
Abstract
Despite recent progress in our understanding of the association between the gut microbiome and inflammatory bowel disease (IBD), the role of microbiome biomarkers in IBD diagnosis remains underexplored. Here we developed a microbiome-based diagnostic test for IBD. By utilization of metagenomic data from 5,979 fecal samples with and without IBD from different geographies and ethnicities, we identified microbiota alterations in IBD and selected ten and nine bacterial species for construction of diagnostic models for ulcerative colitis and Crohn’s disease, respectively. These diagnostic models achieved areas under the curve >0.90 for distinguishing IBD from controls in the discovery cohort, and maintained satisfactory performance in transethnic validation cohorts from eight populations. We further developed a multiplex droplet digital polymerase chain reaction test targeting selected IBD-associated bacterial species, and models based on this test showed numerically higher performance than fecal calprotectin in discriminating ulcerative colitis and Crohn’s disease from controls. Here we discovered universal IBD-associated bacteria and show the potential applicability of a multibacteria biomarker panel as a noninvasive tool for IBD diagnosis.
Similar content being viewed by others


Main
Inflammatory bowel disease (IBD), which includes Crohn’s disease (CD) and ulcerative colitis (UC), is a chronic and relapsing inflammatory disorder of the gastrointestinal (GI) tract. Globally, over 7 million people are estimated to be living with IBD1. Although IBD used to be more prevalent in Western countries, newly industrialized countries have witnessed a rise in incidence over the past few decades largely attributed to the influence of lifestyle and environmental factors2,3,4,5. Delayed diagnosis is often associated with disease progression and intestinal surgery, whereas early diagnosis followed by timely intervention can lead to improved outcomes6,7,8.
Recently, a growing body of literature has shown altered gut microbial composition and metabolic pathways in patients with IBD9,10,11. For instance, the prevalence of proinflammatory bacteria, including adherent-invasive Escherichia coli12, Proteus mirabilis13, Klebsiella pneumoniae14,15 and toxigenic Bacteroides fragilis16, has been reported to be increased, whereas Faecalibacterium prausnitzii, Roseburia inulinivorans, Ruminococcus torques and other short-chain fatty acid-producing bacteria were shown to be diminished in the feces of patients with IBD17,18. Preclinical studies showed that some of these bacterial species play a role in aberrant immune responses and the development of intestinal inflammation19,20. However, the role of microbiome biomarkers in IBD diagnosis remains underexplored.
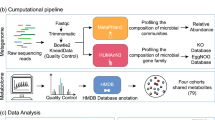
In this study, we aimed to develop a microbiome-based diagnostic test for IBD. We performed comprehensive analyses of metagenomic datasets to assess the predictability of selected bacterial species for IBD diagnosis, constructed diagnostic models using disease-specific species and developed a multiplex droplet digital polymerase chain reaction (m-ddPCR)-based multibacteria biomarker panel for IBD diagnosis (Fig. 1a).

a, Workflow of the study. A total of 5,979 samples, including 1,884 from in-house sequencing datasets and 4,095 from public datasets, were included in this study. The discovery cohort includes 174 patients with CD, 205 with UC and 118 controls. Independent IBD cohorts include 139 patients with UC, 190 with CD and 328 controls. Public datasets include 678 patients with UC, 875 with CD and 1,699 controls. Non-IBD cohorts include 146 patients with IBS, 230 with CA, 372 with CRC, 318 with obesity, 143 with CVD and 364 corresponding controls. FAM and HEX represent fluorescent modification groups of different colors. b, Violin plots showing the Shannon index and observed species of fecal microbiome in patients with UC (n = 205) and CD (n = 174) and controls (n = 118). Data in boxplots are median (center line), 25th and 75th percentiles (box limits) and 5th and 95th percentiles (whiskers). P values were calculated using two-sided Wilcoxon rank-sum test. c, PCoA plot showing the varied microbial composition among groups (174 patients with CD, 205 with UC and 118 controls). Data in boxplots are median (center line), 25th and 75th percentiles (box limits) and 5th and 95th percentiles (whiskers). P values of beta-diversity based on Bray–Curtis distance were calculated with permutational multivariate analysis of variance using 999 permutations (df = 2, R2 = 0.02219, F = 5.606, P < 0.001). d, Stacked bar chart showing the relative abundance of the six most abundant phyla in patients with UC (n = 205) and CD (n = 174) and controls (n = 118). ‘Others’ includes phyla not shown in the figure. *P < 0.05, **P < 0.01, ***P < 0.001. NS, not significant. Illustrations in a were created with BioRender.
