Pt) (Fig. 1a)7,8,10,14,15,16,17. Employing this strategy, several successful and general protocols for the hydrogenation of amides, carboxylic acids, and esters have been described. Prominent examples in the field include the general hydrogenation of amides using [PdRe/Graphite]18 or [PtV/HAP]19, and the more recently described ester-to-ether hydrogenation with [PtMo/ZrO2]20, among others.

a General mechanism of carboxylic acid derivatives hydrogenation and main approach for the design of supported bimetallic nanocatalysts. b Relevance of N-CH2-lactams. c Described general methodologies for the hydrogenative functionalization of cyclic imides. d This work: General synthesis of lactams by a highly selective catalytic hydrogenation of cyclic imides through the use of a bimetallic [AgRe/Al2O3] nanomaterial.
Cyclic imides are a particular kind of carboxylic acid derivative in which a nitrogen atom is directly connected to two carbonyl groups6,10,21 (Fig. 1c). Thus, these compounds present a slightly more electrophilic carbonyl group in comparison with amides, and hence, are more activated towards hydrogenation. The selective reduction of one carbonyl group of cyclic imides via hydrodeoxygenation, such as phthalimides, succinimides, glutarimides or homophthalimides, is a very interesting reaction as it affords lactams, considered privileged scaffolds in medicinal chemistry. In fact, amides have been recently quantified as the most represented functional group in biologically active molecules22 (Fig. 1b). Moreover, in the top 10 of best-selling pharmaceuticals in 2022 three molecules bearing a lactam core with a CH2 directly attached to the N atom can be found (Fig. 1b) (https://www.drugdiscoverytrends.com/50-of-2022s-best-selling-pharmaceuticals/). More specifically, nirmatrelvir23 (4th, commercialized as Paxlovid, 18.900 M of dollars), containing a pyrrolidone moiety and used in combination with ritonavir for the treatment of severe COVID-19, apixaban24 (6th, 18.269 M of dollars) with two piperidone heterocycles and employed as anticoagulant for blood cots treatment, and lenalidomide25 (10th, 9.978 M of dollars) consisting in an isoindolinone core and active as antineoplastic drug for multiple myeloma, achieved outstanding earnings and a large social relevance. Apart from their applications in pharmaceutical industry, lactams are widely used in other chemical industry niches, for example N-methylpyrrolidone (NMP) is widely employed as solvent and polyvinylpyrrolidone as polymer26. In addition, lactams can serve as valuable building blocks for the synthesis of more complex heterocycles27.
Consequently, lactams have awakened a notable interest in the synthetic community and several approaches have been described for their synthesis. More specifically, in the last years an intensive work has been performed for developing multi-step organic methodologies28,29,30,31 or procedures using ad hoc substrates17,32,33 for obtaining isoindolinones and other related lactams. More direct Clemmensen-type reductions of cyclic imides with over stoichiometric reagents such as Zn or Sn in the presence of strong acids34,35, organometallic hydrides36,37 or hydrosilanes38,39, are practical methods for their synthesis. However, these procedures generate important amounts of by-products, being hence less applicable from the sustainability viewpoint. Interestingly, electrochemical methods for the selective reduction of cyclic imides have been recently developed40,41. In this context, the direct hydrogenation of one carbonyl group of cyclic imides is considered a straightforward and green methodology to obtain lactams.
In the last years, important efforts mostly from homogeneous but also from heterogeneous catalysis, have been performed for the hydrogenation of cyclic imides6,10,21. The major challenge here has been the selectivity control, owing to the large variety of valuable products that can be obtained from the hydrogenation of these substrates (Fig. 1c). Several Ru42,43 and Ir44 homogeneous systems have been described to catalyse the general, and in some cases enantioselective, C-N hydrogenation of cyclic imides to afford ω-hydroxamides. In addition, the formal double C-N hydrogenolysis of cyclic imides to afford diols and amines has been recently developed by the groups of Milstein45,46 and Xie47, although in the latter case with a poor selectivity regarding the diol or lactone formation. Interestingly, protocols for the general and selective hydrogenation of cyclic imides to ω-hydroxylactams (cyclic hemiamidals) have been also developed. In the heterogeneous area, the group of McCrindle studied a [Pd/C] system for this transformation that only was active for phthalimide substrates bearing electron-withdrawing groups (EWG) in the nitrogen atom48, while in the homogeneous side the groups of Bergens49 and, more recently, Zhang and Ma44 developed two Ru and Ir systems, respectively. Remarkably, Beller’s group successfully applied ruthenium-50 and cobalt-based51 homogeneous systems for hydrogenative alkoxylation/amination of cyclic imides. Moreover, cyclic amines have been also obtained through a protocol developed by Maj, Agbossou-Niedercorn et al. that employed an unsupported heterogeneous bimetallic Rh/Mo system52.
Regarding the hydrogenation of one carbonyl group of mainly phthalimides and succinimides to isoindolinones and pyrrolidones, there are only two described catalytic systems that can be considered general for this transformation. The first was described by the group of McCrindle in 1977, employing a catalytic system based on [Pd/C] in strong acidic conditions (using trifluoroacetic acid as co-solvent)53. The other was reported by Bruneau and co-workers in 2005, and consisted in a Ru homogeneous catalyst that concomitantly hydrogenated the aromatic ring of phthalimides54. Apart from these examples, a few Ni55,56,57 and Ru58 homogeneous and heterogeneous systems have been also reported to perform this hydrogenation, albeit with a very limited substrate scope. More recently, the group of Palkovits has described a [PtRe/TiO2] material as an active catalyst for the hydrogenation of N-(2-hydroxyethyl)succinimide to the corresponding pyrrolidone59.
In this work, our aim was the design of a heterogeneous nanocatalyst for the general and selective hydrogenation of cyclic imides, mainly phthalimides, to obtain the corresponding lactams (Fig. 1d). In the context of the hydrogenation of carboxylic acid derivatives, materials based on supported species of Pd-Re18,60,61,62,63,64, Pt-Re59,65,66,67, and in minor extent Ni-Re68 or Ir-Re69 have resulted in successful catalysts for the hydrogenation of mainly aliphatic carboxylic acid derivatives. In addition, some examples have been reported dealing with the hydrogenation of these compounds using monometallic Re nanomaterials70,71,72. The role of Re in these materials has been deeply studied, beyond the initial idea of its capacity of acting as an oxophilic species73. These studies point out a certain capacity of this metal to activate hydrogen in its reduced state, and an increase of its reducibility in the presence of Pd, Pt, Ni or Ir. In addition, a common feature of heterogeneous catalysts based on Pd or Pt nanoparticles is its tendency to reduce aromatic rings in the presence of H2, which would be a notable drawback in terms of selectivity for the hydrogenation of phthalimides to isoindolinones, or other relevant imides containing an aromatic ring. In fact, for neither of the described bimetallic combinations containing Re a good tolerance for aromatic rings preservation has been observed. Hence, metals less known to activate H2, such as Ag47,74,75, with a higher tolerance to aromatic rings, can be an interesting alternative in this role. Therefore, in this paper we intend to explore the structural features, catalytic activity, and selectivity of a nanocatalyst based on a AgRe combination, never explored in this chemistry. In this context, we have been able to design a [AgRe/Al2O3] nanomaterial for the general and selective hydrogenation of cyclic imides to lactams showing a notable bimetallic cooperativity (Fig. 1d). Furthermore, we have been able to disclose the exact chemical nature of our material identifying that the formation of a pre-catalyst composed by AgReO4 nanoparticles is key for achieving a highly active and selective nanocatalyst where the cooperativity between Ag and Re is optimal. In addition, we could elucidate the structure of the active species composed by Ag0 nanoparticles and highly dispersed ReOx species formed in situ under the reaction conditions.
Results and discussion
Designing an efficient nanocatalyst for the selective hydrodeoxygenation of N-methylphthalimide to the desired lactam
To start our investigation, we selected the hydrogenation of N-methylphthalimide 1 to the corresponding 2-methylisoindolin-1-one 3 as benchmark reaction (Fig. 2a). As explained before, this is a relevant and challenging transformation as it implies the activation of a cyclic imide towards hydrogenation, but also the control of the desired selectivity towards the C-O mono-hydrogenation of one of the carbonyls with aromatic ring tolerance. Our initial strategy to address this problem consisted in the design of a series of bimetallic nanocatalysts [M1M2/Al2O3] in which M1 is the metal in charge to perform the H2 activation, whereas M2 acts as Lewis acid/oxophilic metal to activate the poorly electrophilic carbonyl and drive the desired selectivity. On the other hand, Al2O3, in γ phase, acts as a support of the active metal species. Considering our willingness to achieve a selective catalytic system with a high synthetic applicability, we decided to explore in first term Ag as M1 metal, because of its known tolerance towards the preservation of aromatic rings in hydrogenations47,74,75. Thus, a series of [AgM2/Al2O3] nanomaterials were synthesized, in which M2 = V, Mo, W, Re, B, Sn or Ga, via wet impregnation followed by calcination at 600 °C under air flow during 3 h (see Methods section for a more detailed information about catalyst preparation). All the materials were prepared with a theoretical [Ag:M2] molar ratio of [1:2] and ICP-AES measurements confirmed a real molar ratio of [1:1.4-1.8] depending on M2 nature (Supplementary Table 5). The bimetallic nanomaterials, as well as the analogous monometallic [Ag/Al2O3], were evaluated at the standard reaction conditions of 6 mol% of Ag, 40 bar of H2, 90 °C, molecular sieves (MS) as additive and n-heptane as solvent during 4.5 h (Fig. 2a). To our delight, under these conditions, [AgRe1.4]-based nanomaterial achieved a promising 61% yield and 95% selectivity to 3 together with 3% yield of hemiamidal 2, while the monometallic [Ag/Al2O3] system just gave the hemiamidal 2 in 12% yield and traces of 3. For the other evaluated alumina-supported [Ag-M21.6-1.8] combinations (M2 = V, Mo, W, B, Sn or Ga), in general lower catalytic activities were observed (Fig. 2a). In the case of [AgW1.8]- and [AgMo1.6]-based nanomaterials, isoindolinone 3 was obtained with good selectivity but very poor yields of 14 and 3%, respectively, while bimetallic nanomaterials composed by M2 = V, B, Sn or Ga gave mainly the corresponding hemiamidal 2 in low to moderate yields (14–36%).

For detailed information about materials preparation see Methods section. a Bimetallic composition and support study. [M1M2/support] materials were prepared with a theoretical [M1:M2] = [1:2] molar ratio (see ICP-AES results in section 4 of the Supplementary Information for the real ratio). For all reactions, 0.25 mmol of 1 and 6 mol% of M1 was added. For M1 and M2 study, reaction time was 4 h 30 min and for support screening was 14 h. b, c Cooperativity studies starting from imide 1 or hemiamidal 2 (0.25 mmol) respectively, nanomaterial (15 mol% of metals), during 4 h 30 min. Metal/s wt% are given between parentheses. d Study of the influence of Re content in the catalyst (wt%). Straight lines correspond to fresh [AgRex/Al2O3] materials and dashed lines to hydrogenated catalysts at 150 °C (i.e., [AgRex/Al2O3]-H2-150) or 280 °C. 0.25 mmol of 1, nanomaterial (6 mol% of Ag) during 4 h 30 min. e Kinetic experiments to study the influence of the Re (wt%) content in [AgRex/Al2O3]-H2-150 materials. All the materials have been hydrogenated at 150 °C (2 °C/min, 2 h) or 280 °C in the case of [Re/Al2O3] material. Selectivity to 3 (blue squares) was calculated considering r0 of conv. 1 (violet squares) and r0 for 3 formation (grey circles). For [Ag/Al2O3] (6 mol% Ag was used), for [Re/Al2O3] (15 mol% Re was used) and for the composite combination of both catalysts [Ag] + [Re] (6 mol% Ag and 9 mol% Re were used, corresponding to a 15 mol% of total metal). Source data are provided as a Source Data file.
At this point we decided to study the influence of M1 nature, in principle in charge of the H2 activation, employing Re as oxophilic metal (M2). With that purpose, several [M1Re/Al2O3] solid systems in which M1 = Pd, Pt, Fe, Co, Ni, Cu or Mn were prepared following the previously commented procedure (see Methods section). In these synthesis, a [M1:Re] molar ratio of [1:2] was also employed, although ICP-AES studies confirmed a real molar ratio of ratio [1:1.2-1.7] depending on the M1 nature (Supplementary Table 6). The bimetallic [M1Re/Al2O3] nanomaterials were evaluated as catalysts for the benchmark hydrogenation of imide 1 at the previously described standard conditions (Fig. 2a and Supplementary Table 1). In these conditions, only materials composed by Pd and Pt resulted active. However, for both catalysts, there was a drastic loss of selectivity to the lactam of interest 3, and important amounts of products coming from aromatic ring hydrogenation (4 and 5) were detected. This observation manifests the unique catalytic activity and chemoselectivity of the nanomaterial based on the alumina-supported Ag-Re combination to mono-hydrogenate a C = O bond of a cyclic imide in the presence of an aromatic ring.
Once established the Ag-Re bimetallic combination as the optimal one, we decided to study the nature of the solid matrix in which the metallic nanoaggregates are stabilized. In the design of heterogeneous nanocatalysts for hydrogenation and, in particular, in the area of carboxylic acid derivatives hydrogenation, the choice of the solid matrix can be critical as it potentially plays several key roles related to metal species dispersion and, hence, stabilisation and formation of the required active sites. In addition, promotion of H2 spillover through the matrix surface and its possible involvement in different mechanistic steps due to its intrinsic acidity are also important aspects. Therefore, we prepared several [AgRe/support]-based materials using as supports SiO2, TiO2, ZrO2, ZnO, CeO2, H-BETA zeolite or MgO with different acid/base properties (see Methods section) and employing a [Ag:Re] theoretical molar ratio of [1:2] (for actual determined [Ag:Re] molar ratio by ICP-AES see Supplementary Table 7). These materials were evaluated for the benchmark reaction at standard conditions but using a longer reaction time of 14 h (Fig. 2a). It is important to note that the best results were obtained employing Al2O3 as support (89% yield of 3 and 99% selectivity), while solid matrices such as TiO2, SiO2, ZrO2, Nb2O5, H-BETA zeolite, ZnO or CeO2, showed very good selectivity but a notable lower activity, giving 3 in yields from 12% to 53%. Interestingly, [AgRe1.6/MgO], containing a highly basic matrix, resulted totally inactive, thus suggesting a significant role of support acid sites for assisting the hydrogenation of 1. The fact that all these [AgRe/support] materials afforded 3 with high selectivity, with the exception of inactive material [AgRe1.6/MgO], indicates that the support is not directly involved in the 2 to 3 step, the Ag-Re bimetallic combination being key for this part of the overall process.
Then, we became interested in exploring the effect that thermal treatment applied during the active nanomaterial synthesis had in the catalytic activity. Thus, we synthesized a series of [AgRex/Al2O3] nanomaterials at the previously essayed conditions, albeit changing calcination at 600 °C by pyrolysis at the same temperature or by reduction at 450 or 150 °C, as well as different combinations of calcination/pyrolysis and reduction (see Methods section). These nanomaterials were catalytically evaluated at the standard reaction conditions for 14 h and the material uniquely calcined was the one that afforded the best results (Supplementary Table 2). Considering this information, nanomaterials obtained after calcination using different temperatures (300–700 °C) and ramps (1–10 °C/min) (see Methods section) were prepared and evaluated at a reaction temperature of 70 °C (Supplementary Table 2). From these essays, it was deduced that the synthesis of the optimal material should be performed by calcination at 500 °C with a 2 °C/min ramp, as the corresponding [AgRe1.4/Al2O3] nanomaterial obtained in these conditions catalyzed the hydrogenation of 1 to 3 with better efficiency than the other materials, a 62% of yield at 70 °C was obtained (Supplementary Table 2, entry 11). To further optimize the synthesis of the active catalyst, two additional Ag-Re nanomaterials were prepared maintaining the theoretical molar ratio of [1:2], but with different metal content (see Methods section). These nanomaterials were catalytically evaluated, and it was confirmed that the originally prepared [AgRe1.4/Al2O3] catalyst with Ag and Re loadings of 4.1 and 10 wt% was the most active one (Supplementary Table 3).
The solvent effect in the hydrogenation of phthalimide 1 to isoindolinone 3 catalyzed by the optimized [AgRe1.4/Al2O3] was also studied (Supplementary Table 4, entries 1–10). Among all the solvents tested, good results were obtained using n-heptane, 2-MeTHF, CPME and toluene, CPME being the one that afforded the best results (68 and 94% yield of 3 at 5 or 16 h, respectively), and, hence, we continued our studies with it. In addition, [AgRe1.4/Al2O3] was also evaluated at lower Ag mol % of 4 and 6 %, giving place to lower 3 yields of 59 and 35% respectively (Supplementary Table 4, entries 11 and 12). The reaction in the absence of molecular sieves was also tested, confirming their relevant role to absorb water and shift the equilibrium towards the isoindoline formation (Supplementary Fig. 7).
