
Abstract
Errors during translation can cause ribosome stalling, leaving incomplete nascent chains attached to large ribosomal subunits. Cells rely on the Ribosome-associated Quality Control (RQC) complex to recognize, process, and remove these aberrant proteins to maintain proteostasis. Despite its importance, the mechanisms by which the RQC orchestrates nascent chain processing and extraction have remained unclear. Here, we present a cryo-EM structure of the RQC complex from budding yeast, revealing how its core components function in nascent chain removal. We show that the Cdc48 ATPase and its Ufd1-Npl4 adaptor are recruited by the Ltn1 E3 ubiquitin ligase to extract ubiquitylated peptides from the 60S ribosome. Additionally, we find that Rqc1 bridges the 60S subunit with ubiquitin and Ltn1, facilitating formation of K48-linked polyubiquitin chains. These findings provide a structural and mechanistic framework for understanding how the RQC complex collaborates to clear stalled translation products, advancing insight into cellular protein quality control.
Similar content being viewed by others


Introduction
All cells depend on accurate protein synthesis to maintain proteostasis. From bacteria to humans, errors during translation can cause ribosome stalling, leading to the production of incomplete, potentially proteotoxic nascent chains and dissociation of the large and small ribosomal subunits1,2,3,4,5. The large subunit retains the aberrant tRNA-conjugated nascent chain, which is recognized, modified, and removed by the Ribosome-associated Quality Control (RQC) complex for its subsequent degradation. Mutations in RQC components are linked to neurological diseases, underscoring the importance of this pathway in maintaining protein homeostasis6,7,8,9.
The eukaryotic RQC complex comprises several conserved factors that work together to detect and resolve stalled translation events. Central to this process is Rqc2, which recognizes aberrant 60S subunits bearing peptidyl-tRNA, initiating RQC complex assembly. Rqc2 also mediates non-templated elongation of nascent polypeptides by recruiting alanine- and threonine-charged tRNAs to the A site of the ribosome, resulting in carboxy-terminal alanine and threonine (CAT) tails10,11,12,13,14,15,16,17. Beyond CAT tail synthesis, Rqc2 serves as a scaffold to recruit other RQC components, including the ubiquitin ligase Ltn1.
Ltn1 is an E3 ligase that ubiquitylates the nascent chain via its C-terminal RING ___domain, typically assembling K48-linked polyubiquitin chains that mark the substrate for degradation18,19. Ltn1’s ligase activity is spatially constrained, operating on exposed lysines within the exit tunnel-proximal portion of the nascent chain. This ubiquitylation is significantly enhanced by the presence of Rqc1, which contributes to K48 chain formation and is required to recruit the downstream Cdc48 complex20,21,22. Although the precise molecular role of Rqc1 remains unclear, evidence suggests that it both regulates Ltn1 activity and acts as a necessary adapter for engaging Cdc4822,23,24.
The Cdc48-Ufd1-Npl4 complex is a highly conserved protein segregase that plays central roles in numerous quality control pathways, including endoplasmic reticulum-associated degradation (ERAD), chromatin-associated degradation, and ribosome-associated quality control25. Cdc48 (p97/VCP in mammals) is an ATP-driven AAA+ ATPase that extracts polyubiquitylated substrates from macromolecular assemblies or membranes, preparing them for proteasomal degradation. In the context of the RQC, the heterodimeric Ufd1-Npl4 complex functions as a ubiquitin-binding adaptor that recognizes K48-linked polyubiquitin chains on stalled nascent chains and recruits them to Cdc48.
Despite extensive genetic, biochemical, and structural studies, critical mechanistic questions about the RQC pathway remain unresolved. Chief among these are how the eponymous Rqc1 component facilitates the recruitment of Cdc48 to stalled 60S subunits, how Ltn1 assembles degradation-competent K48-linked polyubiquitin chains in the context of the ribosome, and how Cdc48 engages and extracts the nascent chain from the ribosomal exit tunnel. In this work, we address these questions by determining cryo-EM structures of the fully assembled RQC complex. Combined with AlphaFold modeling, these structures provide key insights into the organization and interactions of RQC components and provide a structural framework for understanding how stalled nascent chains are marked and extracted for proteasomal degradation.
Results
Architecture of the eukaryotic RQC complex
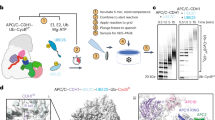
RQC particles were isolated from budding yeast lysates through a rapid one-step affinity purification, as performed previously10,22,26. Cells expressed a C-terminal 3xFLAG tag at the endogenous locus of Rqc1, and the complex was purified by co-immunoprecipitation with anti-FLAG affinity resin (Supplementary Fig. 1). To enrich for Cdc48-bound particles, purifications were supplemented with the non-hydrolyzable ATP analog ADP-BeFx, which traps Cdc48 in its active, substrate-engaged conformation27.
Cryo-EM image processing revealed a subset of particles containing known and novel features associated with 60S particles (Fig. 1, Supplementary Fig. 1, 2, Supplementary Movie 1). The reconstruction is consistent with previous studies that show Rqc2 binding to P- and A-site tRNAs at the exposed 60S-40S interface10,11. Ltn1 contacts Rqc2 at the sarcin-ricin loop and extends around the 60S ribosome to position its catalytic RING ___domain in the vicinity of the stalled nascent chain. Nascent chain density is observed within the exit tunnel and remains connected to the Rqc2-bound P-site tRNA. In addition to these previously established features of the complex, we resolved uncharacterized densities outside of the nascent chain exit tunnel through local refinement. The resulting maps enabled us to place models of Cdc48-Ufd1-Npl4 and Rqc1 in relation to the nascent chain and revealed the complete architecture of the RQC complex (Supplementary Movie 1).

a Composite map of the RQC complex segmented according to their individual components. b Models of RQC components fitted into reconstruction density.