Results
Characterization of gut microbial alteration in IBD
We analyzed fecal metagenomics data from 4,406 samples from 13 IBD cohorts across eight countries and regions, to identify and validate gut microbial biomarkers for IBD diagnosis. Specifically, we utilized in-house sequencing data from Hong Kong as a discovery cohort (Supplementary Table 1) and included three additional independent in-house cohorts from Hong Kong and Australia, as well as nine public datasets from the United States, the Netherlands, mainland China, Spain, Denmark and the United Kingdom as validation cohorts (Supplementary Table 2).
In the discovery cohort, we identified a total of 1,175 taxa (three kingdoms, 14 phyla, 25 classes, 40 orders, 85 families, 226 genera and 788 species). At the species level, we identified totals of 674, 637 and 506 bacterial species in UC, CD and control groups, respectively. We found decreased microbial diversity (median: UC 2.73, CD 2.71, controls 3.08; P < 0.001) and richness (median: UC 82, CD 86.5, controls 95; P < 0.001) in patients with UC and CD compared with controls, but there was no statistically significant difference between UC and CD (Fig. 1b). On principal coordinates analysis (PCoA) based on Bray–Curtis distances, the gut microbiome of patients with UC and CD clustered separately from that of controls, and patients with UC exhibited a greater distance from controls than those with CD (Fig. 1c). The presence of IBD accounted for 2.22% of microbiome variance (P < 0.001), while age and gender contributed 0.28% (P = 0.107) and 0.29% (P = 0.073), respectively (Extended Data Fig. 1). There were significant differences in gut microbial communities at the phylum level between patients with IBD and controls, characterized by a reduction in Firmicutes and an enrichment of Proteobacteria in IBD. Patients with CD had lower levels of Bacteroidetes compared with UC and controls (both P < 0.001; Fig. 1d and Extended Data Fig. 2). We found that patients with IBD harbored reduced microbial diversity compared with controls, and patients with CD and UC showed distinct differences in their gut microbial composition21,22.
Identification of gut microbiome signatures in UC and CD
We next used general linear models, as implemented in microbiome multivariable associations with linear models (MaAsLin2), to identify differentially abundant bacterial species in UC and CD after filtering out low-prevalence species (<10%), followed by adjustment for age and gender. We identified 126 and 161 species differentially abundant in UC and CD, respectively, compared with controls (false discovery rate (FDR) < 0.25). Amongst these, 15 species, including B. fragilis, Veillonella parvula, Tyzzerella nexilis, Clostridium spiroforme, Rothia mucilaginosa and Blautia hansenii, were enriched in UC (FDR ≤ 0.1, coefficient ≤ 0.1) whereas 48 species, including Collinsella aerofaciens, Clostridium leptum, R. torques, Asaccharobacter celatus, Gemmiger formicilis and Fusicatenibacter saccharivorans, were depleted in the gut of patients with UC (FDR < 0.05, coefficient ≤ −0.45; Fig. 2a). In CD, 58 bacterial species were either enriched or depleted compared with controls (FDR < 0.001 and |coefficient| ≥ 0.5). In particular, certain bacterial species with proposed anti-inflammatory properties, including Eubacterium hallii, Blautia obeum, R. inulinivorans and Eubacterium rectale, were depleted in CD (Fig. 2b). In addition, E. coli and some Streptococcus species were enriched in the gut of patients with CD but not UC. Taken together, these data highlighted the presence of disease-specific bacterial species in UC and CD.