Cooperativity studies are key to understand the role of each metal in bimetallic nanocatalysis. Therefore, monometallic materials [Ag/Al2O3] (4.2 wt% Ag) and [Re/Al2O3] (7.5 wt% Re) were prepared (see Methods section 2.2 for more specific information). It is interesting to note that, in the absence of silver, rhenium has a limit of deposition on the support of 7.5 wt%, due to its tendency to sublimate as volatile species under the synthetic conditions employed. These nanomaterials, together with optimal [AgRe1.4/Al2O3] (4.1 wt% Ag and 10.0 wt% Re), were evaluated as catalysts in the catalytic hydrogenation of not only imide 1, but also ω-hydroxylactam 2, in the presence of 15 mol% of total amount of metals and under the optimized reaction conditions for 4.5 h of reaction time (Fig. 2b, c, respectively). As it was expected, [AgRe1.4/Al2O3] afforded the corresponding lactam 3 with high selectivities and good yields with both starting materials (68% and 79% yield of 3 from 1 and 2 respectively with a 97% of selectivity in both cases). Remarkably, for both hydrogenations using [Ag/Al2O3], selectivity to 3 dramatically dropped. In the case of phthalimide 1 hydrogenation, ω-hydroxylactam 2 was the major product (41% yield), but a 21% of phthalide 6, coming from a C-N pathway, was also obtained, while from 2 the major product was phthalimide 1, together with a 14% of phthalide 6. These results indicate an important role of Re as oxophilic metal enhancing C-O selectivity, as well as increasing electrophilicity of the carbonyl from the imide. On the other hand, [Re/Al2O3] obtained at the same conditions than the optimal nanocatalyst was completely inactive towards the hydrogenation of both phthalimide 1 and ω-hydroxylactam 2, hence confirming the necessary synergy between Ag and Re. By contrast, [Re/Al2O3] materials subjected to a hydrogenating treatment at 150, 280 and 600 °C were also evaluated and, interestingly, nanomaterials reduced at 280 and 600 °C resulted active for the hydrogenation of phthalimide 1, affording 3 with good selectivities (98 and 90% respectively), although in lower yields than the AgRe-based material (43 and 34% vs 68%, Fig. 2b). Furthermore, [Re/Al2O3] nanomaterial subjected to a hydrogenating treatment at 280 °C was also active for the hydrogenation of ω-hydroxylactam 2 to isoindolinone 3, although the selectivity dropped to 70%, and a 25% of phthalimide 1 was formed (Fig. 2c). These observations suggest that Re partially reduced species, whose formation could be enhanced by a Ag-Re cooperation in the bimetallic material, could have an important role in the reaction mechanism. Finally, the composite constituted by the physical mixture of [Ag/Al2O3] & [Re/Al2O3] monometallic materials was essayed for the hydrogenation of both phthalimide 1 and ω-hydroxylactam 2, and it was observed that the mixture was less active that the bimetallic material and afforded 3 in lower selectivity and yield (Fig. 2b, c).
With the aim of better understanding our catalytic system and being able to obtain further structure-activity insights, we decided to explore the catalytic activity for the phthalimide 3 hydrogenation of a series of nanomaterials with different [Ag:Re] molar ratios. Following the optimized procedure, [AgRex/Al2O3] nanomaterials in which x = 0.18, 0.35, 0.75, 1 and 2 were prepared, apart from the previously studied [AgRe1.4/Al2O3], maintaining in all the materials a constant Ag charge of 4-4.5 wt% (see Methods section for a more detailed procedure). The Ag-Re nanomaterials were evaluated under the optimized reaction conditions during 4.5 h of reaction time (Fig. 2d and Supplementary Fig. 2) and, to our delight, [AgRe/Al2O3] (with x = 1) showed an improved catalytic activity with respect to [AgRe1.4/Al2O3], affording isoindolinone 3 in 74% of yield, vs a 62% obtained with AgRe1.4 material, and a 97% of selectivity. It is also relevant to note that Ag-Re based materials with x < 1 showed lower catalytic activity and selectivity to the formation of 3. Interestingly, both activity and 3 selectivity are observed to proportionally increase with Re content of [AgRe0.18-1/Al2O3] nanomaterials. On the other hand, while [AgRe1.4/Al2O3] still maintains a good catalytic activity and affords 3 with a high selectivity (97%, the same as the observed with [AgRe/Al2O3]), in the case of [AgRe2/Al2O3] there is an important drop of catalytic activity (3 is obtained with a 20% of yield) and, in minor extent, of selectivity (83%). Hence, these studies reveal that [AgRe/Al2O3], the Ag-Re based material with an equimolar proportion of both metals is the optimal nanocatalyst, the materials with Ag excess being less active and selective to 3, affording hemiamidal 2 in larger amounts. On the other hand, nanomaterials with a Re excess show lower catalytic activity.
To further explore this system, we registered a yield-time kinetic profile using [AgRe/Al2O3] as catalyst under the optimized reaction conditions (Supplementary Fig. 1a). Remarkably, in this experience an induction period of 50 min was clearly observed, which totally disappeared when [AgRe/Al2O3] was reduced at 150 °C under a H2 flow during 2 h before being used as catalyst (named as [AgRe/Al2O3]-H2-150) (Supplementary Fig. 1b). Therefore, this observation clearly indicates that the active catalyst is a reduced Ag-Re based material produced under reaction conditions. However, it is important to highlight that isoindolinone 3 yields were higher when the reaction was performed with unreduced [AgRe/Al2O3] vs [AgRe/Al2O3]-H2-150 (74 vs 51% at 4.5 h and 98 vs 86 % at 14 h, respectively). With all this information in hand, and to further elucidate the nature of our active system, we decided to catalytically evaluate in the benchmark 1 hydrogenation all the series of [AgRex/Al2O3] materials previously subjected to the corresponding hydrogenation treatment at 150 °C ([AgRex/Al2O3]-H2-150) (Fig. 2d dashed lines and Supplementary Fig. 2). In these experiments it was observed that with [AgRex/Al2O3]-H2-150 materials containing Ag in excess, similar results were observed with respect to the unreduced materials regarding 3 yield and selectivity, being these values higher with larger proportions of Re. However, in the case of pre-reduced nanomaterials with equimolar amounts of Ag and Re or Re excess ([AgRe1-2/Al2O3]-H2-150) there is an effect by which 3 yield and selectivity become stable, suggesting that these reduced materials present a more similar structure between them (see Fig. 2d dashed lines, Supplementary Fig. 2 for results at 4.5 h and Supplementary Fig. 6 for results at 14 h). Moreover, yield time kinetic profiles of [AgRex/Al2O3]-H2-150 were also registered and these results are summarized in Fig. 2e (see also Supplementary Figs. 4 and 5) in which the initial rates of 1 conversion, 3 formation and selectivity at low conversions (r0 3 / r0 conv. 1) are represented as a function of Re content. From these results, we can infer an increasing catalytic activity of the materials at larger Re ratios, reaching a maximum at Ag:Re equimolar ratio. By contrast, selectivity at low conversions increases with larger Re content without reaching a maximum. It is interesting to note that [Re/Al2O3]-H2-280 shows slightly higher 3 selectivity at low conversion than [AgRe2/Al2O3]-H2-150, although a much lower catalytic activity. Moreover, a [Ag/Al2O3]-H2-150 & [Re/Al2O3]-H2-150 composite shows very low initial rates (see also Supplementary Fig. 3).
Characterization of the catalysts
The catalytic studies performed up to this point revealed that the previously unreported combination of Ag and Re species supported onto an Al2O3 matrix is essential for the efficiency of the catalytic system. In addition, these studies also showed that, as a function of [AgRex/Al2O3] system composition, different catalytic activities were measured. Moreover, for all the materials it was observed the in situ formation of the active reduced species. Thus, we believe that explaining all these catalytic features through the understanding of not only the structure and composition of both pre-catalysts and active materials, but also the process by which this activation occurs, was essential. To this end, we performed an in-depth characterization of [AgRex/Al2O3] and [AgRex/Al2O3]-H2-150 materials, as well as the corresponding monometallic catalysts.
Firstly, all [AgRex/Al2O3] materials were analysed by X-Ray Powder Diffraction (XRPD) with the aim of identifying crystalline phases and learning about metal dispersion (Fig. 3a and Supplementary Fig. 12). Interestingly, whereas in [AgRex/Al2O3] materials in which x < 0.75, only Al2O3 phases were identified, materials with [AgRe0.75] or higher molar ratio showed diffraction peaks corresponding to AgReO4 planes in the scheelite structure (bulk AgReO4 as reference ICDD No. 00-008-095)76,77,78. Considering the improved catalytic activity of these [AgRex/Al2O3] materials with x ≥ 0.75, [AgReO4] and [AgReO4] in the presence of calcined [Al2O3] at 500 °C were evaluated as catalysts for the hydrogenation of N-methylphthalimide 1 at the optimized reaction conditions (Supplementary Fig. 7). After 14 h, the composite system [AgReO4] + [Al2O3] showed a moderate catalytic activity affording the corresponding lactam 3 in 22%, whereas [AgReO4] by itself only afforded traces of 3, confirming the prominent role of [Al2O3] in the catalytic system. In addition, [AgReO4/Al2O3] was prepared analogously to the [AgRex/Al2O3] materials, by a first step of AgReO4 impregnation, and it was evaluated as catalyst in the optimized 1 hydrogenation. Under these conditions during 14 and 4.5 h, this catalyst afforded 3 in 74 and 62% of yield respectively, vs 94 and 74% previously obtained with [AgRe/Al2O3] (Supplementary Fig. 7). As it could be expected, [AgReO4/Al2O3] XRD pattern shows the peaks at 2θ values typical of Al2O3 and AgReO4 (Fig. 3a). Hence, at this point it was interesting to analyse the XRPD of [AgRe/Al2O3]-H2-150 and [AgReO4/Al2O3]-H2-150. Remarkably, after hydrogenation, AgReO4 patterns disappeared for both materials, and only in the case of [AgRe/Al2O3]-H2-150, broad signals identified as diffraction patterns corresponding to Ag0 and Re0 were detected62,79,80. It is interesting to note that in monometallic materials subjected to a hydrogenating thermal treatment, Ag0 and Re0 broad signals were also observed, although in the case of [Re/Al2O3], Re0 was only detected when the hydrogenation was performed at 280 °C, and no Re reduced species were observed when the material was hydrogenated at lower temperatures (Fig. 3a). The presence of such reduced Re species in [Re/Al2O3]-H2-280 could explain the moderate catalytic activity of this material. In addition, [AgReO4/Al2O3]-H2-150 afforded the corresponding lactam 3 in the same yields than [AgRe/Al2O3]-H2-150. However, kinetic experiments of [AgReO4/Al2O3]-H2-150 revealed some differences in comparison with [AgRe/Al2O3]-H2-150, such as a shorter induction period of 10 min and slower initial rates regarding both 1 conversion and 3 formation (r0 1 = 0.38%/min, r0 3 = 0.29% 3 /min vs r0 1 = 1.05%/min, r0 3 = 0.78 % 3/min) (Fig. 2e).

All thermal reduction treatments were done at the specified T (°C) during 2 h (150 or 280 °C) with a heating rate of 2 °C/min under H2 flow (100 mL/min). a X-Ray Powder Diffraction (XRPD). Ref. code used for AgReO4 (ICDDN 00-008-095). Ref. code used for Ag0 (00-003-0921). Ref. code used for Re0 (00-005-0702). See Supplementary Fig. 11 for assignation of Miller indexes of the more intense diffractions. The detected species are in the black line boxes. b H2-Temp.-Programmed Red. (H2-TPR). c UV-Vis Diffuse Reflectance Spec. (UV-Vis DRS). “After 1 h” indicates that material was measured after having been under reaction conditions for 1 h. d RAMAN spectroscopy. The detected species are in the box with black lines. e High-Angle Annular Dark-Field Scanning Transmission Electron Microscopy (HAADF-STEM) and Energy-Dispersive X-Ray (XEDS) mapping analysis of freshly prepared [AgRe/Al2O3]. f HAADF-STEM and XEDS of [AgRe/Al2O3] previously reduced at 150 °C, named [AgRe/Al2O3]-H2-150. Source data for Fig. 3a–d are provided as a Source Data file.
Then, relevant physicochemical properties of [AgRex/Al2O3] nanomaterials, such as the surface area or the number and strength of accessible Lewis acid sites, were evaluated. Nitrogen adsorption-desorption isotherms were performed for several [AgRex/Al2O3] materials, in comparison with [AgReO4/Al2O3] and [Al2O3], to determine BET surface areas, as well as BJH average pore diameter (Supplementary Table 10). In general, the presence of metal species over [Al2O3] implied materials with lower areas, this effect being more important in the case of materials with a higher Re content (i.e., 175.4 m2/g for [AgRe0.35/Al2O3] and 141.1 m2/g for [AgRe2/Al2O3]) (Supplementary Table 10). Lewis acid sites are believed to have a prominent role in the structure of heterogeneous catalysts active for hydrogenative methodologies with carboxylic acid derivatives10. As commented before, in these transformations the activation of the poorly electrophilic carbonyl group through its coordination with an acid centre, is essential. To determine Lewis acid sites amount and study their strength, Fourier-Transform Infrared spectroscopy of adsorbed pyridine (Pyr-FTIR) was employed (Supplementary Fig. 14 and 15)81. In this experience, the pyridine-Lewis acid adducts were quantified and their strength was determined through the integration of their characteristic band centered at 1450–1455 cm−1 after the pyridine desorption at different temperatures (150 °C = weak sites; 250 °C = medium sites; 350 °C = strong sites)82. We compared Lewis acidity of the [Al2O3] matrix with both monometallic nanomaterials and the optimal catalyst [AgRe/Al2O3]. For [Ag/Al2O3], the number of quantified acid sites either weak, medium or strong was notably lower than for [Al2O3]. On the contrary, in the case of [Re/Al2O3] a higher number of all the types of acid sites was determined in comparison with the matrix. This can be interpreted as Ag species in [Ag/Al2O3] are covering [Al2O3] acid sites, while in [Re/Al2O3] highly valent Re species present an acid nature. Interestingly, for [AgRe/Al2O3] nanomaterial, the amount of acid sites resulted similar to the one quantified for [Re/Al2O3]. This suggests that in the [AgRe/Al2O3] bimetallic nanomaterial, Ag species nature is totally different than in [Ag/Al2O3] and, in fact, this is in agreement with the stabilization of AgReO4 species over the matrix in this nanomaterial. Furthermore, more information about the nature of the acid sites can be inferred considering the exact frequency of the 1450–1455 cm−1 band, as higher frequencies have been associated with stronger acid sites82. Remarkably, for [Re/Al2O3] and [AgRe/Al2O3] nanomaterials the maximum frequency was found at 1455 cm−1, while for [Al2O3] and [Ag/Al2O3] it was found at 1450 cm−1, suggesting that Re species are stronger acid sites (Supplementary Fig. 15).
To better understand the behavior of the bimetallic materials towards hydrogenation, H2-temperature-programmed reduction (TPR) studies of some of the [AgRex/Al2O3] nanomaterials, as well as the monometallic ones was performed (Fig. 3b). As it has been reported for similar Re-based materials66,83, [Re/Al2O3] showed sharp reduction peaks at 312 and 314 °C (Fig. 3b), clearly corresponding to the reduction of ReOx species. On the other hand, [Ag/Al2O3] only showed very low H2 consumptions at 64 and 386 °C (Supplementary Fig. 16), probably due to the previous reduction of silver species. The peak at 64 °C can be assigned to the reduction of Ag2O species, while the one at 386 °C probably belongs to more disperse Ag+ species84,85. In the case of the bimetallic nanomaterials, only the H2 consumption assigned to the reduction of Re species was observed at lower temperatures between 195–254 °C (Fig. 3b). It was very interesting to note that there was a clear relationship between the Ag/Re molar ratio present and the observed reducibility of Re species. The materials with a higher Ag content show reduction peaks maximums at lower temperatures, 195–240 °C for [AgRe0.35/Al2O3] vs 254 °C for [AgRe2/Al2O3]. This points out that Ag is assisting Re reduction and suggests a relevant interaction between both metals.
At this point, we wanted to better understand the nature of Ag and Re species in the bimetallic [AgRex/Al2O3] and [AgRex/Al2O3]-H2-150 nanomaterials. To obtain information regarding the oxidation state of Ag and Re surface species we studied all the series of [AgRex/Al2O3] materials by UV-Vis DRS (Fig. 3c and Supplementary Fig. 17). It has been well stablished in the literature that Ag+ ions and Agδ+ clusters as well as Re7+ species absorb in the 200–300 nm region, while Ag0 and reduced Re species absorb at higher wavelengths between 330–800 nm86,87,88,89,90,91,92. In this study we observed that [AgRex/Al2O3] materials, as well as [AgReO4/Al2O3], [Ag/Al2O3] and [Re/Al2O3] only showed absorption in the 200–300 nm region (Fig. 3c and Supplementary Fig. 17). From this it can be deduced that also [AgRex/Al2O3] materials with x < 0.75, that did not exhibit XRD characteristic signals of AgReO4, present Ag+ and Re7+ species. By contrast, [AgRe/Al2O3]-H2-150 and [AgReO4/Al2O3]-H2-150, as well as the corresponding unreduced materials employed as catalysts during one hour, showed bands in both regions, indicating that there is a mixture of reduced and unreduced species in these materials (Fig. 3c). In the case of the reduced monometallic materials, the band at 330–800 nm region was only observed for [Ag/Al2O3]-H2-150 and neither [Re/Al2O3]-H2-150 nor [Re/Al2O3]-H2-280 displayed absorption in this region (Fig. 3c). This observation allows us to assign the band observed for [AgRe/Al2O3]-H2-150 and [AgReO4/Al2O3]-H2-150 at the 330–800 nm region to Ag0 species, albeit this does not exclude the possibility that Re reduced species are also formed.