Cdc48 interacts with Ltn1 on stalled ribosomes
The Cdc48 AAA+ ATPase is an abundant and essential enzyme, known for its role as a segregase across multiple cellular contexts. Cdc48 subunits comprise an N-terminal ___domain (Cdc48NTD), followed by tandem AAA+ motor domains, D1 and D2, which drive substrate unfolding. Structures of Cdc48 and its human ortholog p97/VCP show that substrates are unfolded by threading them through homo-hexameric assemblies of stacked D1 and D2 rings27,28,29,30. Cdc48 recruitment to specific subcellular localizations and substrates is regulated by its interactions with adaptor proteins, most notably the Ufd1-Npl4 heterodimer. Ufd1-Npl4 binds to K48-linked polyubiquitin chains and feeds substrates into the Cdc48 hexameric central pore to facilitate unfolding31,32. Despite their flexible association, our composite reconstruction places the entire Cdc48-Ufd1-Npl4 complex outside of the exit tunnel (Figs. 1, 2). Focused particle sub-extraction and 2D classification over the density revealed characteristic features of the complex, including the stacked D1 and D2 ATPase rings and a tower-like density belonging to Ufd1-Npl4, which protrudes from the center of the particle28,33 (Fig. 2a and Supplementary Fig. 2). The peripheral Cdc48NTD domains are positioned in the elevated, ‘up’ positions, which are associated with its active state34. Ufd1-Npl4 is positioned near the peptide exit tunnel and appears poised to transfer the ubiquitylated nascent chain from the 60S ribosome to Cdc48 (Fig. 1).

a Positioning of Cdc48-Ufd1-Npl4 relative to the 60S ribosome. Focused view on bottom shows a 2D class average of sub-extracted Cdc48 complexes from RQC particles. b Close up view of the Ltn1-Cdc48 interface at the Ltn1 elbow. Key interaction residues are labeled. Residues mutated to generate the Ltn1AAA mutant are indicated with a red outline. c Silver stained SDS-PAGE of RQC co-IPs expressing WT or Ltn1AAA. d Anti-Cdc48 immunoblot from RQC co-IPs. Anti-FLAG (Rqc1) blot shown as loading control. e Immunoblot of whole cell lysates expressing nonstop substrate (Protein A-nonstop) in Ltn1∆ cells and re-expressed with either wild-type Ltn1 or Ltn1AAA mutant. Anti-Pgk1 blot shown as loading control. In (c–e), each experiment was repeated at least three times as independent biological replicates.
The positioning of Cdc48-Ufd1-Npl4 relative to the 60S is facilitated through direct interactions between Cdc48NTD and Ltn1. The interaction interface is located at the elbow of Ltn1, where its extended structure makes a sharp bend to position its catalytic RING ___domain in the vicinity of the emerging nascent chain. Focused 3D refinement over Ltn1 revealed sufficiently resolved density to place an AlphaFold3 model of the Ltn1-Cdc48NTD interface as a rigid body (Fig. 2b and Supplementary Fig. 3a, b). Residues within four α-helices of Ltn1 form a network of interactions with the N-terminal double φ-barrel and a C-terminal β-barrel sub-domains of Cdc48NTD. Based on the predicted interactions, we performed mutagenesis experiments to test the relevance of the model’s contacting residues. Ltn1 residues R945, Q1022, and R1023 were mutated to alanines on an expression plasmid (R945A, Q1022A, & R1023A, Ltn1AAA) and transformed into Ltn1∆ cells. RQC co-IPs showed a significant reduction of Cdc48 recovery from cells expressing Ltn1AAA compared to wild-type control, while other components of the RQC complex were recovered at similar levels (Fig. 2c, d). To test the functional relevance of the Cdc48-Ltn1 interaction, cells were expressed with a non-stop mRNA substrate and probed for substrate expression35. As expected, Ltn1∆ cells displayed an accumulation of non-stop substrate, and the degradation phenotype was rescued by re-expression of wild-type Ltn1 on a plasmid (Fig. 2e). In contrast, expression of Ltn1AAA in Ltn1∆ cells led to an accumulation of stalled substrate at a milder extent compared to Ltn1∆. Together, these experiments demonstrate that the Ltn1 elbow evolved to recruit Cdc48 to the dissociated 60S particles, and the loss of Cdc48 recruitment partially disrupts substrate degradation.
The direct interaction between Cdc48NTD and Ltn1 establishes that Ltn1 is a Cdc48 adaptor. These adaptors typically contain conserved domains or motifs that define their interaction sites with Cdc48. The Ltn1 helical array interacts with Cdc48NTD in a way that is reminiscent of the VIM and VBM motifs, where an α-helix sits in the groove that separates the φ-barrel and β-barrel subdomains36 (Supplementary Fig. 3c). However, unlike VIM and VBM motifs, which utilize a single α-helix for this interaction, Ltn1 does not share sequence similarity with these motifs and instead uses multiple helices to form a more extensive interface. Several E3 ligases are known to interact directly with the human ortholog of Cdc48 (p97/VCP), including HOIP37, gp7838, and HRD139. These interactions likely drive Cdc48 localization to target 60S particles and couple substrate ubiquitylation directly to their unfolding.
Rqc1 interacts with Ltn1 and ubiquitin
Rqc1 is the least characterized component of the RQC complex despite it being the source of co-IP experiments used for structure determination in previous studies10,11. Previous RQC structures lack Rqc1, presumably due to its dissociation from the particle during cryo-EM specimen preparation. Our cryo-EM specimens prepared using graphene oxide grids likely shielded particles from the hydrophobic air-water interface and preserved Rqc1 on 60S particles. Cryo-EM reconstructions revealed uncharacterized, low local resolution densities ascribed to Rqc1 positioned near the opening of the nascent chain exit tunnel. The density is connected to ES27 of the 25S rRNA (nucleotides 1956-2092), Rpl38, and the Ltn1 RING ___domain (residues 1507-1562, Ltn1RING) (Fig. 3 and Supplementary Fig. 4a–e). Displaying the reconstruction with a low-pass Gaussian filter provided sufficient quality to fit the density associated with Ltn1RING with the AlphaFold predicted structure of the Rqc1 C-terminal ___domain (CTD) as a rigid body (residues 176-682, Rqc1CTD) (Fig. 3a–c and Supplementary Movie 2). By contrast, the other densities ascribed to Rqc1 displayed poorer local resolution likely due to disorder. In support of this, the first 175 residues of Rqc1 (Rqc1NTD) are predicted to be mostly disordered, although we identify structured density corresponding to an internal helix (residues 122-135) that interacts with the 60S protein Rpl38 (Fig. 3a, Supplementary Fig. 4c-e). Despite the predicted disorder, the Rqc1NTD is enriched with polar and basic residues that would be favorable to interact with rRNA and is consistent with a cloud of unstructured density associated with ES27 following the internal helix (Supplementary Fig. 4e).