a,b, Top bacterial species associated with UC (a) and CD (b). Left, lollipop plot showing the coefficient of each species, with disease calculated by MaAsLin2 including adjustment for age and gender. Middle, the phylum of each species is indicated. Right, bar plot demonstrating the proportion of each species present in UC, CD and control groups. c,d, Relative abundance of ten bacterial species biomarkers in UC (n = 205) and controls (n = 118) (c), and of nine bacterial species biomarkers in CD (n = 174) and controls (n = 118) (d) as determined by metagenomics. Data shown as median (center line), 25th and 75th percentiles (box limits) and 5th and 95th percentiles (whiskers). P values were calculated using two-sided Wilcoxon rank-sum test. e,f, Performance of model with ten UC and nine CD bacterial species biomarkers for classification of patients with UC (e) and CD (f) compaerd with controls in discovery cohort. Shaded areas of ROC curves represent the 95% CI of AUC for the test set. g, SHAP values of the ten UC bacterial species biomarkers for each sample. h, SHAP values of the nine CD bacterial species biomarkers for each sample. Each point represents the SHAP value of each biomarker for each sample; the distribution of points indicates the impact of each biomarker on model output. Colors represent relative abundance of the biomarkers (yellow, high; purple, low). i, Correlation between functional dysbiosis scores and probability of disease generated by models based on ten UC and nine CD bacterial species biomarkers. Shaded area corresponds to 95% CI for the regression fit. Correlation coefficient and two-sided P values are given by Spearman correlation. Coeff., coefficient; pos. pred., positive prediction; neg. pred., negative prediction.
Development of metagenomics-based diagnostic models
We next performed a fivefold cross-validation with all discriminative bacterial species to construct diagnostic models. We achieved stable classification performances by utilization of seven and eight bacterial features in UC and CD, respectively (Extended Data Fig. 3). After accounting for functional properties of selected bacteria and optimal distribution of enriched and depleted bacterial species, we selected ten bacterial species as biomarkers for UC (four enriched: Gemella morbillorum, B. hansenii, Actinomyces sp. oral taxon 181 and C. spiroforme; and six depleted: C. leptum, F. saccharivorans, G. formicilis, R. torques, Odoribacter splanchnicus and Bilophila wadsworthia); and nine bacterial species as biomarkers for CD (three enriched: B. fragilis, E. coli and Actinomyces sp. oral taxon 181; and six depleted: R. inulinivorans, B. obeum, Lawsonibacter asaccharolyticus, Roseburia intestinalis, Dorea formicigenerans and Eubacterium sp. CAG: 274) (Fig. 2c,d). Among these, Actinomyces sp. oral taxon 181 and C. spiroforme were first reported to be enriched in UC whereas Actinomyces sp. oral taxon 181, L. asaccharolyticus and Eubacterium sp. CAG: 274 were new bacterial species associated with CD. Using the random forest algorithm, the ten selected bacterial species discriminated patients with UC from controls, with an AUC of 0.95 (95% confidence interval (CI): 0.92–0.98) in the training set and 0.90 in the test set (95% CI: 0.84–0.96; sensitivity 88.06%, specificity 80.95%). F. saccharivorans, C. leptum and G. formicilis were the top three key discriminatory bacterial species in the UC model (Fig. 2e,g). In CD, nine bacterial species showed an AUC of 0.95 (95% CI: 0.92–0.98) and 0.94 (95% CI: 0.89–0.98; sensitivity 88.33%, specificity 89.47%) in discriminating CD from controls in the training and test set, respectively. B. obeum, L. asaccharolyticus, R. inulinivorans (depleted in CD), Actinomyces sp. oral taxon 181 and E. coli (enriched in CD) were the top-ranking bacteria in our model (Fig. 2f,h).