Raman spectroscopy has proved to be a useful technique to study Re=O vibrational modes and, hence, it is interesting to characterize our AgRe bimetallic and monometallic materials through this technique. Raman spectra of [Re/Al2O3] materials under ambient conditions had been previously studied and they showed a main band at 982 cm−1 assigned to isolated ReO4− species, discarding the presence of Re-O-Re linkages that display a typical band around 800 cm−1 89,93,94. In addition, AgReO4 Raman spectra has also been reported to show internal modes at 941, 898 and 863 cm−1 76,78. Thus, we explored [AgRex/Al2O3] materials Raman spectra and, interestingly, with the exception of [AgRe/Al2O3] and [AgReO4/Al2O3], they all showed a band between 966–972 cm−1 that can be assigned to isolated ReO4− species stabilized over Al2O3 matrix (Supplementary Fig. 20 and Fig. 3d). Moreover, this band also was observed at 972 cm−1 for [Re/Al2O3]. Remarkably, the band shifted to higher wavelengths in the materials with higher Re content, indicating the presence of tetrahedral ReO4− species with different environments93,95. In addition, in the [AgRex/Al2O3] materials with x > 0.75 and [AgReO4/Al2O3] the characteristic bands at 941, 898 and 863 cm−1 belonging to AgReO4 were clearly observed (Supplementary Fig. 20 and Fig. 3d). From these data it can be deduced that only in the materials with an equimolar Ag/Re stoichiometry, all Ag+ and Re7+ species present AgReO4 nature, but AgReO4 is present together with ReO4− species in the other [AgRex/Al2O3] materials with x > 0.75. It is also very interesting to remark that the 941 cm−1 band assigned to AgReO4 could also be identified for AgRe materials with different supports (TiO2, Nb2O5, MgO, ZnO, ZrO2 and SiO2) (Supplementary Fig. 19) revealing that AgReO4 formation is not limited to Al2O3. This was also confirmed through the analysis of XRD patterns of these materials (Supplementary Fig. 11). On the other hand, [AgRe/Al2O3]-H2-150 and [AgReO4/Al2O3]-H2-150 Raman spectra was also registered, and they only showed a band at 966 and 968 cm−1 that can be identified as characteristic of ReO4− species stabilized over Al2O3 (Fig. 3d). In the case of [Re/Al2O3]-H2-150 and [Re/Al2O3]-H2-280 a similar band at 966 cm−1 appeared (Fig. 3d). This can be interpreted as in all these reduced materials, Re7+ species of perrhenate nature are present. It should be noted that this experience is valuable to clearly distinguish between ReO4− and AgReO4 species, but it cannot discard the formation of Re reduced species due to the large tendency of Re to become oxidized, as the experimental setup did not allow an in situ hydrogenation of the materials.
Then, a structural and compositional study at nanoscopic and atomic level of the representative materials [AgRe0.35/Al2O3], [AgRe/Al2O3] and [AgRe2/Al2O3], as well as [AgReO4/Al2O3], was performed by high-angle annular dark-field scanning-transmission electron microscopy (HAADF-STEM) and scanning-transmission energy-dispersive X-ray analysis (STEM-XEDS) (Fig. 3e, f, Supplementary Figs. 21–25). The optimal [AgRe/Al2O3] precatalyst showed an intimate contact between Ag and Re species, as well as an equimolar homogeneous distribution of Ag and Re over the support (Fig. 3e and Supplementary Fig. 21a). Moreover, the presence of AgReO4 species could be clearly identified from the analysis of the atomic-resolution STEM-HAADF images. As illustrated in Fig. 3e and Supplementary Fig. 21a, images along specific zone axis of this crystallographic phase could be perfectly detected. In particular, Fig. 3e depicts the features characteristic of AgReO4 along [010] zone axis through the measurement of the corresponding d-spacing values in several axes (e.g. d400 = 0.299 nm and d200 = 0.278 nm) (Fig. 3e and Supplementary Fig. 21a). Similarly, [AgReO4/Al2O3] also showed equimolecular Ag:Re ratios and the presence of AgReO4 species could be also identified (Supplementary Fig. 21b). However, in this material the observed nanoaggregates were in general of larger size. In the case of [AgRe2/Al2O3], we could identify areas with similar Ag and Re atomic content corresponding to AgReO4 species, as well as areas only populated by Re nanoaggregates (Supplementary Figs. 21c and 22). This observation totally fits with the Raman results that showed for this material two kinds of Re7+ perrhenate type species (Supplementary Fig. 20). For [AgRe0.35/Al2O3], the ICP- AES measured atomic fractions were confirmed by STEM-XEDS and an intimate Ag-Re contact was also observed (Supplementary Fig. 23). Consistently with the spectroscopic measurements, in this material no AgReO4 species were identified, and, instead, we could observe the presence of Ag0 nanoparticles and subnanometric ReOx clusters, probably ReO4− type species. It is plausible that Ag-reduced species are formed under the measurement conditions due to the large reducibility of Ag+ species that are not forming AgReO4. To better understand the active nanocatalysts structure, the same materials after a hydrogenating treatment at 150 °C were studied (Fig. 3f and Supplementary Fig. 24). Interestingly, after this process the materials structure was more similar to the one previously observed for [AgRe0.35/Al2O3] as Ag0 nanoparticles and ReOx dispersed nanoclusters (< 1.5 nm) were observed for all the materials (Fig. 3f and Supplementary Fig. 24). This result confirms that a kinetic phenomenon of dispersion and reorganization of Ag and Re species upon H2 contact is occurring. It is interesting to note that for the [AgRex/Al2O3] materials, Ag0 nanoparticles of increasing size were measured with larger Re content. In fact, in [AgRe0.35/Al2O3] Ag0 nanoparticles between 2–10 nm were detected while in [AgRe/Al2O3] and [AgRe2/Al2O3] sizes between 3–30 and 12–33 nm, respectively, were measured. Probably, the presence of isolated ReOx species in [AgRe2/Al2O3] made the reduction and dispersion of AgReO4 nanoaggregates in this material more difficult, which explains the lower catalytic activity of [AgRe2/Al2O3] precatalyst. Remarkably, [AgReO4/Al2O3]-H2-150 was the material that displayed the larger Ag0 nanoparticles (30–80 nm), which could be related with the lower catalytic activity of this material.
To achieve a more in-depth knowledge regarding metals oxidation states in the materials, X-Ray Photoelectronic Spectroscopy (XPS) studies were conducted for [AgRe0.35/Al2O3], [AgRe/Al2O3] and [AgRe2/Al2O3], as well as [AgReO4/Al2O3] and monometallic materials on their fresh and reduced version (Fig. 4a, b, Supplementary Fig. 26 and 27 and Supplementary Tables 11 and 12)96. In the Re 4 f region, for all the fresh materials two main features were identified at 46.0 eV (Re 4f7/2) and 48.4 eV (Re 4f5/2) representative of Re7+. Interestingly, in the reduced bimetallic materials more differences were observed as a function of Ag/Re molar ratio. Specifically, on [AgRe/Al2O3] material in situ reduced at 150 °C and after its use as catalyst, a mixture of Re7+ and Re6+ was detected (Fig. 4a), while for the in situ reduced [AgRe0.35/Al2O3] and [AgRe2/Al2O3] nanocatalysts, Re0 species were additionally observed (Fig. 4b). The simultaneous presence of Re7+, Re6+ and Re0 was also seen for [Re/Al2O3] reduced at 280 °C, albeit only Re7+ was observed in the 150 °C reduced Re nanomaterial (Fig. 4a and Supplementary Fig. 27). In addition, a small fraction of highly dispersed Re species (48.2 and 50.3 eV), probably doping the matrix, were also detected for [Re/Al2O3]-H2-280. These results provide clear evidence about the formation of Re reduced species in the reduction treatment and under reaction conditions. Moreover, it can be deduced that in the monometallic Re nanomaterial higher temperatures are required to lead to a mixture of Re in several oxidation states, and, hence, becoming an active material. It is very notable that the most active [AgRe/Al2O3] nanocatalyst does not display the formation of surface Re0 species.

a Re 4 f XPS spectra of [AgReO4/Al2O3], [AgRe/Al2O3] and [Re/Al2O3] materials (fresh calcined or calcined and then in situ reduced at 150 °C during 2 h under H2 flow or after being used in a reaction cycle). b Re 4 f XPS spectra of [AgRe2/Al2O3] and [AgRe0.35/Al2O3] (fresh calcined or calcined and then in situ reduced at 150 °C during 2 h under H2 flow). c IR-CO spectra acquired at −170 °C for fresh [Re/Al2O3], [AgReO4/Al2O3], [AgRe2/Al2O3], [AgRe/Al2O3], [AgRe0.35/Al2O3], [Ag/Al2O3] materials and [Al2O3] support. d IR-CO spectra acquired at −170 °C for [Re/Al2O3], [AgReO4/Al2O3], [AgRe2/Al2O3], [AgRe/Al2O3], [AgRe0.35/Al2O3], [Ag/Al2O3] materials and [Al2O3] support after being in situ reduced at 150 °C during 2 h under H2 flow. e IR-CO spectra acquired at −170 °C under 3 mbar of CO for [Re/Al2O3], [AgReO4/Al2O3], [AgRe2/Al2O3], [AgRe/Al2O3], [AgRe0.35/Al2O3], [Ag/Al2O3] materials and [Al2O3] support after being in situ reduced at 150 °C during 2 h under H2 flow. Note that in figs. c–e the name of alumina-based materials is shown in abbreviated form. f IR-CO spectra acquired at −170 °C during the first CO stream pulse (8·10−2 mbar) for [Ag/Al2O3], [AgRe0.35/Al2O3], [AgRe/Al2O3] and [AgReO4/Al2O3] materials after being in situ reduced under H2 flow at 150 °C during 2 h. Source data are provided as a Source Data file.
Regarding silver, the characteristic Ag 3 d core level was studied for each of the bimetallic materials and [Ag/Al2O3] (Supplementary Fig. 26). To elucidate Ag oxidation state in each material, a modified Auger parameter (α’) was determined, confirming the presence of Ag+ species in the unreduced materials and Ag0 in the reduced ones, as well as in [AgRe/Al2O3] after reaction (Supplementary Table 11)97. Remarkably, neither for [AgRe2/Al2O3], nor for the reduced version of this nanomaterial, an Auger parameter could be determined due to the low exposure of Ag species. Considering the information given by other techniques, the most probable explanation for this is that AgReO4 species in this unreduced material are covered by Re7+ oxides. This explains the fact that [AgRe2/Al2O3]-H2-150 is more catalytically active than [AgRe2/Al2O3], as the in situ generation of the active species is much more difficult. Metal surface composition of the materials was also analyzed by quantifying Ag/Al and Re/Al surface atomic ratio (Supplementary Table 12). From these data it is interesting to note that the presence of small amounts of Re increases Ag dispersion (1.8% of Ag/Al at [Ag/Al2O3] vs 2.4% at [AgRe0.35/Al2O3]) while the reduction stage in bimetallic materials at 150 °C promotes the dispersion of rhenium, making it more representative at higher Re loads. According to the catalytic results and the Re/Ag ratio on the surface measured by XPS (Supplementary Table 12), the configuration of ca. 5 Re atoms per 1 Ag atom on the surface is ideal to stabilize the active site of the reaction.
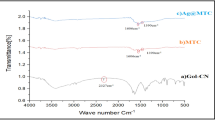
CO adsorption FTIR spectroscopy studies were performed for the most representative bimetallic nanomaterials, as well as the monometallic ones (Fig. 4c–f and Supplementary Figs. 28–33). The contribution of hydroxyls from the Al2O3 support (OH-CO)98 was identified at ~2151 cm−1 (Fig. 4c and Supplementary Fig. 28a). Meanwhile, IR spectra of the fresh [AgRex/Al2O3] (x = 0.35, 1 and 2) samples at low surface coverage of CO showed a band between 2169–2175 cm−1 mainly assigned to Ag+-CO interaction, while for [Ag/Al2O3] it was detected at 2165 cm−1 (Fig. 4c and Supplementary Figs. 28c, 30, 31 and 33)99,100. Interestingly, this signal became less intense and blue-shifted with the presence of increasing amounts of Re in the bimetallic materials. This can be interpreted not only as an indirect prove of the lower surface Ag+ accessibility with Re increasing amounts, but also as a Re modification of surface Ag+ local environment. However, [AgReO4/Al2O3] displayed the band at the same frequency than [AgRe/Al2O3] (2171 cm−1), supporting the influence of Re on the electronic properties of Ag+ (Fig. 4c and Supplementary Figs. 31 and 32).
Upon the in situ reduction at 150 °C of either bimetallic materials or [Ag/Al2O3], the Ag+-CO characteristic band disappeared, confirming the reduction of Ag+ species (Fig. 4d, e and Supplementary Figs. 28c and 30–33). Remarkably, characteristic CO absorption bands on metallic Re (Re0) at 2035 and 1960 cm−1 were observed for reduced bimetallic materials and for [Re/Al2O3] reduced at 280 °C, with the exception of [AgRe/Al2O3]101,102. This result is in good agreement with the Re 4 f XPS spectrum. Furthermore, on the in situ reduced [AgRe0.35/Al2O3] and [AgRe2/Al2O3] bimetallic materials and Ag monometallic, bands at 2344 and 2278 cm−1 associated with CO2 adsorbed on Ag0 (Ag0-OCO) and its isotope species, respectively, were detected. Other features at 1482 and 1395 cm−1 were identified as asymmetric (νas(CO3)) and symmetric (νs(CO3)) stretching of monodentate carbonate, respectively (Fig. 4d, e and Supplementary Figs. 28c–d, 30 and 33)98,103,104. The in situ oxidation of CO mediated by subsurface oxygen that emerges after silver reduction can give place to CO2 that adsorbs to Ag0 and can react with Al2O3 or ReOx to afford CO32−. The absence of these bands in the [AgRe/Al2O3]-H2-150 nanomaterial suggests that Ag oxophilic properties have been modified in this material, probably due to the Re influence in Ag local environment (Fig. 4d, e and Supplementary Fig. 31). Moreover, a shoulder at 2100 cm−1 was observed only for [AgRe/Al2O3]-H2-150 that has been assigned to Ag0-CO species with a modified metallic silver (Fig. 4f).
Finally, to confirm all the acquired data, while better understanding the structural differences among the different bimetallic nanocatalysts, as well as their dynamic reorganization upon reduction, X-Ray absorption spectroscopy (XAS) studies were performed at both the Re L3 and Ag K edges. This technique allowed the comparison of the most representative bimetallic nanomaterials with the monometallic ones and several Ag and Re standards, in order to get information about oxidation states and metals local environment in the bulk. Hence, the characteristic restrictions of other techniques limited to afford information about materials surface, species presenting well-defined crystalline phases or to determined selection rules, were avoided. X-Ray absorption near edge structure (XANES) spectra at the Ag K-edge corresponding to fresh and in situ hydrogenated [AgRex/Al2O3] (x = 0.35, 1 and 2), [AgReO4/Al2O3] and [Ag/Al2O3] materials as well as Ag0, Ag2O and AgReO4 standards is displayed in Fig. 5a, while the Fourier transforms (FT) of the corresponding k2-weighted extended x-ray absorption fine structure (EXAFS) oscillations are reported in the Supplementary Figs. 35 and 36. As it could be expected from the results previously obtained from other characterization techniques, [AgRe/Al2O3], [AgReO4/Al2O3] and [AgRe2/Al2O3] displayed a very similar Ag K-edge XANES spectrum to the one obtained for AgReO4 standard, while [AgRe0.35/Al2O3] showed more similarities with [Ag/Al2O3]. Interestingly, an analysis of Ag K-edge XANES first derivative spectra of the fresh materials revealed that the maximum at 25512 eV shifts to higher energy values with increasing amounts of Re (Supplementary Fig. 34) approaching the AgReO4 bulk reference. At the same time, the FT-EXAFS reveal a systematic Ag-O contribution reduction by decreasing the Re amount. Moreover, differences in the averaged Ag-O radial distances were observed between [AgRe0.35/Al2O3] (1.6 Å, similar to Ag2O) and [AgRe/Al2O3], [AgReO4/Al2O3] and [AgRe2/Al2O3] (1.9 Å, similar to AgReO4) (Supplementary Fig. 35). This suggests that higher Re amounts favor the formation of the ordered AgReO4 phase.

a Ag K-Edge XANES spectra before and after hydrogenation at 150 °C. b Re L3-Edge XANES spectra before and after hydrogenation at 150 °C. Monometallic [Re/Al2O3] (7.5 wt% Re) was hydrogenated at 280 °C. c Re L3-Edge FT-EXAFS spectra before and after hydrogenation at 150 °C. Monometallic [Re/Al2O3] (7.5 wt% Re) was hydrogenated at 280 °C. d In situ XAS-H2-TPR study following the variation of the Ag K-Edge peak at 25518 eV and Re L3-Edge peak at 10542 eV. e In situ XAS-TPR study of [AgRe/Al2O3] catalyst comparing simultaneously the variation of the Ag K-Edge and Re L3-Edge, most relevant species in XANES and FT-EXAFS. f In situ XAS-H2-TPR study of [AgRe0.35/Al2O3] catalyst comparing simultaneously the variation of the Ag K-Edge and Re L3-Edge, most relevant species in XANES and FT-EXAFS. Source data are provided as a Source Data file.
In addition, bimetallic materials were subjected to in situ hydrogenation using a temperature ramp of 4 °C/min up to 150 °C and Ag K-edge XANES spectra were registered during this process. At the end of the reduction treatment, a clear change of the Ag K-edge XANES was observed and, for all the nanomaterials, spectrum similar to that of Ag0 was observed (Fig. 5a). The corresponding FT-EXAFS spectra of the reduced materials also confirmed the presence of Ag-Ag bonds in all of them (Supplementary Fig. 36). FT-EXAFS spectra between 2.5 and 3.5 Å revealed that the Ag-Ag contribution becomes more intense at higher amounts of Re. FT-EXAFS signal intensity can provide information regarding metal dispersion, higher intensities being related with a larger degree of aggregation105,106. Consistently with the results of HAADF-STEM, larger aggregation of Ag0 nanoparticles were detected in the bulk at materials with a higher Re:Ag molar ratio. Remarkably, in both [AgRe/Al2O3] and [AgRe2/Al2O3] nanomaterials reduced in situ at 150 °C, signals assigned to Ag-O bonds were observed. However, while for [AgRe/Al2O3]-H2-150 nanomaterial the signal appears at 1.6 Å, corresponding to Ag2O, for [AgRe2/Al2O3]-H2-150 it is seen at 1.9 Å, typically attributed to AgReO4, revealing that in this material some silver perrhenate species remain after reduction.