a Domain organization of Rqc1 delineating its NTD and CTD. Alpha-helical residue ranges involved with its interactions with Ltn1 and ubiquitin are indicated. b Close up view of Rqc1CTD in complex with ubiquitin and Ltn1RING. Gaussian-filtered map of the Rqc1-Ltn1-ubiquitin portion shown for clarity. c, d Focused views of Rqc1 interactions with Ltn1RING and ubiquitin, respectively. Key interaction residues are labeled. Residues mutated to generated Rqc1Ltn1 or Rqc1Ub are indicated with a red outline. e Immunoblot of RQC co-IPs from wild-type or Rqc1 mutant (Rqc1Ltn1 or Rqc1Ub) backgrounds probed against total ubiquitin. Anti-FLAG (Rqc1) blot shown as loading control. f Same as in (d), but probed with anti-K48-linked poly-ubiquitin antibody. g Silver stained SDS-PAGE of RQC co-IPs reveal a loss of Cdc48 recovery by Rqc1Ltn1 and Rqc1Ub mutants. h Anti-Cdc48 immunoblot from Rqc1 co-IPs. Anti-FLAG (Rqc1) blot shown as loading control. i Immunoblot of whole cell lysate against nonstop substrate (anti-Protein A) in Rqc1∆ cells expressing Rqc1Ltn1 or Rqc1Ub mutants compared to wild-type control. Anti-Pgk1 blot shown as loading control. In (e–i), each experiment was repeated at least three times as independent biological replicates.
The structure of Rqc1 suggests a bipartite mode of interaction such that the internal helix spatially separates the Rqc1NTD and Rqc1CTD to facilitate interactions with the 60S ribosome and Ltn1RING, respectively. To test this model, we expressed a construct of Rqc1 that lacks its NTD, i.e. by deletion of residues 1-175. Expression and co-IP of this construct in Rqc1 single deletion strain (Rqc1∆) failed to recover the 60S or other RQC components, thereby indicating that the Rqc1CTD alone is insufficient for ribosome binding and RQC function (Supplementary Fig. 4f). Interestingly, ES27 and Rpl38 are also accessible on intact 80S ribosomes and would suggest that Rqc1 is capable of binding 80S particles as suggested in a previous study20. However, the specificity for 60S particles is augmented by the interaction between Rqc1 and Ltn1RING on dissociated ribosomes.
Additional low-threshold reconstruction densities were observed connected with Rqc1CTD. These densities displayed poorer local resolution than the Rqc1CTD, suggestive of structural heterogeneity. Considering the role of Rqc1 in facilitating the formation of K48-linked polyubiquitin chains, we explored the possibility that these additional densities are ascribable to ubiquitin and Ltn1RING. AlphaFold 3 was used to generate a predicted complex of Rqc1CTD in complex with ubiquitin and this model was fit well into the corresponding reconstruction density as a rigid body (Fig. 3b, d, Supplementary Fig. 4c and Supplementary Movie 2). A similar exercise was performed to predict the interaction between the Ltn1RING and Rqc1CTD, and the resulting model showed good agreement with the observed density (Fig. 3b, d).
The predicted modeling suggested that residues F214 and D215 of Rqc1 are crucial for its interaction with Ltn1RING (Fig. 3c). We therefore substituted F214 and D215 with alanine to test how disruption of this interface would affect RQC function (F214A & D215A, Rqc1Ltn1). Immunoblots showed that total ubiquitylation within purified RQC complexes was partially reduced in Rqc1Ltn1 co-IPs compared to wild-type control (Fig. 3e). Strikingly, K48-linked polyubiquitylation was virtually undetectable in the mutant (Fig. 3f). The loss of K48-linked chains likely explains why Rqc1 deletions abrogate Cdc48 recruitment to RQC complexes, as previously reported20,22. Consistent with this, Cdc48 recovery was greatly reduced in Rqc1Ltn1 compared to WT control (Fig. 3g, h).
Similar experiments were performed to test the functional significance of the interaction between Rqc1 and ubiquitin. The predicted structure suggested that E637 and E657 of Rqc1 are key residues for this interaction (Fig. 3d). Based on this, cells were expressed with Rqc1 mutants substituting E637 and E657 with alanines (E637A & E657A, Rqc1Ub) in an Rqc1∆ background and used to test whether disrupting this interaction would impair the formation of K48 poly-ubiquitin linkages. Like RqcLtn1, co-IP of Rqc1Ub showed that the recovery of ubiquitin was diminished compared to wild-type control and that K48-linked chains were not detectable (Fig. 3e, f). As expected, Cdc48 recovery in the Rqc1Ub mutant was also greatly reduced compared to WT control (Fig. 3g, h). The loss of K48-linked polyubiquitin and Cdc48 recruitment to RQC in both RqcLtn1 and Rqc1Ub mutants prompted us to test their effects on stalled substrate degradation. As expected, the expression of a nonstop mRNA in these cells led to an accumulation of stalled substrate (Fig. 3i). In essence, Rqc1Ub mutations phenocopy Rqc1Ltn1, thus indicating that the interactions among Rqc1, Ltn1, and ubiquitin are functionally linked.
The interaction between Rqc1CTD and Ltn1RING appears to be conserved from yeast to human. A recent study characterized how the human Rqc1 ortholog, TCF25, imposes K48 linkage specificity on Listerin-mediated ubiquitination40. Sequence and predicted structural alignment of Ltn1RING and Rqc1 suggest that the interaction interface is conserved across species. Specifically, the predicted complexes display moderate-confidence interface scores via AlphaFold 3 and a notable structural overlay of key interacting residues, including Rqc1-F214:Ltn1-F1543 (yeast) and TCF25-F195:Listerin-F1750 (human) (Supplementary Fig. 5).
The ubiquitin molecule associated with Rqc1 is likely appended to the nascent chain, although we do not observe continuous density of the nascent chain outside of the ribosome. Parts of the Rqc1CTD are positioned within 10 Å of the exit tunnel opening and would overlap with the binding of other ribosome-associated factors that are known to engage nascent chains, such as the nascent polypeptide-associated complex (NAC) and signal recognition particle (SRP)41 (Supplementary Fig. 6). The binding of Rqc1 may prevent the association of these factors from interacting with stalled nascent chains.