Alteration of metabolic pathways in IBD
Apart from altered microbial composition and taxonomy, we found that metabolic functions were dysregulated in both UC and CD. Using MaAsLin2 comparison analysis, we identified 545 metabolic pathways consisting of 244 and 315 differential pathways in UC and CD, respectively, compared with controls (FDR < 0.05). Pathways involved in amine and polyamine degradation, and in fatty acid and lipid biosynthesis, were significantly enriched in patients with UC or CD compared with controls23. UC- and CD-enriched bacterial species biomarkers were positively correlated with disease-enriched metabolic pathways, while UC- and CD-depleted bacterial species biomarkers showed negative correlations (Extended Data Fig. 4). New findings from stratified analyses revealed that pathways belonging to amino acid biosynthesis (l-arginine, l-ornithine and l-valine biosynthesis) were mainly contributed by bacterial species depleted in patients with UC, including C. leptum, F. saccharivorans, G. formicilis and R. torques. Depletion of these key bacterial species in UC was also associated with a significant decrease in the abundance of their respective functional pathways (Extended Data Fig. 5a). Similarly, bacterial species, including B. obeum, R. inulinivorans, D. formicigenerans, Eubacterium sp. CAG: 274 and R. intestinalis, also contributed to pathways of amino acid biosynthesis (l-tryptophan biosynthesis), carbohydrate degradation (starch degradation) and cofactor, carrier and vitamin biosynthesis (thiamine phosphate formation from purithiamine and exythiamine) in controls. In CD, there was a shift in the main contributors of these pathways characterized by a predominance of E. coli rather than a diversified bacterial profile (Extended Data Fig. 5b). Variations in bacterial metabolic pathways suggested that altered microbiota-mediated metabolic capabilities may be essential for IBD development.
To examine the role of bacterial biomarkers in mediation of metabolic functions, we developed a dysfunctional score for each subject by calculating median Bray–Curtis dissimilarity to controls using metabolic pathway profiles. Both the distribution and level of dysfunctional scores in patients with UC and CD were different from those of controls, implying dysfunctional changes in IBD (Extended Data Fig. 5c,d). The probability of disease determined by diagnostic models showed a positive correlation with dysfunctional scores in UC and CD, implying that our bacterial biomarkers could reflect the metabolic dysregulations in patients with IBD (Fig. 2i).
Performance of diagnostic models in active and inactive IBD
We next evaluated the accuracy and stability of our multibacteria biomarker panel in IBD with different disease activities. Using Mayo score and Crohn’s disease activity index (CDAI), we categorized patients into active (UC, Mayo >2; CD, CDAI >150) and inactive disease (UC, Mayo ≤2; CD, CDAI ≤150). There was differential abundance of CD- and UC-associated bacterial species in patients with inactive IBD compared with controls (six decreased, four increased in UC; six decreased, three increased in CD). Moreover, levels of some bacterial species changed with disease activity. For instance, the relative abundance of six UC-depleted species was lower in patients with active compared with inactive UC, and R. inulinivorans, B. obeum, L. asaccharolyticus, D. formicigenerans and Eubacterium sp. CAG: 274 were lower in patients with active compared with inactive CD, whereas some disease-enriched bacteria, such as G. morbillorum, were higher in active UC, and B. fragilis, E. coli and Actinomyces sp. oral taxon 181 were higher in active CD, implying that these bacterial species may be involved in disease activity and severity (Extended Data Fig. 6a,b). We next utilized diagnostic models to calculate a probability value for disease risk, and found that disease probability showed no significant difference between inactive and active IBD (Extended Data Fig. 6c,e). Our model was able to distinguish patients with inactive UC and CD from controls, with AUC of 0.89 and 0.84, respectively (Extended Data Fig. 6d,f). These data suggested that bacterial biomarkers may not just be a consequence of inflammation but also contribute to—or possibly reflect—underlying disease pathogenesis.