Considering the large number of Re oxidation states, Re L3-edge XANES resulted a very useful technique in order to shed light on the nanomaterials nature (Fig. 5b). Thus, fresh and in situ reduced AgRe and Re nanomaterials, together with the most relevant rhenium standards (Re0, ReCl3, ReO2, ReCl5, ReO3, AgReO4 and Re2O7) were subjected to this analysis. All the fresh nanocatalysts as well as AgReO4 and Re2O7 exhibited a signal at 10542.5 eV that can be clearly assigned to Re7+ species. It is very interesting to note that in the 10550–10560 eV region another signal of minor intensity was detected, normally attributed to a more outer coordination sphere, and in which differences between the nanomaterials could be observed (Fig. 5b and Supplementary Fig. 37). In fact, for [AgRe/Al2O3], [AgReO4/Al2O3] and [AgRe2/Al2O3] nanomaterials, in which AgReO4 species are present, the maximum of the signal appeared at a higher energy value (10554.7 eV), as it occurs for AgReO4. On the contrary, the nanomaterials in which AgReO4 species are not present, [AgRe0.35/Al2O3] and [Re/Al2O3], showed this signal at a lower energy value (10553.5 eV), more similar to that in the Re2O7 standard. Moreover, additional differences between the nanomaterials were observed through the analysis of the Re L3-edge XANES first derivative spectra (Supplementary Fig. 38) in which a tiny shift of the Re band to higher energy values was observed at higher Ag amounts. FT-EXAFS show that fresh materials, either bimetallic or Re monometallic, exhibit the Re7+-O bonds at 1.37 Å, similarly to AgReO4 and Re2O7 (Fig. 5c).
After the reduction treatment of the Ag-Re bimetallic nanomaterials and [Re/Al2O3] (at 150 °C for the bimetallics and 280 °C for the Re monometallic) important changes in the Re L3-edge XANES and EXAFS spectra were observed. In the first place, the XANES white line intensity decreased and the energy maximum shifted to lower values (10540 eV) (Fig. 5b). In addition, the band in the 10550–10560 eV region completely disappeared. A comparison of these results with the Re L3-edge XANES of the standards allows us to deduce that, after the H2 treatment, the reduction of originally present Re7+ species to an average oxidation state near Re3+ occurs. Probably this average oxidation state corresponds to the combined presence of ReO2 and Re0 species, since Re3+ and Re2+ oxides are not known. Interestingly, a close analysis of the energy value of the Re L3-edge XANES first derivative for each reduced nanomaterial as a function of the Ag content, reveals that the materials with a higher Ag content show the signal shifted to lower energy values and, hence, present more reduced Re species (Supplementary Fig. 39). On the other hand, FT-EXAFS spectra confirmed these observations and afforded more valuable information (Fig. 5c). As it could be expected, FT-EXAFS of the reduced materials showed the disappearance of Re7+-O bonds and a complex mixture of Re-O bonds attributable to ReOx species with an oxidation state <4 was detected. In addition, a global decrease in FT-EXAFS features in intensity was observed upon reduction, which is compatible with a large dispersion of ReOx species, as it has been observed by HAADF-STEM. In agreement with XANES, a low intensity Re-Re contribution become visible for [AgRe0.35/Al2O3]-H2-150, while [AgRe2/Al2O3]-H2-150 shows a larger Re-O contribution, which confirms the higher reducibility of Re in materials with larger Ag amounts. Both XANES and EXAFS also confirmed that [Re/Al2O3] nanomaterial required higher temperatures (280 °C) to get reduced in the same degree as bimetallic materials.
In summary, XAS studies undoubtedly confirmed that [AgRe0.35/Al2O3] fresh material is composed by Ag2O and ReOx species, in which Re is in a + 7 oxidation state, while [AgRe/Al2O3] and [AgRe2/Al2O3] are mainly constituted by AgReO4 species. After reduction at 150 °C, the nature of all the nanomaterials is to get composed by Ag0 nanoparticles and a diversity of ReOx species of which the average oxidation state is +3. Moreover, these experiments have also revealed that changes in the composition, in terms of Ag:Re molar ratio, lead to differences in the electronic properties of both metal centers (Ag and Re). This is reflected in the subtle changes in XANES first derivative of Ag K-edge and Re L3-edge of the fresh materials, that experience an increase in energy values at higher concentrations of the second metal. In the case of reduced materials, the energy levels measured for Re L3-edge XANES first derivative clearly experience a shift to lower values with larger Ag amount.
All this multitechnique characterization study provided with a very precise picture of the AgRe bimetallic materials either in their fresh or reduced state. However, key information regarding the mechanism by which the activation under H2 occurs and how this process is influenced by the nanomaterials nature was still missing. To better understand this, and extract further conclusions, XAS-TPR studies were conducted through the XAS monitoring at increasing temperatures under hydrogen flow (Fig. 5d–f). Firstly, the evolution of the XANES features characteristic of Ag+ (25518 eV) and Re7+ (10542 eV) species were monitored (Fig. 5d). As it was observed by conventional TPR studies, Re7+ reduction required lower temperatures at higher Ag content, confirming the prominent role of Ag to mediate Re reduction. On the other hand, the opposite effect was observed in the case of Ag, whose reduction appeared to be hampered by the presence of a higher Re content. It is important to remark that for the three AgRe materials studied, the required temperature for silver reduction was in the 25–93 °C range and, hence, this would justify the observed induction period for the fresh nanomaterials when the reaction was conducted at 90 °C. Thus, Ag+ species reduction would be the first required activation process to form the Ag0 in charge of mediating the first reaction step.
To better understand this phenomenon, the evolution of the most representative species of Ag K-edge and Re L3-edge from both XANES and EXAFS was jointly analysed for [AgRe0.35/Al2O3] and [AgRe/Al2O3] (Fig. 5e, f). The specific selection of these materials allowed us to understand the possible relevance of having AgReO4 species stabilized in the unactivated catalyst, as it is the case in [AgRe/Al2O3], or presenting Ag2O and Re7+ species on its surface, as it occurs in [AgRe0.35/Al2O3]. Moreover, the fact that AgRe0.35 nanomaterial was a less active catalyst despite showing a better tendency to the reduction of both Ag and Re species, was an important point to investigate. A simultaneous comparison of Ag+ and Re7+ species evolution in [AgRe/Al2O3] and [AgRe0.35/Al2O3] through XANES (Fig. 5e, f, up) clearly showed an induction period for the Ag+ reduction in both materials of 18 and 5 min, respectively. From this point, a fast consumption of Ag+ species was observed throughout the disappearance of the 25518 eV signal from Ag K-edge XANES and the simultaneous appearance of Ag-Ag bonds (2.6 Å) at FT-EXAFS. In the case of [AgRe0.35/Al2O3], once there was a complete reduction of Ag+ species, a clear disappearance of Re7+ species started. This was observed following the evolution of 10542 eV Re L3-edge signal of XANES and Re-O (1.38 Å) bonds from FT-EXAFS, in a sequential manner (Fig. 5f). However, for the optimal nanocatalyst [AgRe/Al2O3], a different trend was observed (Fig. 5e). Here, Ag+ reduction progressed through two different slopes, the first one being faster. Re7+ reduction only started at the end of the first slope. These experimental observations could be explained through a phenomenon in which after the first reduction of Ag+ to Ag0, there is a complementary process of Ag0 in situ oxidation in the ReOx interface that promotes Re reduction. From a mechanistic perspective, in [AgRe0.35/Al2O3], Ag and Re reduction proceed through two sequential steps. On the contrary, in the optimal [AgRe/Al2O3] nanomaterial, there is a cooperative reduction process between Ag and Re which could be key to explain the improved catalytic properties of this material in the hydrogenation of imides. This cooperation is probably a consequence of the presence of AgReO4 species, implying a more direct Ag and Re interaction.
Proposal of the nanomaterials structure and plausible reaction mechanism
At this point, considering all the information obtained from the catalytic and characterization studies, it was possible to perform a rationalisation of the Ag-Re nanomaterials structure through the definition of several relevant catalytically active sites (Fig. 6a). Active site I was defined to be constituted by Ag0 nanoparticles stabilized over the γ-Al2O3 support. On the other hand, active sites II and III implied the presence of Re, being sites II constituted by Ag0 nanoparticles and ReOx species of a wide range of oxidation states (Re0, Re4+, Re6+ and Re7+) over γ-Al2O3. Active sites III were proposed to be uniquely composed by Re species combining high-valent and low-valent oxidation states over the γ-Al2O3 support. As it was clearly demonstrated through catalytic and characterization studies, active sites I are only able to mediate the hydrogenation of N-methylphthalimide 1 to ω-hydroxylactam 2, while II and III can catalyze the formation of isoindolinone 3 from 1. As it has been demonstrated along the paper, the Ag-Re combination, present in active site II, is optimum for this transformation. However, it has been also described that [Re/Al2O3]-H2-280 shows a moderate catalytic activity. This can be explained by the presence of active sites III in this material, in which Re low-valent species are able to perform H2 activation, while Re7+ species with highly acidic properties and an oxophilic character can activate the substrate. In addition, an inactive site IV, under our reaction conditions, constituted by Re7+ species over the γ-Al2O3 can be defined.

a Plausible types of catalytic active sites. b Schematic representation of [AgRe0.35/Al2O3], [AgRe/Al2O3] and [AgRe2/Al2O3] nanomaterials before and after hydrogenation process. c Proposed reaction mechanism for the hydrogenation of 1 to 3. HV = High valent, LV = Low valent.
Identifying precatalytic nanomaterials structure is key for understanding and rationalising which of these active sites are present in the active materials (Fig. 6b). The discussion here will be focused in the most studied materials [AgRe0.35/Al2O3], [AgRe/Al2O3], and [AgRe2/Al2O3]. For the AgRe0.35 material, it was stablished through several characterization techniques (DRX, Raman, HAADF-HRSTEM, and XAS) that this precatalyst was composed by Ag2O nanoparticles and Re7+ oxides highly dispersed on the surface of the support and with a perrhenate geometry. In the case of AgRe optimal precatalyst, it was clearly demonstrated (DRX, DR-UV-Vis, Raman, HAADF-HRSTEM, and XAS) that uniquely AgReO4 nanoparticles are stabilized over γ-Al2O3 in this material. However, [AgRe2/Al2O3] shows these AgReO4 species together with Re7+ oxides of perrhenate geometry.
As it has been exposed, these materials need to be activated through a reductive process by which there is a first step of Ag+ reduction to Ag0, followed by Re7+ perrhenate species reduction to ReOx and/or Re0 species. XAS-TPR studies clearly demonstrated that Ag:Re molar ratio was key to define the tendency towards reduction, both Ag and Re species being more easily reducible in materials with an excess of Ag with respect to Re. Thus, in [AgRe0.35/Al2O3] nanomaterial, Ag+ centers are more accessible and, hence, more easily reducible and they mediate Re species reduction at lower temperatures. However, as it has been demonstrated through catalytic studies, this material is rather an inactive catalyst, being the most efficient catalysts those mainly containing AgReO4 species in their structure. Interestingly, XAS-TPR studies provided additional insights regarding how the reductive activation process occurs. In fact, it was observed that for [AgRe/Al2O3] optimal nanocatalyst, Ag+ reduction proceeded through two phases coupled with Re7+ reduction in a cooperative manner, which suggests that this process can give place to Ag and Re reduced species with a more intimate contact and, hence, with a suitable structure to establish catalytical synergies. Moreover, the deep characterization also revealed other differential and characteristic trends of this optimal nanocatalyst in its reduced form. IR-CO spectroscopy showed that Ag0 species in this material present a more metallic or less oxophilic nature in comparison with silver reduced species of the other nanomaterials. On the other hand, both IR-CO and XPS techniques could not detect Re0 species in the AgRe reduced material. This observation would suggest that an optimal catalytic efficiency is achieved with the combination of Ag0 and ReOx surface species. However, as it is the case for [Re/Al2O3]-H2-280, this catalysis can also operate through the cooperation of Re0 and oxophilic Re species, although in a less efficient way.
With all this information in hand, active sites for each activated nanomaterial can be defined (Fig. 6b). For the optimal [AgRe/Al2O3]-H2-150, the main presence of active sites II, formed in a cooperative manner, is proposed. In addition, the large dispersion of Re species makes feasible to propose the additional presence of active sites III, formed through the activation of sites IV. In the case of [AgRe0.35/Al2O3]-H2-150 with a silver excess, a high presence of active sites I is proposed, that would explain the lower selectivity to 3 observed. In addition, a moderate number of active sites II, formed through a sequential reduction, and III is proposed. Finally, for [AgRe2/Al2O3]-H2-150 it is obvious that the reduction and dispersion of both Ag and Re species is more difficult due to the presence of an excess of ReO4− species, which explains the low activity of the fresh material. Here, it is proposed the presence of mainly active sites II and III, as well as inactive site IV.
Therefore, a plausible mechanism for the N-methylphthalimide 1 hydrogenation to the corresponding isoindolinone 3 using as catalyst [AgRe/Al2O3], can be proposed (Fig. 6c). The first step would be the activation of the nanomaterial consisting in the cooperative hydrogenation of AgReO4 species to Ag0 nanoparticles and ReOx dispersed clusters of mixed valences. Then, the hydrogenation of the corresponding phthalimide 1 to the hemiamidal 2 would occur through several possible intermediates implying mainly active sites II but also I or III. On one hand, Ag0 nanoparticles have shown to be able to activate H2 to form the hemiamidal 2 in cooperation with both Re oxophilic species (site II) or γ-Al2O3 acid sites (site I) able to coordinate and activate the carbonyl group. On the other hand, sites III involving a combination of Re high and low valent species, would also offer the suitable properties to mediate this transformation, although this is probably a minor via. The next step would require the C-OH hydrogenolysis to give place to the isoindolinone 3 and, in this case, also different routes can be proposed. The main one involves sites II in which there is a cooperation between Ag0 centers activating H2 and ReOx species promoting OH or, more probable, H2O elimination. It is important to remark that [Ag/Al2O3] could not mediate this step, which suggests that Re species stronger acidity results key here. In addition, the same transformation is feasible to occur at sites III performing the H2 activation with Re low valent species.
Recycling studies and heterogeneous nature of the bimetallic [AgRe/Al2O3] nanocatalyst
Re-based catalysts are generally known to present a limited stability due to Re leaching tendency in polar solvents, as well as the high volatility of Re oxides (Re2O7; b.p. = 363 °C)87. Therefore, at this point, it was very interesting to perform stability studies to confirm the heterogeneous nature of our AgRe bimetallic system. Firstly, a filtration test was performed (Supplementary Fig. 9), which confirmed that after catalyst removal at 20 min no additional conversion of 1 occurred. Furthermore, ICP-AES of the filtrated material after being used as catalyst for 16 h confirmed its stability, as 4.0 wt% Ag and 7.0 wt% Re were measured, being 4.2 wt% Ag and 7.5 wt% Re the original data (see section 3.2 of the Supplementary Information). Moreover, possible metal content in the filtered reaction mixture was also discarded by ICP-AES (see section 3.2 of the Supplementary Information). In addition, elemental analysis of the used material revealed that a 3 wt% of carbon was present, which indicates the presence of organic compounds attached to the material surface, probably due to Re oxophilicity. Hence, considering all this data, we attempted to explore the reusability of [AgRe/Al2O3] nanocatalyst. With the aim of avoiding the presence of any organic residue and regenerating the active catalytic species, a calcination treatment at 300 °C between each reaction cycle was performed. To our delight, [AgRe/Al2O3] system could be used until five consecutive reaction cycles without apparent loss of activity and selectivity (Supplementary Fig. 10). Furthermore, we could perform these tests scaling up our usual reaction conditions (x15), which indicates the large robustness of our catalytic system (for details on the experimental procedure see Methods section).
To better understand the influence of the reaction conditions and the reactivation treatment in [AgRe/Al2O3] structure, we characterized both the used and reactivated systems by XRD and UV-Vis DRS (Supplementary Figs. 13 and 18). In both techniques we could observe the formation of Ag0 nanoparticles in the used materials which disappeared after the calcination treatment at 300 °C. In addition, XRD confirmed the formation of AgReO4 species after the 300 °C calcination. Interestingly, HAADF-STEM and STEM-XEDS analysis also showed that the nanostructure of the used [AgRe/Al2O3] in one reaction cycle was very similar to the one observed for [AgRe/Al2O3]-H2-150, as Ag0 nanoparticles (3–10 nm) and subnanometric ReOx species were observed (Supplementary Fig. 25). Therefore, all these studies confirm the good stability of [AgRe/Al2O3] nanocatalyst, suggesting that the deposition of AgReO4 species is a good strategy for improving Re based catalysts stability.
Comparison of the catalytic activity of [AgRe/Al2O3] with well-known nanocatalysts active for hydrodeoxygenation reactions of carboxylic acid derivatives
In the last years, a few interesting catalytic protocols have been described for performing the challenging hydrogenation or hydrogenative functionalization of carboxylic acid derivatives employing heterogeneous catalysis19,59,70,107. One of the main problems of this kind of reactions employing nanocatalysts has been to achieve a good tolerance to the preservation of aromatic rings. Remarkably, some of the described systems, such as [PtV/HAP]19, [Re/TiO2]70 and [Au/TiO2]107 have been demonstrated to be successful in this sense. Therefore, at this point we decided to perform a comparison of the catalytical activity of these described nanocatalysts (see section 1.1 of Supplementary Information for their preparation procedure) with our optimal [AgRe/Al2O3] in the hydrogenation of N-methylphthalimide 1 at 90 °C, under 40 bar of H2 during 5 h and employing a 10 mol% of metal (Supplementary Fig. 8). Furthermore, we also decided to include in this study [PtRe/TiO2] nanomaterial59, as it has been described by the group of Palkovits as an active catalyst for the monohydrogenation of one succinimide. The comparison revealed that both catalysts containing Pt, [PtV/HAP] and [PtRe/TiO2], afforded total conversions but were not selective towards the preservation of the aromatic ring of the phthalimide 1, and no lactam 3 was formed. In the case of [Au/TiO2], the aromatic ring was not reduced but a mixture of hemiamidal 2, lactam 3 and phthalide 6 was formed. Finally, [Re/TiO2] gave place to lactam 3 in good selectivities but showed a much lower catalytic activity than [AgRe/Al2O3]. It is also important to remark that this Re-based material needs to be hydrogenated at 700 °C to be active, whereas our [AgRe/Al2O3] optimal nanocatalyst is more efficient when is hydrogenated in situ.