Rqc1 facilitates K48-linked polyubiquitylation by Ltn1
Rqc1 facilitates the formation of K48-linked ubiquitin chains by Ltn1 through an unknown mechanism21. The interactions among Rqc1CTD with Ltn1RING and ubiquitin rationalize how this occurs. RING ___domain E3 ligases directly transfer ubiquitin between the ubiquitin-conjugated E2 (E2-Ub, donors) and their substrates (acceptors). The specificity of polyubiquitin linkages requires the proper positioning of the acceptor ubiquitin to the active site of E2-Ub42,43. This led us to hypothesize that Rqc1 functions to position the acceptor ubiquitin in a specific orientation that favors K48-linkages with the donor ubiquitin from E2-Ub. We therefore extended our analysis by using AlphaFold3 to generate a predicted structure containing of Rqc1CTD, Ltn1RING, and the acceptor ubiquitin in complex with E2-Ub (Rad6-ubiquitin). This produced a high-confidence prediction of the complex, and the fitting of the complex into our experimental density containing Rqc1CTD, Ltn1RING, and acceptor ubiquitin showed that the E2 enzyme and donor ubiquitin were positioned in a solvent-accessible region free of clashes (Fig. 4a and Supplementary Fig. 7). This modeling indicates that the Rqc1-bound acceptor ubiquitin is oriented such that its K48 residue becomes the only accessible lysine proximal to the active site cysteine of the E2 and the C-terminal glycine of the donor ubiquitin (Fig. 4b). The model also agrees with other experimentally determined structures of other RING domains bound to E2-Ub complexes (Supplementary Fig. 7b). Together, these results suggest that Rqc1 holds the acceptor ubiquitin in a defined position to facilitate the specific assembly of K48-linked polyubiquitin chains on stalled nascent chains. As the nascent chain becomes elongated by CAT tails, the ubiquitin chain is presumably pushed toward Cdc48-Ufd1-Npl4, where Npl4 would be in position to unfold the initiator ubiquitin and feed it into the Cdc48 central pore (Fig. 4c). Consistent with this, the distance separating the acceptor ubiquitin on Rqc1 and Npl4 is approximately 50 Å, and it is conceivable that the extension of polyubiquitin chains at ~25 Å per ubiquitin is sufficient to bridge its gap with Cdc48.

a Model of the RQC complex including the predicted model of E2 (Rad6) and its accompanying donor ubiquitin. b Close up view of Rqc1CTD, acceptor ubiquitin, and Ltn1RING fitted in cryo-EM reconstruction density with the predicted orientation of E2 bound to donor ubiquitin. Key interaction residues between E2, donor ubiquitin (G76), and acceptor ubiquitin (K48) are labeled. The positions of other lysines on the acceptor ubiquitin are also labeled for spatial context. K11 and K27 are also inaccessible for ubiquitylation but are not labeled due to obscured views. Reconstruction density is low-pass filtered for clarity. c Schematic of nascent chain extension, ubiquitylation, and extraction by the RQC complex.
Discussion
Our study provides a comprehensive structure of the RQC complex containing all major components, including Ltn1, Rqc2, Rqc1, and the Cdc48-Ufd1-Npl4 complex. The structure establishes the mechanistic basis for Cdc48 recruitment to RQC substrates via a direct interaction with Ltn1, and this interaction couples substrate ubiquitylation with their extraction from 60S ribosomes. Additionally, Rqc1 plays a critical role in positioning Ltn1RING and ubiquitin to facilitate the formation of K48-linked polyubiquitin chains. The addition of CAT tails by Rqc2 is likely required to extend specific lysine residues to reach the RING ___domain of Ltn113,14. In yeast, this CAT-tailing process is terminated by the peptidyl-tRNA nuclease Vms144,45,46. Given that Vms1 interacts directly with Cdc4847, it remains to be determined whether this interaction plays a functional role in the RQC pathway. It is also curious why Cdc48-Ufd1-Npl4 is needed to remove the nascent chain, i.e. why the nascent chain does not simply diffuse out of the ribosome following peptidyl-tRNA cleavage. In addition to extracting substrates for proteasomal degradation, the pulling action of Cdc48 may help position the nascent chain for more efficient Vms1-mediated peptidyl-tRNA hydrolysis. Other possibilities are that Vms1-mediated cleavage leaves unusual CCA trinucleotide signatures on the nascent chain that may not be readily diffusible through the 60S exit tunnel48, or that the pulling force from Cdc48 alters the composition of CAT tails to favor nascent chain release49.
Methods
Cell growth & lysis
Yeast cells (S. cerevisiae strain BY4741) were grown in either rich media or synthetic dropout media and harvested at OD600 of ~1.2. The cells were pelleted at 4500 x g for 6 min, washed in ice cold water, pelleted again at 4000 x g for 6 min, then resuspended in lysis buffer (50 mM HEPES-KOH pH 6.8, 150 mM KOAc, 2 mM Mg(OAc)2, 1 mM CaCl2, 0.2 M sorbitol, and supplemented with cOmplete protease inhibitor cocktail (Roche)). Cell droplets were frozen in liquid nitrogen, lysed into powder using a Freezer/Mill® (SPEX SamplePrep), and stored at −80 °C until use. A list of yeast strains used in this study is provided in Supplementary Table 1.
Plasmid construction
To generate the wild-type RQC1 expression construct (pRQC1_WT), the RQC1 gene including its endogenous promoter, a C-terminal Flag tag, and the kanMX selection marker was PCR-amplified from a previously constructed yeast strain10. All RQC1-related plasmids used in this study were derived from pRQC1_WT by introducing site-specific mutations using the megaprimer PCR mutagenesis method, as described in Liu and Naismith50 and Tseng et al.51. Briefly, primers containing regions homologous to the target site in the pRS315 backbone were used to amplify the entire plasmid. For RQC1 mutants, the corresponding fragments containing the desired mutations were first generated by PCR and subsequently used as megaprimers to amplify the full plasmid using pRQC1_WT as the template. Plasmids pLTN1_WT and pLTN1_Ltn1AAA were constructed using the same approach. A list of all primers used in this study is provided in Table S1.
RQC immunoprecipitation
RQC particles were purified from cells expressing a C-terminal 3xFLAG tag at the endogenous locus of Rqc1, as previously described10,22,26. Lysed yeast powder was thawed on ice and resuspended in immunoprecipitation buffer (95 mM KCl, 5 mM NH4Cl, 50 mM HEPES (pH 7.5), 1 mM DTT, 15 mM Mg(OAc)2, 0.5 mM CaCl2, supplemented with cOmplete protease inhibitors (Roche)). Supernatant was clarified by centrifugation and incubated with anti-FLAG M2 agarose resin (Sigma) overnight at 4 °C. The resin was washed thoroughly with buffer containing ADP-BeFx, and the RQC particles were eluted with excess 3xFLAG peptide for 30 min at 4 °C. Purified samples were analyzed by silver stain SDS-PAGE and negative stain electron microscopy and used for cryo-EM experiments and immunoblot analyses.