Validation of diagnostic models in independent cohorts
Next, we analyzed two independent datasets from Hong Kong (139 UC, 139 controls; 92 CD, 108 controls) and Australia (98 CD, 81 controls) and three public datasets from the United States (53 UC, 68 CD, 34 controls), the Netherlands (23 UC, 20 CD, 22 controls) and mainland China (25 UC, 15 controls; 48 CD, 54 controls). Both the abundance and prevalence of bacterial biomarkers in the independent cohorts and public datasets were consistent with those reported in the discovery cohort (Extended Data Fig. 7a,b). Specifically, our diagnostic models showed satisfactory performances in classifying IBD from controls, with AUCs of 0.81 (95% CI: 0.76–0.86) for UC (Fig. 3a) and 0.83 (95% CI: 0.77–0.89) for CD in the Hong Kong cohorts (Fig. 3b) and 0.73 (95% CI: 0.65–0.80) for CD in the Australian cohort (Fig. 3c). Using datasets from United States, the Netherlands and mainland China, the diagnostic model achieved AUCs of 0.85, 0.87 and 0.82, respectively, for UC diagnosis (Fig. 3d and Extended Data Fig. 7c), and AUCs of 0.89, 0.86 and 0.97, respectively, for CD diagnosis (Fig. 3e and Extended Data Fig. 7c). Bacteria ecological network analysis showed co-occurring correlations between depleted bacterial species in IBD, and stable coexcluding correlations among disease-depleted and -enriched species in almost all cohorts (Extended Data Fig. 7d,e). To further validate our models, we utilized additional metagenomic datasets from the United States, the Netherlands, Spain, Denmark and the United Kingdom (577 UC, 739 CD, 1,574 controls). By integration of data from all IBD cohorts and adjusting for geography and ethnicity, the multibacteria biomarker panel remained significantly different between IBD and controls (Fig. 3f,g). The UC model maintained an overall AUC of 0.82, and the CD model achieved an AUC of 0.76, in all validation cohorts (Fig. 3h,i). Taken together, these results highlighted the robustness of diagnostic performance of our multibacteria biomarker panel across different regions and ethnicities.

a, Performance of model with ten UC selected bacterial species biomarkers for classification of patients with UC versus controls in Hong Kong validation cohort. b,c, Performance of model with nine CD selected bacterial species biomarkers for classification of patints with CD versus controls in Hong Kong (b) and Australia validation cohorts (c). d,e, Performance of model with the selected bacterial species biomarkers for classification of patients with UC (d) or CD (e) versus controls in the three downloaded public datasets. f, Associations among disease group, geography, ethnicity and the relative abundance of ten UC bacterial species biomarkers were calculated by MaAsLin2 in cohorts with UC patients and controls. g, Associations among disease group, geography, ethnicity and the relative abundance of the nine CD bacterial species biomarkers were calculated by MaAsLin2 in cohorts with CD patients and controls. Positive and negative associations are colored red and blue, respectively. Significant associations (FDR < 0.05) are marked with a plus sign for positive associations and a minus for negative associations. FDR was computed by Benjamini–Hochberg correction. h, Performance of model with the selected bacterial species biomarkers for classification of patients with UC (n = 817) versus controls (n = 1,746) in all UC validation cohorts. i, Performance of model with the selected bacterial species biomarkers for classification of patients with CD (n = 1,065) versus controls (n = 1,873) in all CD validation cohorts. j, Model performance in distinguishing treated and treatment-naive patients with UC from controls in two downloaded public datasets. k, Model performance in distinguishing treated and treatment-naive patients with CD from controls in two downloaded public datasets. l, Model performance in distinguishing patients with UC from controls, compared using fecal calprotectin test in two downloaded public datasets. m, Model performance in distinguishing patients with CD from controls, compared using fecal calprotectin test in two downloaded public datasets. Shaded areas of ROC curves represent 95% CI of the AUC for each cohort.
Given that the drugs used to induce and maintain disease remission in IBD can alter the gut microbiome, we tested whether our models would be affected by treatment. Using metagenomic data from two public datasets (United States and the Netherlands)21 whereby treatment data were available for IBD cases, the UC model could discriminate treatment-naive (n = 14) and treated-UC patients (n = 62) from controls (n = 56), with AUCs of 0.74 and 0.89, respectively, while the CD model discriminated treatment-naive (n = 20) and treated-CD patients (n = 66) from controls (n = 56), with AUCs of 0.89 and 0.88, respectively, suggesting that the performance of the multibacteria biomarker panel is unlikely to be influenced by treatment (Fig. 3j,k).