Scope of the reaction
Once demonstrated the good activity and recyclability of our designed bimetallic [AgRe/Al2O3] nanocatalyst for the selective hydrogenation of imide 1 to lactam 3, we became interested in evaluating its potential applicability for the hydrogenation of a wide range of cyclic imides to the desired lactams. In our study, not only symmetrical cyclic imides (Fig. 7), but also more challenging non-symmetrical substrates (Fig. 8), in which the regioselectivity towards the hydrogenation of one carbonyl fragment is a key point, were employed.

[a] Unless otherwise noted, standard conditions: cyclic imide (0.25 mmol), [AgRe/Al2O3] (6 mol% Ag), 4 Å MS (45 mg), anhydrous CPME (1 mL) and 40 bar of H2 at 90 °C during 16 h. Yields of isolated products are given between brackets. [b] Run at 120 °C. [c] Run with 70 bar of H2. [d] Run during 36 h. [e] Run at 160 °C. [f] Run with 10 mol% Ag.

[a] Unless otherwise noted, standard conditions: cyclic imide (0.25 mmol), [AgRe/Al2O3] (6 mol% Ag), 4 Å MS (45 mg), anhydrous CPME (1 mL) and 70 bar of H2 at 100 °C during 16 h. Yields of isolated products are given between brackets and regioisomeric ratios (calculated by 1H NMR) are given between parentheses. In the cases in which two different regioisomers are formed, only the structure of the major one is shown. [b] Run at 120 °C. [c] Run with 50 bar of H2. [d] Run during 36 h. [e] Run at 160 °C. [f] Run with 10 mol% Ag. [g] Run at 140 °C.
Among the symmetrical cyclic imides studied (Fig. 7), several N-alkyl, aryl or heteroaryl substituted imides, including aromatic ring substituted phthalimides, as well as succinimides and glutarimides, were subjected to the reaction conditions for obtaining the desired lactams. To ensure a total conversion of the imide and/or to achieve the best yield of the desired lactam, in selected cases, some reaction parameters (temperature, reaction time, mol% Ag, etc…) were modified (see footnote of Fig. 7 for detailed information). Apart from N-methylisoindolinone 3 (96% isolated yield), other N-alkyl isoindolinones (7–9) were obtained in high yields (68–81%) from the corresponding phthalimides bearing a longer carbon chain or more hindered alkyl substitution (n-Bu, i-Pr and Cy). In addition, N-alkyl substituted phthalimides with a ketone or a carboxylic acid functional group in their structure were successfully hydrogenated giving place to lactams 10 and 11 (79% and 71%, respectively) that underwent the concomitant hydrogenation of the carbonyl or acid group. It should be noted that in these cases our AgRe system was able to mediate both hydrogenations, and no catalyst poisoning was observed.
Then, N-phenyl phthalimides with a diversity of substituents in ortho, meta and para positions (-Cl, -Br, -MeO, -OH) were tested (Fig. 7), and the corresponding isoindolinones 12 to 17 could be isolated in moderate to very good yields (49–93%). As it could be expected, some degree of debromination was observed in the case of compound 14. In addition, the corresponding lactams 18 and 19, with 1,3-benzodioxolane and naphthyl substitution, were obtained with 56% and 75% yield, respectively. Here, it is interesting to emphasize the excellent C-O selectivity of our catalytic system towards N-aryl substituted imides hydrogenation, more prone to undergo C-N hydrogenolysis, since anilines are good leaving groups. At this point, it was interesting to explore the activity of our developed system in the hydrogenation of N-benzylphthalimide and its analogues with ortho-, meta- and para substitution in the aryl ring (Fig. 7). Gratifyingly, either electron-donating groups (EDG), such as -Me or -BPin, or electron-withdrawing groups (EWG), such as CF3 or F, were well tolerated affording lactams 20 to 24 in yields from 48 to 97%. Herein, it is very remarkable that a labile pinacol boronic ester (BPin) group, with a large potential of further functionalization, was tolerated under the applied catalytic conditions, affording compound 22 in a 48% of isolated yield. Moreover, a N-benzylphthalimide with a 1,3-benzodioxolane scaffold afforded almost quantitative yields of the corresponding lactam 25 (98%). None of the explored N-benzylphthalimides afforded by-products coming from the hydrogenolysis of the sensitive benzyl bond. Furthermore, homobenzylic isoindolinones 26 and 27 could be also isolated in very good yields (97–99%) from the corresponding cyclic imides.
With the aim of further expanding the applicability of our catalytic protocol to the hydrogenation of more complex cyclic imides, the hydrogenation of several heterocycle containing phthalimides was evaluated with [AgRe/Al2O3] nanocatalyst (Fig. 7). Remarkably, N-substituted lactams 28–32 bearing heterocyclic fragments such as tetrahydrofuran, electron-rich furan, thiophene or indole as well as an electron-poor pyridine were efficiently accessed in 38 to 76% isolated yields. No products coming from degradation or (over)hydrogenation, very common for heterocycles in the presence of heterogeneous catalysis, were detected.
After demonstrating the wide applicability of our catalytic protocol for the effective hydrogenation of a diversity of N-substituted phthalimides to the desired lactams, we were interested in studying the hydrogenation of phthalimide substrates with symmetrical aromatic ring substitution and/or a NH-free position (Fig. 7). To our delight, N-methyl halogen-substituted phthalimides featuring F or Cl atoms in several positions of the benzene ring, yielded the desired halogen-containing lactams 33–36 in quantitative yields (91–98%). In addition, several NH-free cyclic imides, including unsubstituted phthalimide as the most fundamental one, afforded the corresponding isoindolinones 37–39 with moderate to very good yields (49–93%). The easy functionalization of this class of lactams in order to obtain more complex organic molecules indicates the high potential of our protocol. In fact, Fosun Farma recently designed a synthetic route for an antineoplastic agent with activity against lung cancer in which 1-isoindolinone 37 was the starting material and it should be produced in kg scale. However, isoindolinone 37 has been traditionally synthesized using a Clemmensen-type reduction of phthalimide that requires over-stoichiometric Sn, large concentrations of strong acid and high temperatures with a limited yield around 50%108. By employing more modern methods, phthalimide was reduced to isoindolinone 37 employing homogeneous catalysis57, heterogeneous systems requiring harsh reaction conditions55 or through an electrocatalytic strategy41. Notably, our strategy afforded lactam 37 almost quantitatively (93% isolated yield). In addition, 2,3-naphthalenedicarboximide was converted to lactam 39 in 66% of isolated yield.
Finally, apart from evaluating the monohydrogenation of symmetrically substituted phthalimides, aliphatic cyclic imides such as succinimides (5-membered) and glutarimides (6-membered) were also subjected to the developed catalytic protocol (Fig. 7). To our delight, the desired 2-pyrrolidone compounds 40–44, including the NH-free 2-pyrrolidone 44, were obtained with yields from 54 to 89%, as well as a two NH-free 2-piperidones 45 and 46 obtained with 61 and 91% yield, respectively. The synthesis of these relevant scaffolds further proves the good applicability of the developed methodology.
At this point, we became interested in evaluating the application of our catalytic system towards the reduction of non-symmetrical imides, mainly aromatic-ring substituted phthalimides and homophthalimides. The selective hydrogenation of this imides class implies an additional challenge related with the control of the regioselectivity towards the hydrogenation of one of the non-equivalent carbonyls (Fig. 8). Indeed, up to date only two reports based on homogeneous catalysis studied such type of hydrogenations in non-symmetrically substituted cyclic imides50,51. Hence, the exploration of the behavior of our catalytic protocol towards this kind of substrates is an interesting extension of our work.
First, several N-methyl and NH-free 3-substituted phthalimides bearing EWG (fluoro) or EDG (Me, OMe, OH and NH2) were employed as starting materials (Fig. 8). The substitution in 3-position presented, in general, a strong effect in the regioselectivity, obtaining in most of the cases a main regioisomer in which the more distal carbonyl group had been hydrogenated. Thus, for 3-substituted phthalimides with non-coordinating groups of diverse electronical character, such as F and Me, the less hindered C = O bond was selectively hydrogenated. Regioisomer mixtures of (13:1) and (10:1) ratios were respectively obtained, with isolated yields of 98% and 90%, being lactams 47 and 48 the major regioisomers. In the case of electron-donating groups (OMe and OH), very good regioselectivity was observed in the case of the methoxy substituent, obtaining lactam 49 as the major regioisomer of a (8.6:1) mixture ratio, in a 96% yield. Interestingly, in the presence of a hydroxy group, the regioselectivity was shifted, observing the formation of lactam 50 as the major regioisomer with a (1.7:1) regioisomeric mixture ratio. Notably, the enhanced coordinating properties of the OH group caused that a less electrophilic and more hindered C = O position reacted, contrary to the observed for other imides. In the case of N-unsubstituted substrates, the influence of polar groups was evidenced, and in fact, harder reaction conditions were needed to obtain lactam 51 as the major regioisomer with a worst regioisomeric ratio (3.3:1) compared to its N-methyl analog 47. Interestingly, the hydrogenation of 3-amino-substituted phthalimide uniquely afforded lactam 52, coming from the reduction of the most electrophilic and exposed C = O functionality, with a 49% yield.
As expected, in the case of 4-substituted N-Me and NH phthalimides, a minor influence of the substituent in the regioselectivity was observed (Fig. 8). Thus, lactams 53–58 were obtained as the major regioisomer of mixtures with 2.9-1.3:1 ratios. Interestingly, in these 4-substituted imides the electronic character of the substituents was key. For electron-withdrawing groups F and Br, the most electrophilic and more distal carbonyl was the one mainly hydrogenated, affording 53 and 54 lactams as the main compounds. On the other hand, for phthalimides substituted in 4 position with electron-donating groups (OMe, NH2 or OH), the most proximal carbonyl to the substituent was the one hydrogenated, affording mainly lactams 55–58. This is probably due to the mesomeric effect by which the proximal carbonyl is the most electrophilic one.
Finally, the hydrogenation of homophthalimides, characterized by presenting two chemically distinguishable carbonyls, was explored with our [AgRe/Al2O3] nanocatalyst (Fig. 8). For all these substrates, the hydrogenation of the carbonyl group directly bonded to the methylene fragment was the only observed. This is easily explained if it is considered that this carbonyl is in equilibrium with the corresponding enol, and hence, its hydrogenation is favored through the formation of an enamide. In fact, for many homophthalimides we have been able to modulate the reaction conditions for synthesizing either the corresponding isoquinolinone, with an enamide scaffold and a high interest for pharmaceutical industry109,110, or the iso(3,4-dihydro)quinolinone. This was the case of the N-methyl, phenyl, benzyl and 2,6-difluorobenzyl homophthalimides, for which a fine tuning of the reaction conditions allowed to isolate the corresponding isoquinolinones 59, 61, 65 and 68 in yields of 48–89% or the derived iso(3,4-dihydro)quinolinones 60, 62, 66 and 69 in 57–89%. On the other hand, homophthalimides with N-phenyl substitution containing a chlorine or methoxy groups in the benzene ring, only allowed to obtain the corresponding isoquinolinones 63 and 64 in 57 and 89% of yield, respectively. This was also the case of the NH free homophthalimide, that afforded isoquinolinone 70 in a 74 % of yield, highly improving previously reported synthetic procedures for this molecule111. On the contrary, for the N-benzyl homophthalimide with 3,5-bis(trifluoromethyl) substitution, the corresponding iso(3,4-dihydro)quinolinone 67 was the only compound obtained in a 68%. Similarly, when a non-symmetrical 3,3-disubstituted succinimide was subjected to our reaction conditions, the less hindered carbonyl bond was reduced affording pyrrolidone 71 with a moderate isolated yield (44%), but total selectivity.
In summary, we have presented a nanostructured catalyst able to promote a widely general and selective hydrodeoxygenation of cyclic imides to lactams. An exhaustive design of the system has revealed that the optimal nanomaterial is constituted by a combination of Ag and Re species in an equimolar ratio and stabilized onto a γ-Al2O3 matrix. Notably, a deep characterization using a battery of complementary techniques has allowed to identify the most relevant active sites of the nanocatalyst, revealing that the highest catalytical efficiencies are achieved through the in situ hydrogenative activation of the [AgRe/Al2O3] nanomaterial. Thus, the optimal AgRe catalyst is constituted by well-defined and non-innocent AgReO4 species that, under our reaction conditions, evolve to Ag0 nanoparticles and ReOx nanoaggregates homogeneously dispersed over the support crystallites. Interestingly, we have disclosed that [AgRe/Al2O3] nanomaterial presents a suitable Ag and Re molar ratio not only to achieve better bimetallic cooperation, but also to form the active species with the appropriate electronic and structural properties of both metal species. Furthermore, the optimal bimetallic AgRe catalyst has proved its robustness through its repeated use up to five catalytic cycles. The wide synthetic applicability of the catalytic protocol has been demonstrated through the synthesis of a wide range of lactams (>60) with a full tolerance to the presence of aromatic rings and an interesting regioselectivity.
Methods
General Information
All the chemicals were obtained from commercial sources and were used without further purification otherwise indicated. 4 Å Molecular sieves (MS) were activated at 300 °C under vacuum for 3 h before use. Metallic precursors, solid supports used for nanomaterials preparation were purchased from Sigma-Aldrich ([Ag(acac)], [VO(acac)2], [MoO2(acac)2], [NH4ReO4], [(NH4)6H2W12O40·xH2O], [H3BO3], [Bu4Sn], [Ga(NO3)3·xH2O], [Pd(acac)2], [Pt(acac)2], [Ni(acac)2], [Fe(acac)3], [Co(acac)3], [Cu(acac)2], [Mn(acac)3], SiO2, ZnO, CeO2, TiO2, ZrO2, Nb2O5 and MgO), ABCR ([AgReO4] and γ-Al2O3, ref. AB255279) and Zeolyst (H-BETA CP811, [Si/Al] molar ratio = 12.5). 4 Å Molecular sieves (MS), and all the screened solvents (anhydrous n-heptane, anhydrous THF, 2-MeTHF, 1,4-dioxane, anhydrous CPME, anhydrous toluene, α,α,α-trifluorotoluene, and MeOH) were purchased from Sigma-Aldrich. Reference high-purity compounds were also commercially available, i.e. [Ag foil] (Sigma-Aldrich), [Ag2O] (Sigma-Aldrich), [ReCl3] (Sigma-Aldrich), [ReO2] (Sigma-Aldrich), [ReCl5] (ABCR), [ReO3] (ABCR), [Re2O7] (ABCR), [AgReO4] (ABCR), and [Re powder] (ABCR).
Reaction monitoring and characterization methods for organic molecules
Gas chromatography (GC)
Gas chromatography (GC) was used to monitor the reaction outcome. The analyses were performed in a Bruker 430-GC equipped with a 25 m capillary column of 5% phenylmethyl silicone, using n-dodecane as the internal standard and N2 as carrier gas.
Preparative thin layer chromatography (PTLC) and column chromatography
All the imide hydrogenation products were isolated by silica column chromatography or preparative thin layer chromatography (PTLC). For PTLC, thin layer chromatography plates UniplateTM (20×20 cm) from Miles Scientific were used and the organic compounds were extracted with EtOAc stirring for 2 h. When column chromatography was chosen, it was used Silica Gel Merck 60 with 0.04-0.06 mm of particle size (Merck 109385 reference) using different eluent mixtures (n-hexane/EtOAc or CH2Cl2/MeOH).
Gas chromatography-mass spectrometry (GC-MS)
All the isolated organic products were characterized by gas-chromatography-mass spectrometry (GC-MS), to ensure the structure of the obtained molecules and determine their structural fragmentations. The analyses were acquired on Agilent 6890 Network gas chromatograph equipped with an HP-5 column (30 m, 0.32 mm, 0.25 μm) coupled to an Agilent 5973 Network mass selective detector.
Nuclear magnetic resonance (NMR)
Nuclear magnetic resonance (NMR) is the reference technique to characterize organic molecules. In this work, all the synthesized imides and the obtained lactams were properly characterized with monodimensional 1H, 13C, (and 19F if applicable) and DEPT. When required, also bidimensional spectra were acquired to ensure the molecule structure (HSQC and HMBC). Spectra were recorded on a Bruker 300 or Bruker 400 spectrometer. All chemical shifts (δ) are reported in parts per million (ppm) and coupling constants (J) in hertz (Hz). Abbreviations used in the reported NMR experiments: d, doublet; dd, double doublet; s, singlet; t, triplet; q, quartet; quintuplet, sextet, septet, m, multiplet. All chemical shifts are reported relative to residual proton solvents: i.e., to CDCl3 peaks (δ 7.26 ppm for 1H NMR and δ 77.0 ppm for 13C NMR), CD3CN (δ 2.13 ppm for 1H NMR and δ 118.26 ppm for 13C NMR), DMSO-d6 peaks (δ 2.50 ppm for 1H NMR and δ 39.5 ppm for 13C NMR) or MeOD (δ 4.87 ppm for 1H NMR and δ 49.0 ppm for 13C NMR).