Electron microscopy
4 μl of purified RQC particles were cross-linked using 0.15% glutaraldehyde and applied to either Quantifoil R2/2 or R1.2/1.3 grids (SPT Labtech) deposited with a fresh homemade graphene oxide layer, as previously described52. Grids were plunge frozen in liquid ethane using a Mk. II Vitrobot (Thermo Fisher Scientific) with a wait time of 25 s and −1 mm offset. Datasets were collected across six separate sessions on a Titan Krios G3 (Thermo Fisher Scientific) operating at 300 kV, equipped with a post-GIF K3 direct detector (Gatan, Inc.). Settings used for data collection are summarized in Supplementary Table 2. Images from five sessions were recorded using SerialEM software53 at a nominal magnification of 81,000X in counting mode, corresponding to a pixel size of 1.058 Å with a total dose between 35-37 electrons/Ų, and 40 frames per movie. The sixth session was recorded in super-resolution mode, corresponding to a pixel size of 0.529 Å with a total dose of approximately 50 electrons/Å2, and 56 frames per movie. Super-resolution movies were 2x Fourier binned for downstream processing. All movies were set in SerialEM to be recorded between −0.8 to −1.5 μm underfocus with a 3 × 3 multishot array and either 2 exposures per hole (R1.2/1.3 grids) or 4 exposures per hole (R2/2 grids).
Image processing and 3D reconstruction
Six datasets were collected and processed. Four datasets were merged into a single group containing 28,302 micrographs, while the remaining two datasets were merged into another group containing 12,667 micrographs. The two groups were processed separately. After data collection, movies were subjected to patch motion correction and patch CTF estimation using cryoSPARC54.
For the first merged dataset, 28,302 micrographs were curated by filtering for exposures with an average defocus between −0.5 and −2.7 μm, CTF fit resolution better than 13.7 Å, and average intensity values ranging from −543 to 346.4. Defocus tilt angles between −9.3 and 28.5 were also included. After manually removing low-quality micrographs, 26,672 micrographs were retained. A total of 1,416,254 particles were initially picked from 5000 micrographs using a blob picker with a particle diameter constraint to 200–360 Å. Particles were extracted with a box size of 640 pixels and Fourier-cropped to 128 pixels, then subjected to several rounds of 2D classification. High-quality 2D classes were subsequently used as templates for particle picking. After template-based particle picking and manual inspection, 8,338,295 raw particles were extracted with a box size of 640 pixels and Fourier-cropped to 128 pixels for further rounds of 2D classification to exclude non-particles (e.g., non-protein images, ice, and other artifacts). After 2D classification, 699,664 particles were retained and used for ab-initio reconstruction followed by heterogeneous refinement with five classes. One of these classes, consisting of 239,021 particles, contained recognizable RQC features (e.g., Ltn1, Rqc2, and Cdc48) and was subjected to additional 3D classification and heterogeneous refinement. A total of 170,995 particles from this dataset were retained for combination with particles from the second dataset.
For the second merged dataset, micrographs were curated with CTF fit resolution better than 9.7 Å and average intensity values between −399.6 and 191.5, resulting in 12,187 curated micrographs. Template picking was performed using templates derived from the first dataset, resulting in the selection of 1,879,959 particles after manual inspection. These particles were extracted with a box size of 640 pixels and Fourier-cropped to 128 pixels, followed by several rounds of 2D classification, yielding 841,042 particles. These particles underwent heterogeneous refinement with five classes, and one class, containing 332,448 particles with RQC features, was subjected to additional 3D classification and heterogeneous refinement. A total of 260,017 particles were retained and combined with the 170,995 particles from the first dataset. Combined particles underwent two rounds of homogeneous refinement and 3D variability analysis (3DVA)55 with a mask surrounding the Ltn1RING and Ltn1RWD domains as well as parts of the 60S ribosomal subunit, with resolution filtered to 15 Å. A 3D variability display was generated using 20 clusters.
A total of 180,785 particles were re-extracted with a box size of 640 pixels and Fourier cropped to 256 pixels. Two additional rounds of homogeneous refinement and 3D variability analysis were performed: the first round applied a mask around the Ltn1RING and Ltn1RWD domains and the 60S subunit, with resolution filtered to 8 Å using 10 clusters; the second round used a mask surrounding the entire structure except for Cdc48, with resolution filtered to 8 Å and 10 clusters. After this step, 159,575 particles were retained for local refinement with a mask around Ltn1 and Rqc1. These particles were re-extracted with a box size of 640 pixels and Fourier-cropped to 320 pixels. Subsequent rounds of non-uniform refinement and 3D classification (10 classes, resolution filtered to 8 Å) yielded 121,159 particles for another round of non-uniform refinement and local refinement, during which particle subtraction was used to remove densities not associated with Ltn1 and Rqc1. After additional 3D classification with 10 classes at 10 Å resolution, particles containing Cdc48 densities were selected. A total of 77,542 particles were extracted with a box size of 640 pixels for non-uniform and local refinement, with a mask around Ltn1 and Rqc1 while subtracting other densities. This produced a density map of Ltn1 and Cdc48 at 6.6 Å resolution.
To perform Cdc48-focused sub-extraction, the 77,542 particles were recentered over Cdc48 and reextracted with a box size of 256 pixels for 2D classification to obtain high-resolution 2D classes. A total of 28,262 particles from classes containing well-resolved Cdc48 features were then selected and were re-extracted with a box size of 640 pixels to cover the entire particle. These particles were recentered over the 60S ribosomal subunit, re-extracted with a box size of 640 pixels, and processed through a final round of non-uniform refinement, achieving a 3.2 Å resolution map of the entire complex.
To improve the local resolution of Rqc1CTD, a total of 209,041 particles were selected from 3D variability analysis (3DVA) following homogeneous refinement of combined particles. This analysis used a mask encompassing the Ltn1RING and Ltn1RWD domains along with the 60S subunit, with the resolution filtered to 15 Å and divided into 10 clusters. Of these, five clusters exhibited density features consistent with Rqc1 and were subjected to further homogeneous refinement and 3DVA. A mask surrounding Rqc1, the Ltn1RING, and the 60S subunit was applied, with resolution filtered to 13 Å across six clusters, retaining 197,360 particles. These particles were re-extracted with a box size of 640 pixels and Fourier-cropped to 320 pixels, then processed through homogeneous refinement and another round of 3DVA using a mask focused on Rqc1, with resolution filtered to 6 Å and analyzed across eight clusters. From these, a single class with 14,039 particles was identified with clear secondary structure density consistent with Rqc1 features. The selected particles were re-extracted with a box size of 640 pixels and subjected to local refinement with a mask around Rqc1, producing a 3.7 Å resolution map.