For comparison of our models with a commonly used IBD screening test, fecal calprotectin, we utilized data from two public datasets (United States and the Netherlands)21 for which fecal calprotectin data were available (46 UC, 65 CD, 42 controls). Our diagnostic models had a numerically higher AUC than fecal calprotectin in the diagnosis of UC (AUC = 0.85 versus 0.81) and CD (AUC = 0.87 versus 0.79) (Fig. 3l,m). The multibacteria biomarker panel also showed a higher sensitivity (72 versus 54% for CD, 67 versus 57% for UC) and specificity (95 versus 86% for CD, 88 versus 86% for UC) than fecal calprotectin.
IBD specificity of diagnostic models in transethnic cohorts
In light of shared microbiota alterations across various diseases24, it is important to verify disease specificity for bacterial biomarkers. Hence, we assessed several in-house non-IBD disease metagenomic datasets from Hong Kong comprising subjects with various GI diseases (n = 439), including colorectal cancer (CRC, n = 160), colorectal adenoma (CA, n = 162), irritable bowel syndrome (IBS, diarrhea subtype, IBS-D, n = 117) and non-GI diseases (n = 291): obesity (n = 148) and cardiovascular disease (CVD, n = 143). Among UC bacterial biomarkers, depletion in R. torques and enrichment in C. spiroforme were unique to patients with UC compared with all other non-IBD diseases (Extended Data Fig. 8a). Among CD bacterial biomarkers, depletion of R. inulinivorans and B. obeum and an increase for B. fragilis and E. coli were specifically associated with CD (Extended Data Fig. 8b). To validate our models in an internationally diverse non-IBD cohort, we included 843 extra metagenomic data from non-IBD cohorts (212 CRC, 68 CA, 29 IBS-D, 170 obesity and 364 controls) from Austria, France, Germany, Japan, the United States and Denmark (Fig. 4a). The probability of disease generated by our models showed significant differences between IBD and non-IBD subjects (Fig. 4b,d). The UC diagnostic model discriminated patients with UC from non-IBD subjects with an AUC of 0.78 (Fig. 4c), whereas the CD diagnostic model distinguished CD from non-IBD subjects with an AUC of 0.72 (Fig. 4e). Overall, these results suggested that our multibacteria panel is specific to UC and CD.

a, Composition of international multidisease datasets from different countries and regions. b, Comparison of the probability of disease generated by the UC model based on ten UC bacterial species biomarkers in controls (n = 2,391) and in patients with CVD (n = 143), obesity (n = 318), CA (n = 230), CRC (n = 372), IBS-D (n = 146) and UC (n = 817). Data in boxplots show the median (center line), 25th and 75th percentiles (box limits) and 5th and 95th percentiles (whiskers). c, Performance of UC model in classification of patients with UC (n = 817) versus other non-IBD subjects (n = 3,600). d, Comparison of the probability of disease generated by the CD model based on nine CD bacterial species biomarkers in controls (n = 2,391) and in patients with CVD (n = 143), obesity (n = 318), CA (n = 230), CRC (n = 372), IBS-D (n = 146) and CD (n = 1,065). Data in boxplots show the median (center line), 25th and 75th percentiles (box limits) and 5th and 95th percentiles (whiskers). e, Performance of CD model in classification of patients with CD (n = 1,065) versus other non-IBD subjects (n = 3,600). Boxplots represent the minimum (Q1), median (Q3) and maximum. P values were calculated using the two-sided Wilcoxon rank-sum test. Shaded areas of ROC curves represent 95% CI of the AUC for each cohort.
Development of a general IBD model
Because there is an unmet need for universal biomarkers to differentiate IBD from non-IBD subjects, we developed an IBD model using a total of 18 bacterial species identified for UC and CD (UC, ten species; CD, nine species; one species overlapping in both UC and CD). The discriminative power of the IBD model achieved an AUC of 0.91 (95% CI: 0.85–0.97) with a sensitivity of 92% and specificity of 75% in the discovery cohort, and an AUC of 0.81 (95% CI: 0.80–0.82) with a sensitivity of 78% and specificity of 70% in the validation cohort (controls, n = 2,027; IBD, n = 1,882). In the international multidisease cohort (IBD, n = 1,882; non-IBD, n = 3,600), which included subjects with both GI and non-GI diseases, our model could differentiate IBD from non-IBD with an AUC of 0.77 (95% CI: 0.75–0.78) (Extended Data Fig. 9a,b). In a pilot cohort, we performed direct comparison of our multibacteria biomarker panel with fecal calprotectin in samples from the above-mentioned in-house cohorts (36 patients with UC, 36 with CD and 36 with IBS). Our multibacteria biomarker panel (AUC = 0.91; sensitivity, 79%; specificity, 92%) showed numerically higher performance than fecal calprotectin (AUC = 0.86; sensitivity, 68%; specificity, 89%) in distinguishing patients with IBD from those with IBS (Extended Data Fig. 9c).