Ultra-performance liquid chromatography high-resolution mass spectroscopy (UPLC-HRMS)
When available, the characterization given in the literature was used for comparison; otherwise, UPLC-HRMS experiments were additionally done to confirm the novel structure. HRMS measurements of all non-described organic isolated products were performed using the electrospray ionization technique in UPLC equipment.
Materials characterization techniques
X-ray powder diffraction (XRPD)
All the catalysts were measured with X-ray powder diffraction (XRPD) to identify the crystalline phases. Powder XRD measurements were performed in Bragg-Brentano geometry using a PANalytical CUBIX diffractometer equipped with an X-Celerator detector using Cu Kα (λ1 = 1.5406 Å, λ2 = 1.5444 Å, I2/I1 = 0.5) radiation. The applied tube voltage and intensity were 45 kV and 40 mA, respectively. The length of the goniometer arm is 200 mm, and a fixed divergence slit with a 1/8° aperture was applied. The scanning range was from 3.5° to 90.0° (2θ), with a step of 0.020° (2θ) and an acquisition time of 35 s per step. The measurement was performed at 25 °C while the sample was rotated at 0.5 revolutions per second.
Inductively coupled plasma-atomic emission spectroscopy (ICP-AES)
The amount of metal (wt%) contained in the fresh and used catalysts was determined by inductively coupled plasma-atomic emission spectroscopy (ICP-AES) using Varian 715-ES after the dissolution of the solid samples in HCl/HNO3 solution (3:1 vol).
Fourier-transform infrared (FTIR) spectroscopy of adsorbed pyridine
To quantify the acidity of the solid catalysts, the monitoring of the adsorption-desorption process of pyridine through infrared spectroscopy is an interesting tool. Both, Brönsted acid sites (pyridinium ion band at 1555 cm−1) and Lewis acid sites (band at 1450 cm−1) can be distinguished. Nevertheless, in this work, it is only possible to study the nature and quantify the Lewis acid sites, due to the necessary preparation of the pellet by mixing the catalyst with chromatography SiO2 (50 wt%). With this goal, the concentration of Lewis Acid Sites [LAS] (mmol/g) was calculated according to the methodology proposed by Emeis81 where A is the integrated absorbance of FTIR signals, r is the radius of the pellet, and w is its weight (Eq. 1).
Moreover, the strength distribution can also be studied by performing the desorption at different temperatures, being stronger these remaining adsorbed at higher temperatures. In our experiments, FTIR measurements were performed in a Nicolet Is-10 Thermo FT-infrared spectrophotometer. Self-supported pellets (10 mg) were degassed under vacuum (10−2 Pa) at 300 °C for 12 h. Pyridine was then fluxed into the cell (650 Pa) and when equilibrium was achieved, the cell was degassed at a desired experiment temperature and cooled down to room temperature. At this point, FTIR was acquired. This sequence was carried out at 150 °C (weak strength), 250 °C (medium strength), and 350 °C (high strength). In all cases, a spectrum was collected under vacuum before pyridine adsorption and was used as background. This background was subtracted from each spectrum, and the absorbance was normalized to weight before calculations.
Elemental analysis (EA)
With elemental analysis (EA) it is possible to determine the carbon, hydrogen, nitrogen, and sulfur of the catalyst by means of a combustion process. In this work, it was used to quantify the carbon present after the catalytic hydrogenation reaction. EA was carried out in a EURO EA elemental analysis from Eurovector. Experimentally, the samples were introduced in an Sn capsule into an oven at 1020 °C. He was used as carrier gas (140 mL/min).
N2 Physisorption
Nitrogen adsorption technique by the application of the Brunauer-Emmet-Teller (BET) method, was used to determine the surface area of the catalysts. Nitrogen adsorption isotherms were registered using an ASAP 2420 apparatus (Micromeritics) at −196 °C (77 K). Before the analysis, 200 mg of the sample (sieve fraction 200–400 μm) were degassed at 673 K and ∼5×10−6 bar overnight. The specific surface area was determined by applying the Brunauer−Emmett−Teller (BET) equation in the relative pressure range (P/P0) of 0.05-0.25 of the isotherms.
H2-Temperature programmed reduction (H2-TPR)
The study of the effect of H2 over the catalysts at a programmed temperature is used to characterize several physicochemical properties. In this work, H2-temperature programmed reduction (H2-TPR) was performed to study the effect of Ag and Re in their reducibility, at different Ag/Re molar relationships. H2-TPR experiments were carried out in Micrometrics Autochem 2910 equipped with a Thermal Conductivity Detector (TCD) detector, using 50 mg of freshly calcined catalyst, and increasing the temperature from room temperature to 800 °C with a heating ramp of 10 °C/min under 10% H2/Ar (vol%) and a constant flow rate of 50 mL/min.
High-angle annular dark-field scanning-transmission electron microscopy (HAADF-STEM) and scanning-transmission energy-dispersive X-ray analysis (STEM-XEDS)
The samples were prepared by physical contact with a Cu grid and measured in a double aberration corrected, monochromated, Titan Themis 60–300 kV with a probe convergence angle of 21.4 mrad working at 200 kV. This microscope allows recording STEM images with subangstrom (0.8 Å) resolution in the different STEM imaging modes. It is also equipped with a high sensitivity, large XEDS Super-X system, which integrates 4 detectors providing a total solid angle of collection of roughly 0.8 srad. Electron beam currents were adjusted both during imaging and analytical STEM measurements to minimize beam damage effects while preserving signals with high S/N ratios.
Raman spectroscopy
Raman spectroscopy is a reference characterization method to obtain knowledge about the vibration of the chemical bonds at the surface, thus providing access to the structure of crystalline solids and amorphous materials. Complementary to XRD, with Raman spectroscopy it was possible to gain insight into the structure, changes in composition, or phase transitions in both Ag and Re. Raman spectroscopy experiments were performed with a Renishaw “in via” spectrometer equipped with a Leika DM LM microscope and a 314 nm, 514 nm and 785 nm HPNIR laser diode as the excitation source. Multiple Raman scans were performed between 100 and 2500 cm−1 with a ~ 4 cm−1 resolution. When indicated, some of the samples were ex situ reduced before measurement by the following procedure: A quartz tube containing the material was introduced into a tubular furnace with a temperature controller, and a system to allow gas flow to enter and exit were connected. Once the system was set up, the furnace was programmed for reaching to the desired final temperature (150 or 280 °C) with a programmed heating ramp of 2 °C/min. All the heating processes were done under inert gas flow (N2 or Ar, 100 mL/min). When arrived at the maximum temperature, the gas was changed to H2 (100 mL/min), and the reductive treatment was maintained during 2 h. Then, the furnace was switched off, and the catalyst was cooled to room temperature under inert gas flow (N2 or Ar).
IR-CO adsorption spectroscopy (IR-CO)
Infrared spectra of the CO titration (IR-CO) were recorded with a Nicolet iS50 FTIR spectrometer equipped with a DTGS detector. A transmission cell with KRS-5 (Thallium-iodide bromide) windows and a resolution of 4 cm−1 was used. Samples were pressed into self-supported wafers. Initially, the calcined sample was measured and subsequently reduced in situ under H2 gas flow (10 ml/min) at 150 °C (2 °C/min) for 2 h. After the reduction, the cell was evacuated (10−4 mbar) at the same temperature for 1.5 h in a dynamic vacuum. Finally, the cell was cooled to −170 °C and the CO was dosed to identify the main bands associated with Ag and Re species on the surface. The binding strength of CO with surface sites during cell evacuation was studied. Spectra were collected after each CO dosing and during evacuation. Omnic 9.1 software was used for spectra processing.
UV-Vis diffuse reflectance spectroscopy (UV-Vis DRS)
Diffuse reflectance (UV-vis) spectra were obtained on a Cary 5000 (Agilent Technology) instrument equipped with a Harrick “Praying Mantis” cell, using BaSO4 as a reference for reflectance.
X-ray photoelectron spectroscopy (XPS)
The surface composition and chemical state of Re and Ag were determined by X-ray photoelectron spectroscopy (XPS). The photoelectron spectra were recorded with a PHOIBOS 150 MCD-9 analyzer from SPECS and a non-monochromatic Al Kα X-ray energy of 1486.60 eV under vacuum conditions (10−9 mbar) and 25 °C. The binding energy has been corrected by reference to the Al 2p component at 74.2 eV112. The relative quantification of Re 4 f and Ag 3 d, and data analysis was performed with the CasaXPS software (Casa Software Ltd) using the NIST library113. The catalysts were reduced in situ at 150 °C and only the [Re/Al2O3] catalyst was also reduced in situ at 280 °C under 100 ml/min of H2 for 2 h. After reduction and reaction, a new spectrum is taken to compare the chemical state and composition of the surface.
X-ray absorption spectroscopy (XAS)
X-ray absorption spectra were recorded at the Ag K-edge (255564 eV) and Re L3-edge (10585 eV) at the CLÆSS (BL22) of the ALBA synchrotron light source (Spain)114. The wiggler source at this beamline was monochromatized using a double crystal Si(311) monochromator, while the higher harmonics were rejected by choosing appropriated angles for the collimating and focusing mirrors. Reference high-purity compounds, i.e. Ag foil (Sigma-Aldrich, 99.99%), Ag2O (99.0%), ReCl3 (Sigma-Aldrich), ReO2 (Sigma-Aldrich, 99.7%), ReCl5 (ABCR, 99.9%), ReO3 (ABCR, 99.9%), Re2O7 (ABCR, 99.9%) and AgReO4 (ABCR, 99.99%), were pressed into pellets (S = 1.33 cm2), with optimized thickness after dilution in cellulose, and measured in transmission mode. Re powder (ABCR, 99.99%) was diluted in powder boron nitride and reduced at 600 °C in situ before its measure. Several XAS measurements were collected to ensure reproducibility and statistics (5 for Ag and 3 for Re). The averaged spectra were treated by the Athena software package.
In situ temperature-programmed reduction monitored by X-ray absorption spectroscopy (TPR-XAS) experiments were performed to monitor the evolution (under hydrogenative conditions) of Ag and Re species as a function of temperature, thereby assessing the stability of monometallic, and bimetallic AgReO4 species towards reductive processes. All the measurements were done in transmission mode, and two different pellets of bimetallic catalysts were measured (Ag-K edge and Re-L3 edge). Samples were prepared by mixing with mortar the calculated amount of catalyst with powder boron nitride (100–150 mg total) for 1 h, and pelletizing (S = 1.33 cm2) the mixture. Then, the samples were introduced into the cell, equipped with a gas-dosing system and temperature controller. Once sealed, the cell was purged with He for 5 min (500 mL/min) at room temperature and atmospheric pressure. To start the experiment, several spectra were collected as initial states, five for Ag-K edge and three for Re-L3 edge measurements. After that, the gas was changed to H2 (100 mL/min), and the same spectra were acquired for comparison. Then, all the catalysts were heated with a heating rate of 4 °C/min from 25 °C to 150 °C, and this temperature was held for 90 min. For the monometallic Re catalyst, the final temperature was set at 280 °C. Finally, the samples were cooled down under H2, and once arrived at 25 °C, three or five additional XAS spectra were collected for Re-L3 and Ag-K edges, respectively, and used as the final catalysts state (labelled as [AgRex/Al2O3]-H2-150 or [Re/Al2O3]-H2-280 (7.5 wt% Re). XAS spectra were acquired during all the experiment.
General and specific procedures for the preparation of solid nanomaterials
General procedure for the preparation of the solid materials
A 100 mL round-bottom flask containing a stirring bar was sequentially charged with the metal precursors [notation: M1 stands for 0.4 mmol M1 precursor/g support, see specific procedures below for more information] and acetone (50 mL) as solvent under air atmosphere and darkness. After 10 min of stirring, the support (1 g) was added, and the flask was stirred for 4 h at room temperature. Once completed the impregnation, the solvent was distilled under vacuum using a rotary evaporator. Finally, the solid was homogenized and dispersed with a mortar, and a thermal process was carried out to generate the nanostructure of the desired heterogeneous nanomaterial. When desired, the reduction step of the material was conducted by the following procedure: A quartz tube containing the material was introduced into a tubular furnace with a temperature controller, and a system to allow gas flow to enter and exit were connected. Once the system was set up, the furnace was programmed for reaching to the desired final temperature with a programmed heating ramp. All the heating processes were done under inert gas flow (N2 or Ar, 100 mL/min). When arrived at the maximum temperature, the gas was changed to H2 (100 mL/min), and the reductive treatment was maintained during the denoted time. Then, the furnace was switched off, and the catalyst was cooled to room temperature under inert gas flow (N2 or Ar). After each synthesis, the real metal content (wt%) was determined by ICP-AES. This result was used to named either monometallic [M/support] materials as well as bimetallic [M1M2x/support] materials, in which case was used to calculate the real molar relationship between both involved metals. Note that, due to rhenium tendency to form volatile species at high temperatures under aerobic conditions, the amount of metal deposited onto the solid matrix after temperature treatment step is always less than expected, hence, after preparation of the material by adding the theoretical amount of rhenium, the real wt% amount of rhenium deposited was determined by ICP-AES. When necessary, a slightly excess of rhenium precursor is added in order to reach approximately the desired wt% of the metal in the final material.
Study of the influence of the variation of metal M2
[AgM2x/Al2O3] bimetallic materials were prepared by adding 0.4 mmol of [Ag(acac)] and 0.8 mmol of M2 precursor, and then were calcined under air flow at 600 °C during 3 h with a heating rate of 2 °C/min. Used M2 precursor: [VO(acac)2], [MoO2(acac)2], [(NH4)6H2W12O40·xH2O], [NH4ReO4], [H3BO3], [Bu4Sn] and [Ga(NO3)3·xH2O]. For the preparation of monometallic material [Ag/Al2O3] (4.2 wt% Ag) (0.4 mmol [Ag(acac)] was employed (see Fig. 2a). After the preparation process, the real metal content was determined by ICP-AES (see Supplementary Table 5).
Study of the influence of the variation of metal M1
[M1Rex/Al2O3] bimetallic materials were prepared by adding 0.4 mmol of M1 precursor and 0.8 mmol of [NH4ReO4], and then were calcined under air flow at 600 °C during 3 h with a heating rate of 2 °C/min. Used M1 precursor: [Ag(acac)], [Pd(acac)2], [Pt(acac)2], [Ni(acac)2], [Fe(acac)3], [Ni(acac)2], [Co(acac)3], [Cu(acac)2] and [Mn(acac)3] (see Fig. 2a). After the preparation process, the real metal content was determined by ICP-AES (see Supplementary Table 6).
Study of the influence of the variation of support
[AgRex/support] bimetallic materials were prepared by adding 0.4 mmol of [Ag(acac)] and 0.8 mmol of [NH4ReO4], and then were calcined under airflow at 600 °C during 3 h with a heating rate of 2 °C/min. Used solid supports (for some of them a previous activation step was done): nanopowder Al2O3, nanopowder SiO2, ZrO2 (activated at 240 °C under air flow, 3 °C/min, 2 h), nanopowder ZnO, nanopowder CeO2 (activated at 500 °C under air flow, 5 °C/min, 2 h), nanopowder MgO (activated at 500 °C under air flow, 5 °C/min, 2 h), nanopowder TiO2, H-BETA (Si/Al = 12.5) (activated at 300 °C under vacuum, 3 h) and Nb2O5 (see Fig. 2a). After the preparation process, the real metal content was determined by ICP-AES (see Supplementary Table 7).
Study of the influence of the thermal treatment applied in the synthesis of [AgRe1.4/Al2O3] material
In this part of the optimization, in order to ensure the veracity of the experiments, all the thermal treatments were done with the same starting batch (5 ×1 g batch of [AgRe1.4/Al2O3]; i.e. 0.4 mmol Ag precursor and 0.8 mmol Re precursor/g Al2O3 by synthesis).
General treatment
Calcination of the impregnated material was conducted under air flow at 600 °C as described above. Additional pyrolysis and hydrogenation processes were also tested. The pyrolysis process was done in the same way as calcination but under N2 flow [600 °C, heating ramp of 2 °C/min, 3 h]. For the hydrogenative treatment under H2 flow (100 mL/min), the heating rate (2 °C/min) and the treatment time (3 h) were maintained, but in this case two different reduction temperatures (150 °C and 450 °C) were employed. Furthermore, diverse combinations of calcination or pyrolysis treatment (both at 600 °C) in addition to a hydrogenation step (at 150 °C or 450 °C) were also performed, being the general procedures identical (see Supplementary Table 2).
Calcination temperature
After determining that calcination is the best thermal treatment, different additional temperatures were tested (300 °C, 400 °C, 500 °C and 700 °C). In all the cases, the heating rate was 2 °C/min and the treatment time was established at 3 h (see Supplementary Table 2).
Heating rate
Finally, once established 500 °C as the best calcination temperature, several additional heating rates were investigated (1 °C/min, 5 °C/min and 10 °C/min). In all the cases, the treatment time at the maximum temperature (500 °C) was established at 3 h (see Supplementary Table 2).
Study of the influence of wt% metal loading in the [AgxRey/Al2O3] material
Two additional bimetallic materials were prepared maintaining a [Ag:Re] theoretical molar ratio [1:2], applying a calcination step under air flow at 500 °C during 3 h with a heating rate of 2 °C/min. A material [AgRe1.6/Al2O3], with a half metal content of both metals Ag and Re, was synthetized using 0.2 mmol of [Ag(acac)] and 0.4 mmol of [NH4ReO4] (2.4 wt% Ag and 5.9 wt% Re determined from ICP-AES) and a material [AgRe1.2/Al2O3], with a double metal content of both metals Ag and Re, was synthetized using 0.8 mmol of [Ag(acac)] and 1.2 mmol of [NH4ReO4] (6.2 wt% Ag and 13.0 wt% Re determined from ICP-AES). Both materials, were compared with the [AgRe1.4/Al2O3] bimetallic system (see Supplementary Table 3).