Structure prediction
Alphafold3 predictions were carried out through the AlphaFold3 web server (https://golgi.sandbox.google.com/about)56. For the Cdc48 (UniProt P25694) and Ltn1(UniProt Q04781) interaction (Supplementary Fig. 3a), the input sequence for Cdc48 was L36-G196, and the input sequence for Ltn1 was L933-E1074. For the Rpl38 (UniProt P49167) and Rqc1 (UniProt Q05468) interaction, the input sequence for Rpl38 was M1-L78, and the input sequence for Rqc1 was D120-Y143 (Supplementary Fig. 4c).
For the interaction between the Ltn1 RING ___domain (UniProt Q04781) and Rqc1 (UniProt Q05468), the input sequence for Ltn1’s RING ___domain was E1507-R1562, and the input sequence for Rqc1 was G144-G723 (Supplementary Fig. 4c). For the interaction between ubiquitin (UniProt P0CG63) and Rqc1 (UniProt Q05468), the input sequence for ubiquitin was M1-G76, and the input sequence for Rqc1 was G144-G723 (Supplementary Fig. 4c).
For the interactions among Ltn1 RING ___domain (UniProt Q04781), donor and acceptor ubiquitins (UniProt P0CG63), Rad6 (UniProt P06104), and Rqc1(UniProt Q05468), the input sequence for Ltn1’s RING ___domain was E1507-R1562, for Ubiquitin was M1-G76, for Rad6 was M1-D172, and for Rqc1 was G144-G723 (Supplementary Fig. 7a).
Model fitting and structure visualization
Rigid body fittings were done in UCSF Chimera57 or UCSF ChimeraX58. Extraribosomal densities were distinguished by computing difference maps between cryoEM reconstructions (EMDB entries EMD-15431, EMD-15433 & EMD-15434) and reference yeast 60S maps from crystal structure coordinates (PDB entry 8AGW)11.
For the Cdc48-Ltn1 interface, the predicted structural model consisting of the N-terminal ___domain of Cdc48 and the elbow region of Ltn1 was fitted as a rigid body in ChimeraX. For fitting of the Cdc48 hexamer, the structure of the Cdc48-Ufd1/Npl4 complex with the Cdc48NTD in the ‘up’ conformation was also fitted as a rigid body in ChimeraX (PDB ID: 8DAU59). For the subcomplex containing Rqc1CTD, acceptor Ub, E2 (Rad6), donor Ub, and Ltn1RING, we manually guided the fitting of Ltn1RING into its density, and then used UCSF Chimera to refine the rigid body fitting.
To generate the composite model, we docked PDB entries 8AGW11 and 6OA928, along with AlphaFold 3-predicted complexes of Ltn1RING–Rqc1CTD,Rqc1CTD–ubiquitin, Cdc48NTD-Ltn1, and Rpl38-Rqc1NTD into the consensus map. The overlapping Rqc1 segments were merged, and the Ltn1RING ___domain was fitted as a rigid body using UCSF Chimera and the linker regions were built into the reconstruction density using Coot60. The Ltn1 elbow region in PDB 8AGW was replaced with the predicted structure of Ltn1 bound to Cdc48NTD. The Rpl38 component in PDB 8AGW was replaced by the predicted interaction between Rpl38 and Rqc1 residues 120-143 based on the fitted model.
All figures were prepared with UCSF Chimera57, UCSF ChimeraX58, Pymol (https://www.pymol.org), and intermolecular interactions were analyzed using PDBePISA61. To improve visual clarity for some figures, a Gaussian low-pass filter in UCSF ChimeraX was applied to the maps. Figures using filtered map are indicated accordingly in the figure legends.
Protein A-Nonstop substrate degradation/accumulation assay
The Protein-A nonstop plasmid (pAV184) was generously provided by Ambro van Hoof35. Yeast was grown to saturation overnight in the corresponding selection synthetic-raffinose media, then back diluted to an OD600 of approximately ~0.15 and grown for 3-4 h to reach early log phase (A600 ∼0.3–0.5). At this point, 1% galactose was added to induce expression of the GAL1 promoter for 3 h and then harvested. Cells were lysed by boiling for 5 ~ 10 min in Laemmli buffer and then prepared for gel electrophoresis experiments.
Immunoblots
Samples were separated by SDS-PAGE, followed by electrophoretic transfer onto polyvinylidene difluoride membrane (Bio-Rad). Membranes were blocked with 5% nonfat milk in TBST (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.1% Tween-20) for 30-60 min at room temperature. All uncropped gels and blots used in this study are provided in the Source Data file.
For general ubiquitin and anti-K48 linkage-specific ubiquitin immunoblots, yeast cells expressing a stalling substrate reporter were used. The reporter contains a 12x polyarginine, including pairs of the difficult-to-decode CGA codon, inserted between the coding regions of GFP and RFP, as previously used10. The membrane was incubated overnight at 4 °C with the anti-K48-linkage specific ubiquitin (Sigma-Aldrich; 1:6000 dilution) or anti-ubiquitin (ABclonal; 1:6000 dilution). Membranes were washed in TBST, followed by incubation with secondary antibody, donkey α-rabbit conjugated to horseradish peroxidase (HRP, GE Biosciences; 1:10,000 dilution). Antibodies were detected using the SuperSignal West Dura Chemiluminescent Substrate (Thermos Scientific). The membranes were thoroughly washed with TBST and incubated for 1 h at room temperature with anti-FLAG clone M2 (ABclonal; 1:5000 dilution). Membranes were washed in TBST, followed by incubation with secondary antibody IRDye 680RD Goat Anti-Rabbit (LI-COR 926-68071; 1:20,000 dilution) for 1 h at room temperature.
For anti-Cdc48 and anti-FLAG immunoblots, the membranes were incubated for 1 h at room temperature with anti-Cdc48 (gift from Thomas Sommer; 1:5000 dilution). Membranes were washed in TBST, followed by incubation with secondary antibody IRDye 680RD Goat AntiRabbit (LI-COR 926-68071; 1:20,000 dilution) for 1 h at room temperature. The membranes were thoroughly washed with TBST and incubated for 1 h at room temperature with anti-Flag (Sigma; 1:5000 dilution). Membranes were washed in TBST, followed by incubation with secondary IRDye 800CW goat anti-mouse (LI-COR 926-32210; 1:10,000 dilution) for 1 h at room temperature. Membranes were washed in TBST and digitized using an Odyssey CLx scanner (LI-COR).
For immunoblots against nonstop Protein A substrate, cells were expressed with the pAV184 plasmid as described above. The membrane was incubated for overnight at 4o C or 1 h at room temperature with anti-protein A antibody (Sigma; 1:10,000 dilution). Membranes were washed in TBST, followed by incubation with secondary antibody, donkey α-rabbit conjugated to horseradish peroxidase (HRP, GE Biosciences; 1:10,000 dilution). Antibodies were detected using the SuperSignal West Dura Chemiluminescent Substrate (Thermos Scientific). The membranes were thoroughly wash with TBST and incubated for 1 h at room temperature with anti-Pgk1 (Thermos Scientific; 1:5000 dilution). Membranes were washed in TBST, followed by incubation with secondary IRDye 800CW Goat Anti-Mouse (LI-COR 926- 32210; 1:10,000 dilution) for 1 h at room temperature. Membranes were washed in TBST and digitized using an Odyssey CLx scanner (LI-COR).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The cryo-EM maps of RQC complex are deposited to the Electron Microscopy Data Bank with the accession codes EMD-70444 (consensus reconstruction with Cdc48 density) [https://www.ebi.ac.uk/emdb/EMD-70444], EMD-48529 (Ltn1-Cdc48 focused reconstruction) [https://www.ebi.ac.uk/emdb/EMD-48529], and EMD-48530 (Rqc1 focused reconstruction) [https://www.ebi.ac.uk/emdb/EMD-48530]. The model of the composite structure is deposited to the Protein Data Bank with the PDB accession code 9OFV. Source data are provided with this paper.
References
Inada, T. Quality controls induced by aberrant translation. Nucleic Acids Res. 48, 1084–1096 (2020).
Brandman, O. & Hegde, R. S. Ribosome-associated protein quality control. Nat. Struct. Mol. Biol. 23, 7 (2016).
Filbeck, S., Cerullo, F., Pfeffer, S. & Joazeiro, C. A. P. Ribosome-associated quality control (RQC) mechanisms from bacteria to humans. Mol. Cell 82, 1451 (2022).
Defenouillère, Q. & Fromont-Racine, M. The ribosome-bound quality control complex: from aberrant peptide clearance to proteostasis maintenance. Curr. Genetics 1–9 https://doi.org/10.1007/s00294-017-0708-5 (2017).
Joazeiro, C. A. P. Mechanisms and functions of ribosome-associated protein quality control. Nat. Rev. Mol. Cell Biol. 20, 368–383 (2019).
Chu, J. et al. A mouse forward genetics screen identifies LISTERIN as an E3 ubiquitin ligase involved in neurodegeneration. Proc. Natl. Acad. Sci. USA 106, 2097–2103 (2009).
Martin, P. B. et al. NEMF mutations that impair ribosome-associated quality control are associated with neuromuscular disease. Nat. Commun. 2020 11:1 11, 1–12 (2020).
Kimonis, V. E., Fulchiero, E., Vesa, J. & Watts, G. VCP disease associated with myopathy, Paget disease of bone and frontotemporal dementia: review of a unique disorder. Biochim Biophys. Acta. 1782, 744–748 (2008).
Ahmed, A. et al. Biallelic loss-of-function variants in NEMF cause central nervous system impairment and axonal polyneuropathy. Hum. Genet 140, 579–592 (2021).
Shen, P. S. et al. Rqc2p and 60S ribosomal subunits mediate mRNA-independent elongation of nascent chains. Science (1979) 347, 75–78 (2015).
Tesina, P. et al. Molecular basis of eIF5A-dependent CAT tailing in eukaryotic ribosome-associated quality control. Mol. Cell 83, 607–621.e4 (2023).
Sitron, C. S. & Brandman, O. CAT tails drive degradation of stalled polypeptides on and off the ribosome. Nat. Struct. Mol. Biol. 2019 26:6 26, 450–459 (2019).
Kostova, K. K. et al. CAT-tailing as a fail-safe mechanism for efficient degradation of stalled nascent polypeptides. Science (1979) 357, 414–417 (2017).
Osuna, B. A., Howard, C. J., KC, S., Frost, A. & Weinberg, D. E. In vitro analysis of RQC activities provides insights into the mechanism and function of CAT tailing. Elife 6, e27949–e27949 (2017).
Yonashiro, R. et al. The Rqc2/Tae2 subunit of the ribosome-associated quality control (RQC) complex marks ribosome-stalled nascent polypeptide chains for aggregation. Elife 5, e11794–e11794 (2016).
Choe, Y.-J. et al. Failure of RQC machinery causes protein aggregation and proteotoxic stress. Nature 531, 191–195 (2016).
Howard, C. J. & Frost, A. Ribosome-associated quality control and CAT tailing. Crit. Rev. Biochem Mol. Biol. 56, 603–620 (2021).
Bengtson, M. H. & Joazeiro, C. A. P. Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature 467, 470–473 (2010).
Shao, S., Von der Malsburg, K. & Hegde, R. S. Listerin-dependent nascent protein ubiquitination relies on ribosome subunit dissociation. Mol. Cell 50, 637–648 (2013).
Defenouillere, Q. et al. Cdc48-associated complex bound to 60S particles is required for the clearance of aberrant translation products. Proc. Natl. Acad. Sci. 110, 5046–5051 (2013).
Kuroha, K., Zinoviev, A., Hellen, C. U. T. & Pestova, T. V. Release of Ubiquitinated and Non-ubiquitinated Nascent Chains from Stalled Mammalian Ribosomal Complexes by ANKZF1 and Ptrh1. Mol. Cell 72, 286–302.e8 (2018).
Brandman, O. et al. A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell 151, (2012).
Verma, R., Oania, R. S., Kolawa, N. J. & Deshaies, R. J. Cdc48/p97 promotes degradation of aberrant nascent polypeptides bound to the ribosome. Elife 2, e00308–e00308 (2013).
Defenouillère, Q. et al. Rqc1 and Ltn1 prevent C-terminal alanine-threonine tail (CAT-tail)-induced protein aggregation by efficient recruitment of Cdc48 on stalled 60S subunits. J. Biol. Chem. 291, 12245–12253 (2016).
van den Boom, J. & Meyer, H. VCP/p97-Mediated Unfolding as a Principle in Protein Homeostasis and Signaling. Mol. Cell 69, 182–194 (2018).
Cooney, I. et al. Lysate-to-grid: Rapid isolation of native complexes from budding yeast for Cryo-EM imaging. Bio. Protoc. 13, e4596–e4596 (2023).
Cooney, I. et al. Structure of the Cdc48 segregase in the act of unfolding an authentic substrate. Science (1979) 365, 502–505 (2019).