Study of metal cooperativity
[AgRe1.4/Al2O3] bimetallic material was prepared by adding 0.4 mmol of [Ag(acac)] and 0.8 mmol of [NH4ReO4] precursors. For the preparation of monometallic materials: [Ag/Al2O3] (4.2 wt% Ag) (0.4 mmol [Ag(acac)] and [Re/Al2O3] (7.5 wt% Re) (0.8 or 1.2 mmol [NH4ReO4]) were respectively used. Note that in the absence of silver, the preparation of rhenium monometallic material has a limit of wt% Re deposition in the support due to its tendency to sublimate as volatile species at high temperatures under aerobic conditions. All the catalysts were calcined under air flow at 500 °C during 3 h with a heating rate of 2 °C/min. Additionally, when stated, the monometallic Re catalyst was also ex situ reduced under H2 flow (100 mL/min) at 280 or 600 °C during 2 h, with a heating rate of 2 °C/min (see Fig. 2b).
Study of the influence of the molar ratio [Ag:Re] composition
Bimetallic materials were prepared, maintaining constant the Ag amount (0.4 mmol [Ag(acac)]/g Al2O3), while modifying Re precursor quantity (see below). For the preparation of monometallic materials: [Ag/Al2O3] (4.2 wt% Ag) (0.4 mmol [Ag(acac)] and [Re/Al2O3] (7.5 wt% Re) (0.8 or 1.2 mmol [NH4ReO4]) were respectively used. All the materials were calcined under air flow at 500 °C during 3 h with a heating rate of 2 °C/min. Additionally, when stated, monometallic Re material was ex-situ reduced under H2 flow (100 mL/min) at 280 °C during 2 h with a heating rate of 2 °C/min. After the preparation process, the real metal content determined by ICP-AES was used to calculate the real Ag:Re molar ratio to label the catalyst as [AgRex/Al2O3] due to the Re tendency to form volatile species at high temperatures under aerobic conditions (see Supplementary Table 8).
From this point, for performing [AgRex/Al2O3] materials preparation the initial weighted mmol of [NH4ReO4] for each material was: ([AgRe0.18], 0.1 mmol); ([AgRe0.35], 0.2 mmol); ([AgRe0.75], 0.4 mmol); ([AgRe], 0.6 mmol); ([AgRe1.4], 0.8 mmol); ([AgRe2], 3.2 mmol). For the preparation of the optimal [AgRe/Al2O3] material, [Ag(acac)] (0.4 mmol) and [NH4ReO4] (0.6 mmol) were dissolved in 50 mL of acetone and maintained under agitation 10 min. Then, Al2O3 (1 g) was added and the wet impregnation process was maintained during 4 h at room temperature. Then, the solvent was distilled under vacuum with a rotary evaporator, the solid was ground and calcinated in flow at 500 °C during 3 h with a heating ramp of 2 °C/min.
General and specific procedures for the catalytic studies
General procedure for catalytic studies of the hydrogenation of N-methylphthalimide (1) or 3-hydroxy-2-methylisoindolinone (2)
To a 8 mL vial with a stirring bar, N-methylphthalimide 1 (0.25 mmol, 40.3 mg) or 3-hydroxy-2-methylisoindolinone 2 (0.25 mmol, 40.8 mg), monometallic or bimetallic solid nanomaterial (adding the corresponding amount in function of the metal wt% measured by ICP-AES), n-dodecane (20 µL) as an internal standard, 4 Å MS (40–50 mg) and anhydrous CPME (1 mL) as solvent were added sequentially. Afterward, the reaction vial was closed with a screw cap with a septum, and up to seven reaction vials were placed in an aluminum carousel and introduced into a 300 mL autoclave. Then, each septum was perforated with a needle, and the autoclave was closed and pressurized with H2. After three purges with 20 bar of H2, the desired pressure was charged, and the autoclave was placed into an aluminum block preheated at the reaction temperature. Once the reaction was completed, the autoclave was cooled down with the help of an ice bath, carefully depressurized and opened. Finally, the reaction mixture was diluted with EtOAc, centrifuged and the supernatant was analyzed by GC.
General procedure for kinetic studies of the hydrogenation of N-methylphthalimide (1) or 3-hydroxy-2-methylisoindolionone (2)
To a 25 mL Teflon-covered stainless steel autoclave with a magnet with X shape, N-methylphthalimide 1 (0.75 mmol, 120.9 mg) or 3-hydroxy-2-methylisoindolinone 2 (0.75 mmol, 122.3 mg), the corresponding nanomaterial, n-dodecane (60 or 30 µL, respectively) as an internal standard, 4 Å MS (150 mg) and anhydrous CPME (3 mL) as solvent were added sequentially. The autoclave was closed and pressurized with H2. After three purges with 20 bar of H2, the desired pressure was charged, and the autoclave was placed into an aluminum block preheated at 90 °C. Periodically, aliquots of 50 µL were taken at different reaction times, diluted with EtOAc, centrifuged and the supernatant analyzed by GC. All the materials were calcined under air flow at 500 °C during 3 h using a heating ramp of 2 °C/min. Before each kinetic experiment, the materials were reduced with H2 flow as follows: Under N2 or Ar flow (100 mL/min), the catalysts were heated up to 150 °C (or 280 °C if indicated) with a ramp of 2 °C/min, once accomplished this temperature, the gas flow was changed to H2 during 2 h (100 mL/min). Finally, the catalysts were cooled under N2 or Ar flow (60 mL/min) overnight. Before kinetic studies, it was checked that identical results were obtained in both types of autoclaves (300 mL and 25 mL autoclave).
Procedure for filtration and leaching test of [AgRe/Al2O3] nanomaterial
The heterogeneous nature of the reaction was demonstrated by performing filtration and leaching tests. The filtration test was conducted as commented above for the realization of the kinetic study. Once the reaction started, three initial aliquots were taken at 5 min, 10 min, and 20 min to control the reaction. After this time, the autoclave was cooled with the help of an ice bath, carefully depressurized, and opened. The reaction mixture was transferred into a vial, centrifuged, and finally transferred into a new autoclave previously charged with 4 Å MS (150 mg) and a stirring bar. The autoclave was closed and pressurized with H2. After three purges with 20 bar of H2, the desired pressure was charged, and the autoclave was placed into an aluminum block preheated at 90 °C. Periodically, aliquots of 50 µL were taken at different reaction times, diluted with EtOAc, centrifuged and the supernatant analyzed by GC (see Supplementary Fig. 9). The same autoclave-opening procedure was done without catalyst removal, and the reaction continued with a similar yield.
A similar experiment was done in the 300 mL autoclave. Four different reactions were conducted as commented above for the catalytic studies in the hydrogenation reaction of N-methylphthalimide 1. After 3 h, the autoclave was cooled with the help of an ice bath, carefully depressurized, and opened. An aliquot of 50 µL was taken from each reaction, diluted with EtOAc, and studied by GC. The reaction mixture of two reactions was transferred into a vial, centrifuged, and decanted into a new vial charged with a stirring bar and 4 Å MS (45–50 mg). Then, the four reaction vials were placed in an aluminum carousel and introduced again into a 300 mL autoclave. After closing and pressuring the autoclave, the reaction started again and was conducted overnight. Finally, each reaction was diluted with EtOAc and studied by GC-MS; only the reactions in which the catalyst material was not removed promoted in the hydrogenation of 1.
Procedure for the recycling studies of [AgRe/Al2O3] system
Recycling experiments for the [AgRe/Al2O3] material in the hydrogenation of N-methylphthalimide 1 were carried out by scaling up the general reaction procedure mentioned above. A 25 mL vial containing a stirring bar was sequentially charged with N-methylphthalimide 1 (3.75 mmol, 603.0 mg), [AgRe/Al2O3] (6 mol% Ag), n-dodecane (300 µL) as an internal standard, 4 Å MS (750 mg) and anhydrous CPME (15 mL) were added sequentially. Afterwards, the reaction vial was closed with a screw cap performed with a needle and placed into the 300 mL autoclave. Then, the autoclave was closed and pressurized with H2. After three purges with 20 bar of H2, 50 bar of H2 were charged and the autoclave was placed into an aluminium block preheated at 100 °C. After 5 h, the autoclave was cooled down with the help of an ice bath, carefully depressurized, and opened. To quantify the reaction outcome, an aliquot of 50 µL of the reaction mixture was diluted with EtOAc, centrifuged and the supernatant was analysed by GC. The used catalyst was filtered under vacuum, washed with EtOAc (200 mL) and acetone (50 mL), and dried overnight in an oven at 100 °C. Finally, calcination of the material in air flow was performed at 300 °C during 3 h (using a heating ramp of 2 °C/min). At this point, the metal content of the material catalyst was determined by ICP-AES, dissolving a known quantity of it with aqua regia (HCl:HNO3, 3:1 in volume). The procedure was the same during the five catalytic cycles, recalculating the quantity of all the reagents as a function of the recovered catalyst (see Supplementary Fig. 10).
Data availability
Additional experimental procedures and optimization of reaction conditions, kinetic and mechanistic studies, compound and material characterization data, and NMR spectra of isolated compounds have been included in the Supplementary Information. Source data are provided with this paper.
References
Climent, M. J., Corma, A. & Iborra, S. Mono- and multisite solid catalysts in cascade reactions for chemical process intensification. ChemSusChem 2, 500–506 (2009).
Li, Z. et al. Well-defined materials for heterogeneous catalysis: From nanoparticles to isolated single-atom sites. Chem. Rev. 120, 623–682 (2020).
Zaera, F. Designing sites in heterogeneous catalysis: Are we reaching selectivities competitive with those of homogeneous catalysts? Chem. Rev. 122, 8594–8757 (2022).
Sheldon, R. A. E factors, green chemistry and catalysis: an odyssey. Chem. Commun. 3352–3365 (2008).
Sheldon, R. A. Fundamentals of green chemistry: efficiency in reaction design. Chem. Soc. Rev. 41, 1437–1451 (2012).
Dub, P. A. & Ikariya, T. Catalytic reductive transformations of carboxylic and carbonic acid derivatives using molecular hydrogen. ACS Catal. 2, 1718–1741 (2012).
Smith, A. M. & Whyman, R. Review of methods for the catalytic hydrogenation of carboxamides. Chem. Rev. 114, 5477–5510 (2014).
Pritchard, J., Filonenko, G. A., van Putten, R., Hensen, E. J. M. & Pidko, E. A. Heterogeneous and homogeneous catalysis for the hydrogenation of carboxylic acid derivatives: history, advances and future directions. Chem. Soc. Rev. 44, 3808–3833 (2015).
Cabrero-Antonino, J. R., Adam, R. & Beller, M. Catalytic reductive N-alkylations using CO2 and carboxylic acid derivatives: Recent progress and developments. Angew. Chem. Int. Ed. 58, 12820–12838 (2019).
Cabrero-Antonino, J. R., Adam, R., Papa, V. & Beller, M. Homogeneous and heterogeneous catalytic reduction of amides and related compounds using molecular hydrogen. Nat. Commun. 11, 3893 (2020).
Lluna-Galán, C., Izquierdo-Aranda, L., Adam, R. & Cabrero-Antonino, J. R. Catalytic reductive alcohol etherifications with carbonyl-based compounds or CO2 and related transformations for the synthesis of ether derivatives. ChemSusChem 14, 3744–3784 (2021).
Qu, R., Junge, K. & Beller, M. Hydrogenation of carboxylic acids, esters, and related compounds over heterogeneous catalysts: a step toward sustainable and carbon-neutral processes. Chem. Rev. 123, 1103–1165 (2023).
Kemnitz, C. R. & Loewen, M. J. “Amide resonance” correlates with a breadth of C−N rotation barriers. J. Am. Chem. Soc. 129, 2521–2528 (2007).
Deshpande, V. M., Patterson, W. R. & Narasimhan, C. S. Studies on ruthenium-tin boride catalysts I. Characterization. J. Catal. 121, 165–173 (1990).
Hirosawa, C., Wakasa, N. & Fuchikami, T. Hydrogenation of amides by the use of bimetallic catalysts consisting of group 8 to 10, and group 6 or 7 metals. Tetrahedron Lett. 37, 6749–6752 (1996).
Pouilloux, Y., Autin, F., Guimon, C. & Barrault, J. Hydrogenation of fatty esters over ruthenium–tin catalysts; characterization and identification of active centers. J. Catal. 176, 215–224 (1998).
Hidajat, M. J., Yun, G.-N. & Hwang, D.-W. Highly selective and stable ZnO-supported bimetallic RuSn catalyst for the hydrogenation of octanoic acid to octanol. Mol. Catal. 512, 111770 (2021).
Stein, M. & Breit, B. Catalytic hydrogenation of amides to amines under mild conditions. Angew. Chem. Int. Ed. 52, 2231–2234 (2013).
Mitsudome, T. et al. Mild hydrogenation of amides to amines over a platinum-vanadium bimetallic catalyst. Angew. Chem. Int. Ed. 56, 9381–9385 (2017).
Sakoda, K., Yamaguchi, S., Mitsudome, T. & Mizugaki, T. Selective hydrodeoxygenation of esters to unsymmetrical ethers over a zirconium oxide-supported Pt–Mo catalyst. JACS Au 2, 665–672 (2022).
Yuan, Y.-C., Bruneau, C. & Gramage-Doria, R. Merging transition-metal catalysis with phthalimides: A new entry to useful building blocks. Synthesis 50, 4216–4228 (2018).
Ertl, P., Altmann, E. & McKenna, J. M. The most common functional groups in bioactive molecules and how their popularity has evolved over time. J. Med. Chem. 63, 8408–8418 (2020).
Lai, C.-C. & Hsueh, P.-R. Coronavirus disease 2019 rebounds following nirmatrelvir/ritonavir treatment. J. Med. Virol. 95, e28430 (2023).
Byon, W., Garonzik, S., Boyd, R. A. & Frost, C. E. Apixaban: A clinical pharmacokinetic and pharmacodynamic review. Clin. Pharmacokinet. 58, 1265–1279 (2019).
Kotla, V. et al. Mechanism of action of lenalidomide in hematological malignancies. J. Hematol. Oncol. 2, 36 (2009).
Haaf, F., Sanner, A. & Straub, F. Polymers of N-vinylpyrrolidone: synthesis, characterization and uses. Polym. J. 17, 143–152 (1985).
Szostak, M. & Aubé, J. Chemistry of bridged lactams and related heterocycles. Chem. Rev. 113, 5701–5765 (2013).
Lawrence, N. J., Liddle, J., Bushell, S. M. & Jackson, D. A. A three-component coupling process based on vicarious nucleophilic substitution (VNSAR)−alkylation reactions: An approach to indoprofen and derivatives. J. Org. Chem. 67, 457–464 (2002).
Comins, D. L., Schilling, S. & Zhang, Y. Asymmetric synthesis of 3-substituted isoindolinones: application to the total synthesis of (+)-lennoxamine. Org. Lett. 7, 95–98 (2005).
Foster, R. W., Tame, C. J., Hailes, H. C. & Sheppard, T. D. Highly regioselective synthesis of substituted isoindolinones via ruthenium-catalyzed alkyne cyclotrimerizations. Adv. Synth. Catal. 355, 2353–2360 (2013).
Gao, W., Chen, M.-W., Ding, Q. & Peng, Y. Catalytic Asymmetric Synthesis of Isoindolinones. Chem. Asian J. 14, 1306–1322 (2019).
Kuninobu, Y., Tokunaga, Y., Kawata, A. & Takai, K. Insertion of polar and nonpolar unsaturated molecules into carbon−rhenium bonds generated by C−H bond activation: synthesis of phthalimidine and indene derivatives. J. Am. Chem. Soc. 128, 202–209 (2006).
Mancuso, R. et al. Divergent palladium iodide catalyzed multicomponent carbonylative approaches to functionalized isoindolinone and isobenzofuranimine derivatives. J. Org. Chem. 79, 3506–3518 (2014).
Norman, M. H., Minick, D. J. & Rigdon, G. C. Effect of linking bridge modifications on the antipsychotic profile of some phthalimide and isoindolinone derivatives. J. Med. Chem. 39, 149–157 (1996).
Arizpe, A., Sayago, F. J., Jiménez, A. I., Ordóñez, M. & Cativiela, C. Synthesis of phosphoproline derivatives with an octahydroisoindole structure. Eur. J. Org. Chem. 2011, 6732–6738 (2011).
Pierce, J. G., Waller, D. L. & Wipf, P. Synthesis of functionalized isoindolinones: Addition of in situ generated organoalanes to acyliminium ions. J. Organomet. Chem. 692, 4618–4629 (2007).
Xu, K., Zhang, S., Hu, Y., Zha, Z. & Wang, Z. Asymmetric michael reaction catalyzed by proline lithium salt: Efficient synthesis of L-proline and isoindoloisoquinolinone derivatives. Chem. Eur. J. 19, 3573–3578 (2013).
Das, S. et al. Selective catalytic monoreduction of phthalimides and imidazolidine-2,4-diones. Angew. Chem. Int. Ed. 50, 9180–9184 (2011).
Ding, G. et al. Potassium hydroxide-catalyzed chemoselective reduction of cyclic imides with hydrosilanes: synthesis of ω-hydroxylactams and lactams. Adv. Synth. Catal. 358, 1241–1250 (2016).
Bai, Y. et al. Electroselective and controlled reduction of cyclic imides to hydroxylactams and lactams. Org. Lett. 23, 2298–2302 (2021).
Hayashi, K., Griffin, J., Harper, K. C., Kawamata, Y. & Baran, P. S. Chemoselective (hetero)arene electroreduction enabled by rapid alternating polarity. J. Am. Chem. Soc. 144, 5762–5768 (2022).