Twomey, E. C. et al. Substrate processing by the Cdc48 ATPase complex is initiated by ubiquitin unfolding. Science (1979) 365, eaax1033–eaax1033 (2019).
Xu, Y. et al. Active conformation of the p97-p47 unfoldase complex. https://doi.org/10.1038/s41467-022-30318-3.
Pan, M. et al. Mechanistic insight into substrate processing and allosteric inhibition of human p97. https://doi.org/10.1038/s41594-021-00617-2.
Williams, C., Dong, K. C., Arkinson, C. & Martin, A. The Ufd1 cofactor determines the linkage specificity of polyubiquitin chain engagement by the AAA+ ATPase Cdc48. Mol. Cell 83, 759–769.e7 (2023).
Ji, Z. et al. Translocation of polyubiquitinated protein substrates by the hexameric Cdc48 ATPase. Mol. Cell 82, 570–584.e8 (2022).
Lee, H. G., Lemmon, A. A. & Lima, C. D. SUMO enhances unfolding of SUMO-polyubiquitin-modified substrates by the Ufd1/Npl4/Cdc48 complex. Proc. Natl. Acad. Sci. USA 120 (2023).
Banerjee, S. et al. 2.3 Å resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science (1979) 351, 871–875 (2016).
Wilson, M. A., Meaux, S. & Van Hoof, A. A genomic screen in yeast reveals novel aspects of nonstop mRNA metabolism. Genetics 177, 773 (2007).
Braxton, J. R. & Southworth, D. R. Structural insights of the p97/VCP AAA+ ATPase: How adapter interactions coordinate diverse cellular functionality. J. Biol. Chem. 299, 105182 (2023).
Schaeffer, V. et al. Binding of OTULIN to the PUB Domain of HOIP Controls NF-κB Signaling. Mol. Cell 54, 349–361 (2014).
Zhong, X. et al. AAA ATPase p97/valosin-containing protein interacts with gp78, a ubiquitin ligase for endoplasmic reticulum-associated degradation. J. Biol. Chem. 279, 45676–45684 (2004).
Ye, Y. et al. Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. USA 102, 14132–14138 (2005).
Abaeva, I. S., Bulakhov, A. G., Hellen, C. U. T. & Pestova, T. V. The ribosome-associated quality control factor TCF25 imposes K48 specificity on Listerin-mediated ubiquitination of nascent chains by binding and specifically orienting the acceptor ubiquitin. Genes Dev https://doi.org/10.1101/GAD.352389.124 (2025).
Jomaa, A. et al. Mechanism of signal sequence handover from NAC to SRP on ribosomes during ER-protein targeting. Science 375, 839 (2022).
Nakasone, M. A. et al. Structure of UBE2K–Ub/E3/polyUb reveals mechanisms of K48-linked Ub chain extension. Nat. Chem. Biol. 2022 18:4 18, 422–431 (2022).
Pan, M. et al. Structural insights into Ubr1 mediated N-degron polyubiquitination. Nature 600, 334 (2021).
Verma, R. et al. Vms1 and ANKZF1 peptidyl-tRNA hydrolases release nascent chains from stalled ribosomes. Nature 557, 446–451 (2018).
Zurita Rendón, O. et al. Vms1p is a release factor for the ribosome-associated quality control complex. Nat. Commun. 9, 2197–2197 (2018).
Izawa, T. et al. Cytosolic Protein Vms1 Links Ribosome Quality Control to Mitochondrial and Cellular Homeostasis In Brief Cytosolic Protein Vms1 Links Ribosome Quality Control to Mitochondrial and Cellular Homeostasis. Cell 171, 890–903 (2017).
Heo, J.-M. et al. A stress-responsive system for mitochondrial protein degradation. Mol. Cell 40, 465–480 (2010).
Yip, M. C. J. et al. Mechanism for recycling tRNAs on stalled ribosomes. Nat. Struct. Mol. Biol. 26, 343–349 (2019).
Khan, D., Vinayak, A. A., Sitron, C. S. & Brandman, O. Mechanochemical forces regulate the composition and fate of stalled nascent chains. bioRxiv 2024.08.02.606406 (2024).
Liu, H. & Naismith, J. H. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 8, 91 (2008).
Tseng, W. C., Lin, J. W., Wei, T. Y. & Fang, T. Y. A novel megaprimed and ligase-free, PCR-based, site-directed mutagenesis method. Anal. Biochem 375, 376–378 (2008).
Boland, A. et al. Cryo-EM structure of a separase-securin complex at near-atomic resolution. Nat. Struct. Mol. Biol. 24, 414 (2017).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. https://doi.org/10.1016/j.jsb.2005.07.007 (2005).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 2017 14:3 14, 290–296 (2017).
Punjani, A. & Fleet, D. J. 3D variability analysis: Resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM. J. Struct. Biol. 213, (2021).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nat. 2024 630:8016 630, 493–500 (2024).
Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput Chem. 25, 1605–1612 (2004).
Meng, E. C. et al. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Lee, H. G., Lemmon, A. A. & Lima, C. D. SUMO enhances unfolding of SUMO-polyubiquitin-modified substrates by the Ufd1/Npl4/Cdc48 complex. Proc. Natl. Acad. Sci. USA 120, e2213703120 (2023).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr D. Biol. Crystallogr 66, 486–501 (2010).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007).
Acknowledgements
This work was supported by funding from the National Institutes of Health (grant number R35 GM133772 to P.S.S.). We thank Ambro van Hoof for providing the Protein A-nonstop reporter plasmid (pAV184) and Thomas Sommer for the anti-Cdc48 antibody. We acknowledge David Belnap and Barbie Ganser-Pornillos the University of Utah Arnold and Mabel Beckman Center for CryoEM for cryo-EM support and the University of Utah Center for High Performance Computing for computational support. We thank Emily Parnell, Ahmed Abouelghar, and Mi-Young Jeong for technical assistance, and Adam Frost, Michael Stewart, and Janet Iwasa for helpful discussions about the manuscript.
Author information
Authors and Affiliations
Contributions
Methodology: W.L., T.S., P.S.S. Investigation: W.L., T.S., P.S.S. Visualization: W.L., P.S.S. Supervision: P.S.S. Writing—original draft: W.L., P.S.S. Writing—review & editing: W.L., P.S.S. Funding acquisition: P.S.S. Project administration: P.S.S.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Toshifumi Inada and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, W., Scheel, T. & Shen, P.S. Mechanism of nascent chain removal by the ribosome-associated quality control complex. Nat Commun 16, 5792 (2025). https://doi.org/10.1038/s41467-025-61235-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-61235-w