Ito, M., Sakaguchi, A., Kobayashi, C. & Ikariya, T. Chemoselective hydrogenation of imides catalyzed by Cp*Ru(PN) complexes and its application to the asymmetric synthesis of paroxetine. J. Am. Chem. Soc. 129, 290–291 (2007).
Ito, M., Kobayashi, C., Himizu, A. & Ikariya, T. Highly enantioselective hydrogenative desymmetrization of bicyclic imides leading to multiply functionalized chiral cyclic compounds. J. Am. Chem. Soc. 132, 11414–11415 (2010).
Wu, C., Wang, J., Zhang, X., Zhang, R. & Ma, B. Highly chemoselective hydrogenation of cyclic imides to ω-hydroxylactams or ω-hydroxyamides catalyzed by iridium catalysts. Org. Chem. Front. 8, 6530–6534 (2021).
Kumar, A., Janes, T., Espinosa-Jalapa, N. A. & Milstein, D. Selective hydrogenation of cyclic imides to diols and amines and its application in the development of a liquid organic hydrogen carrier. J. Am. Chem. Soc. 140, 7453–7457 (2018).
Das, U. K., Janes, T., Kumar, A. & Milstein, D. Manganese catalyzed selective hydrogenation of cyclic imides to diols and amines. Green. Chem. 22, 3079–3082 (2020).
Liu, X. et al. Recyclable silver-catalyzed selective hydrogenation of imides to primary amines via dual C–N bond cleavage. Org. Lett. 25, 3066–3071 (2023).
McAlees, A. J. & McCrindle, R. Catalytic hydrogenations of cyclic imides and ayhydrides. J. Chem. Soc. C 2425–2435 https://doi.org/10.1039/J39690002425 (1969).
Takebayashi, S., John, J. M. & Bergens, S. H. Desymmetrization of meso-cyclic imides via enantioselective monohydrogenation. J. Am. Chem. Soc. 132, 12832–12834 (2010).
Cabrero-Antonino, J. R., Sorribes, I., Junge, K. & Beller, M. Selective ruthenium-catalyzed reductive alkoxylation and amination of cyclic imides. Angew. Chem. Int. Ed. 55, 387–391 (2016).
Cabrero-Antonino, J. R. et al. Unprecedented selective homogeneous cobalt-catalysed reductive alkoxylation of cyclic imides under mild conditions. Chem. Sci. 8, 5536–5546 (2017).
Maj, A. M., Suisse, I., Pinault, N., Robert, N. & Agbossou-Niedercorn, F. Efficient catalytic hydrogenation of N-unsubstituted cyclic imides to cyclic amines. ChemCatChem 6, 2621–2625 (2014).
McAlees, A. J., McCrindle, R. & Sneddon, D. W. Promotion of the catalytic hydrogenation of phthalimide and substituted phthalimides over palladium. J. Chem. Soc., Perkin Trans. 1, 2038–2040 (1977).
Aoun, R., Renaud, J.-L., Dixneuf, P. H. & Bruneau, C. Concomitant monoreduction and hydrogenation of unsaturated cyclic imides to lactams catalyzed by ruthenium compounds. Angew. Chem. Int. Ed. 44, 2021–2023 (2005).
Adkins, H. & Cramer, H. I. The use of nickel as a catalyst for hydrogenation. J. Am. Chem. Soc. 52, 4349–4358 (1930).
Paden, J. H. & Adkins, H. The synthesis of pyrrolidines, piperidines and hexahydroazepines1,2. J. Am. Chem. Soc. 58, 2487–2499 (1936).
Arévalo, A., Ovando-Segovia, S., Flores-Alamo, M. & Garcı́a, J. J. Selective C═O reduction in phthalimide with nickel(0) compounds. Organometallics 32, 2939–2943 (2013).
Patton, D. E. & Drago, R. S. Regenerable N-alkylamide hydroperoxide for catalytic substrate oxidation. J. Chem. Soc., Perkin Trans. 1, 1611–1615 (1993).
Haus, M. O. et al. Correlating the synthesis, structure, and catalytic performance of Pt–Re/TiO2 for the aqueous-phase hydrogenation of carboxylic acid derivatives. ACS Catal. 11, 5119–5134 (2021).
Ly, B. K. et al. Effect of addition mode of Re in bimetallic Pd–Re/TiO2 catalysts upon the selective aqueous-phase hydrogenation of succinic acid to 1,4-butanediol. Top. Catal. 55, 466–473 (2012).
Takeda, Y., Nakagawa, Y. & Tomishige, K. Selective hydrogenation of higher saturated carboxylic acids to alcohols using a ReOx–Pd/SiO2 catalyst. Cat. Sci. Technol. 2, 2221–2223 (2012).
Takeda, Y., Tamura, M., Nakagawa, Y., Okumura, K. & Tomishige, K. Characterization of Re–Pd/SiO2 catalysts for hydrogenation of stearic acid. ACS Catal. 5, 7034–7047 (2015).
Kammert, J. D. et al. Reduction of propionic acid over a Pd-promoted ReOx/SiO2 catalyst probed by X-ray absorption spectroscopy and transient kinetic analysis. ACS Sus. Chem. Eng. 6, 12353–12366 (2018).
Ullrich, J. & Breit, B. Selective hydrogenation of carboxylic acids to alcohols or alkanes employing a heterogeneous catalyst. ACS Catal. 8, 785–789 (2018).
Manyar, H. G. et al. Highly selective and efficient hydrogenation of carboxylic acids to alcohols using titania supported Pt catalysts. Chem. Commun. 46, 6279–6281 (2010).
Burch, R. et al. Catalytic hydrogenation of tertiary amides at low temperatures and pressures using bimetallic Pt/Re-based catalysts. J. Catal. 283, 89–97 (2011).
Suknev, A. et al. The nature of active sites in Pt–ReOX/TiO2 catalysts for selective hydrogenation of carboxylic acids to alcohols. J. Ener. Chem. 24, 646–654 (2015).
Liu, K. et al. Supported nickel–rhenium catalysts for selective hydrogenation of methyl esters to alcohols. Chem. Commun. 53, 9761–9764 (2017).
Keels, J. M. et al. Aqueous-phase hydrogenation of succinic acid using bimetallic Ir–Re/C catalysts prepared by strong electrostatic adsorption. ACS Catal. 8, 6486–6494 (2018).
Toyao, T. et al. Rhenium-loaded TiO2: A highly versatile and chemoselective catalyst for the hydrogenation of carboxylic acid derivatives and the N-methylation of amines using H2 and CO2. Chem. Eur. J. 23, 14848–14859 (2017).
Toyao, T. et al. TiO2-supported Re as a general and chemoselective heterogeneous catalyst for hydrogenation of carboxylic acids to alcohols. Chem. Eur. J. 23, 1001–1006 (2017).
Toyao, T. et al. Mechanistic study of the selective hydrogenation of carboxylic acid derivatives over supported rhenium catalysts. Cat. Sci. Technol. 9, 5413–5424 (2019).
Gothe, M. L. et al. Rhenium – a tuneable player in tailored hydrogenation catalysis. Eur. J. Inorg. Chem. 2021, 4043–4065 (2021).
Shimizu, K.-i, Miyamoto, Y. & Satsuma, A. Size- and support-dependent silver cluster catalysis for chemoselective hydrogenation of nitroaromatics. J. Catal. 270, 86–94 (2010).
Xie, Y., Hu, P., Bendikov, T. & Milstein, D. Heterogeneously catalyzed selective hydrogenation of amides to alcohols and amines. Cat. Sci. Technol. 8, 2784–2788 (2018).
Otto, J. W., Vassiliou, J. K., Porter, R. F. & Ruoff, A. L. Raman study of AgReO4 in the scheelite structure under pressure. Phys. Rev. B 44, 9223–9227 (1991).
Brütsch, L. & Feldmann, C. Synthesis and morphology of AgReO4 plates, rods, and stars. Z. anorg. allg. Chem. 643, 789–792 (2017).
Wang, J. et al. Friction behavior of silver perrhenate in oil as lubricating additive for use at elevated temperatures. Materials 12, 31288441 (2019).
Goudarzi, M., Mir, N., Mousavi-Kamazani, M., Bagheri, S. & Salavati-Niasari, M. Biosynthesis and characterization of silver nanoparticles prepared from two novel natural precursors by facile thermal decomposition methods. Sci. Rep. 6, 32539 (2016).
Ali, M. H. et al. Analysis of crystallographic structures and properties of silver nanoparticles synthesized using PKL extract and nanoscale characterization techniques. ACS Omega 8, 28133–28142 (2023).
Emeis, C. A. Determination of integrated molar extinction coefficients for infrared absorption bands of pyridine adsorbed on solid acid catalysts. J. Catal. 141, 347–354 (1993).
Parry, E. P. An infrared study of pyridine adsorbed on acidic solids. Characterization of surface acidity. J. Catal. 2, 371–379 (1963).
Thompson, S. T. & Lamb, H. H. Palladium–rhenium catalysts for selective hydrogenation of furfural: evidence for an optimum surface composition. ACS Catal. 6, 7438–7447 (2016).
Son, I. H., Kim, M. C., Koh, H. L. & Kim, K.-L. On the promotion of Ag/γ-Al2O3 by Cs for the SCR of NO by C3H6. Catal. Lett. 75, 191–197 (2001).
Wang, F. et al. Nanosize effect of Al2O3 in Ag/Al2O3 catalyst for the selective catalytic oxidation of ammonia. ACS Catal. 8, 2670–2682 (2018).
She, X. & Flytzani-Stephanopoulos, M. The role of AgOAl species in silver–alumina catalysts for the selective catalytic reduction of NOx with methane. J. Catal. 237, 79–93 (2006).
Lwin, S. et al. Surface ReOx sites on Al2O3 and their molecular structure–reactivity relationships for olefin metathesis. ACS Catal. 5, 1432–1444 (2015).
López-Hernández, I. et al. Evaluation of the silver species nature in Ag-ITQ2 zeolites by the CO oxidation reaction. Catal. Today 345, 22–26 (2020).
Zhang, B., Lwin, S., Xiang, S., Frenkel, A. I. & Wachs, I. E. Tuning the number of active sites and turnover frequencies by surface modification of supported ReO4/(SiO2–Al2O3) catalysts for olefin metathesis. ACS Catal. 11, 2412–2421 (2021).
Zhang, B. & Wachs, I. E. Identifying the catalytic active site for propylene metathesis by supported ReOx catalysts. ACS Catal. 11, 1962–1976 (2021).
Izquierdo-Aranda, L., Adam, R. & Cabrero-Antonino, J. R. Silver supported nanoparticles on [Mg4Al-LDH] as an efficient catalyst for the α-alkylation of nitriles, oxindoles and other carboxylic acid derivatives with alcohols. ChemSusChem 16, e202300818 (2023).
Yang, X. et al. γ-Al2O3 supported silver nanoparticle applied in C3H8-SCR: Nanosphere and nanoflake. Catal. Commun. 176, 106634 (2023).
Hardcastle, F. D., Wachs, I. E., Horsley, J. A. & Via, G. H. The structure of surface rhenium oxide on alumina from laser raman spectroscopy and x-ray absorption near-edge spectroscopy. J. Mol. Catal. 46, 15–36 (1988).
Vuurman, M. A., Stufkens, D. J., Oskam, A. & Wachs, I. E. Structural determination of surface rhenium oxide on various oxide supports (Al2O3, ZrO2, TiO2 and SiO2). J. Mol. Catal. 76, 263–285 (1992).
Okal, J., Kępiński, L., Krajczyk, L. & Tylus, W. Oxidation and redispersion of a low-loaded Re/γ-Al2O3 catalyst. J. Catal. 219, 362–371 (2003).
Greiner, M. T. et al. The oxidation of rhenium and identification of rhenium oxides during catalytic partial oxidation of ethylene: An in-situ XPS study. Z. Phys. Chem. 228, 521–541 (2014).
Weightman, P. & Andrews, P. T. The influence of the 4 d bandwidth on the M4,5N4,5N4,5 Auger spectra of Ag in MgAg and AlAg alloys. J. Phys. C: Solid State Phys. 13, 3529 (1980).
Hadjiivanov, K. I. & Vayssilov, G. N. in Adv. Catal. Vol. 47 307-511 (Academic Press, 2002).
Hadjiivanov, K. & Knözinger, H. Low-temperature CO adsorption on Ag+/SiO2 and Ag−ZSM-5: An FTIR study. J. Phys. Chem. B 102, 10936–10940 (1998).
Gravejat, P. et al. Heats of adsorption of linear and bridged CO species adsorbed on a 3% Ag/Al2O3 catalyst using in situ FTIR spectroscopy under adsorption equilibrium. J. Phys. Chem. C. 111, 9496–9503 (2007).
Anderson, J. A., Chong, F. K. & Rochester, C. H. IR study of CO adsorption on Pt, Re and Pt–Re/Al2O3 catalysts before and after coking. J. Mol. Catal. A: Chem. 140, 65–80 (1999).
Chong, F. K., Anderson, J. A. & Rochester, C. H. Effects of oxidation/reduction and oxychlorination/reduction cycles on CO adsorption by Pt–Re/Al2O3 catalysts. J. Catal. 190, 327–337 (2000).
Force, E. L. & Bell, A. T. Infrared spectra of adsorbed species present during the oxidation of ethylene over silver. J. Catal. 38, 440–460 (1975).
Yamamoto, M. et al. Photocatalytic reduction of CO2 with water promoted by Ag clusters in Ag/Ga2O3 photocatalysts. J. Mater. Chem. A 3, 16810–16816 (2015).
Srabionyan, V. V. et al. EXAFS study of size dependence of atomic structure in palladium nanoparticles. J. Phys. Chem. Solids 75, 470–476 (2014).
Padmos, J. D., Boudreau, R. T. M., Weaver, D. F. & Zhang, P. Impact of protecting ligands on surface structure and antibacterial activity of silver nanoparticles. Langmuir 31, 3745–3752 (2015).
Coeck, R. et al. Gold and silver-catalyzed reductive amination of aromatic carboxylic acids to benzylic amines. ACS Catal. 11, 7672–7684 (2021).
Hao, L. et al. Development of kilogram-scale synthesis of EGFR inhibitor EAI045. Org. Process Res. Dev. 23, 397–402 (2019).
Nishiyama, T. et al. Total synthesis of two 8-oxoprotoberberine alkaloids: alangiumkaloids A and B. Eur. J. Org. Chem. 2018, 673–678 (2018).
Shang, X.-F. et al. Biologically active isoquinoline alkaloids covering 2014–2018. Med. Res. Rev. 40, 2212–2289 (2020).
Jangir, R., Gadre, S. R. & Argade, N. P. Facile synthesis of the isoquinoline alkaloids doryanine and oxyhydrastinine. Synthesis 46, 1954–1956 (2014).
He, H., Alberti, K., Barr, T. L. & Klinowski, J. ESCA studies of aluminophosphate molecular sieves. J. Phys. Chem. 97, 13703–13707 (1993).
NIST, NIST X-ray Photoelectron Spectroscopy (XPS) Database, Version 3.5, (n.d.). https://srdata.nist.gov/xps/Default.aspx.
Simonelli, L. et al. CLÆSS: The hard X-ray absorption beamline of the ALBA CELLS synchrotron. Cogent Phys. 3, 1231987 (2016).
Acknowledgements
This work was supported by Generalitat Valenciana through SEJI (Subvencions Excelencia Juniors Investigadors, grants SEJI/2019/006 and SEJI/2020/013), PROMETEO/2021/077 and CIAICO/2021/2138 programs and a program from “La Caixa” Foundation (ID 100010434), fellowship code LCF/BQ/PI18/11630023. The work was also funded by Spanish Government via MICINN with RETOS I + D + I program (PID2019-109656RA-I0 / AEI / 10.13039/501100011033), grants CNS2023-144172, PID2020-113006-RB-I00, PID2021-1262350B-C31, TED2021-130191B-C44 and TED2021-130756B-C32 funded by MICIU/AEI/10.13039/501100011033 and European Union NextGenerationEU/PRTR. Financial support by the Spanish Ministry of Science and Innovation (CEX2021-001230-S grant funded by MCIN/AEI/10.13039/501100011033) is also gratefully acknowledged. This study forms part of the Advanced Materials program supported by MCIN with funding from European Union Next Generation EU (PRTR-C17.11) and Generalitat Valenciana (grant MFA/2022/016). R. A. and J. R. C.-A. thank to MICINN (Spanish Government) for two Ramón y Cajal contracts (ref. RYC2020-029493-I and RYC-2017-22717). XAS experiments were performed at BL22-CLÆSS beamline at ALBA Synchrotron with the collaboration of ALBA staff (proposal 2022075907). ELECMI-ICTS facilities (proposal ELC402023) are also acknowledged for electron microscopy experiments.
Author information
Authors and Affiliations
Contributions
C.L.-G. performed all the work related to materials preparation and characterization, conducted the catalytic experiments, the isolation of lactam compounds and the interpretation of data results, and also collaborated in the writing of the manuscript. J. C. A.-D. developed the synthesis and isolation of the imide starting materials. D. G. and P. C. performed the IR-CO and XPS experiments and interpreted the results. R. S. and J. J. C. performed the HR-STEM measurements and STEM-XEDS analysis, and performed its interpretation. L. S. assisted in XAS Synchrotron ALBA experiments and helped in the interpretation of the data obtained. R. A. and J. R. C.-A. designed and supervised the project, interpreted all the data obtained and wrote the manuscript. All the authors revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Konstantin Khivantsev and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lluna-Galán, C., Arango-Daza, J.C., Gómez, D. et al. Building lactams by highly selective hydrodeoxygenation of cyclic imides using an alumina-supported AgRe bimetallic nanocatalyst. Nat Commun 16, 4119 (2025). https://doi.org/10.1038/s41467-025-59514-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59514-7