Development of m-ddPCR-based multibacteria biomarker panel
To translate the metagenome-derived multibacteria biomarker panel into a simple and affordable clinical tool, we developed a m-ddPCR-based method to quantify selected bacterial species in fecal samples. Three reactions were designed to measure the abundance of bacterial species and to ensure there was no cross-reaction among the primers and probes of targeted species (Fig. 5a). We quantified the abundance of bacterial species by m-ddPCR and correlated this with with the abundance of species generated from metagenomics in UC (205 UC, 84 controls) and CD (172 CD, 86 controls). Quantification by metagenomic sequencing and m-ddPCR showed strong correlations (Spearman r = 0.34–0.73 for UC biomarkers and 0.49–0.94 for CD biomarkers), suggesting that both measurements are reliable and consistent (Fig. 5b,c). From m-ddPCR results, we identified significant differences in the six depleted bacterial species and one enriched bacterial species in UC compared with controls, while the other three enriched bacterial species showed increasing trends in patients with UC compared with controls (Fig. 6a). The random forest diagnostic model constructed using m-ddPCR data yielded an AUC of 0.88 (sensitivity, 85.0%; specificity, 81.8%) for UC diagnosis in the discovery cohort (Fig. 6b). In CD, we also identified significant differences in the abundance of six depleted and three enriched bacterial species in CD compared with controls (Fig. 6c). Using m-ddPCR data from the multibacteria biomarker panel, we constructed a random forest diagnostic model that showed an AUC of 0.87 for CD (sensitivity, 90.2%; specificity, 76.0%) (Fig. 6d). Furthermore, the UC model achieved an AUC of 0.89 (Fig. 6b) while the CD model achieved AUCs of 0.75 and 0.73 (Fig. 6d) in the independent validation cohorts. We compared the probability of disease values derived from the metagenomic model and m-ddPCR model and found that differences in values between the two models were −0.03 (95% CI: −0.05 to −0.01) in UC and −0.07 (95% CI: −0.08 to −0.05) in CD (Extended Data Fig. 10), indicating that the findings from m-ddPCR were consistent with those derived from metagenomics.

a, Panel design of m-ddPCR for UC and CD bacterial species biomarkers. b,c, Correlation between the abundance of ten UC (b) and nine CD bacterial species biomarkers (c), as determined by metagenomics and m-ddPCR. Shaded areas correspond to 95% CI for the regression fit. The correlation coefficient and two-sided P value are given by Spearman correlation.

a, Relative abundance of ten bacterial species biomarkers in UC and controls in the discovery cohort (205 UC, 84 controls). b, Diagnostic performance of UC model with ten bacterial species biomarkers, as determined by m-ddPCR in the discovery cohort (test set, n = 62) and the Hong Kong (HK) cohort (n = 108). c, Relative abundance of nine bacterial species biomarkers in CD and control groups in the discovery cohort (172 CD; 86 controls). Gray diamonds represent mean values. d, Diagnostic performance of CD model with nine bacterial species biomarkers, as determined by m-ddPCR in the discovery cohort (test set, n = 66), the Hong Kong cohort (n = 153) and Australia (AUS) cohort (n = 177). e, Diagnostic performance of fecal calprotectin and UC model with ten bacterial species biomarkers, as determined by m-ddPCR in the Canada cohort (left; 100 UC, 53 Controls) and Taiwan cohort (right; 40 UC, 40 controls). f, Diagnostic performance of fecal calprotectin and CD model with ten bacterial species biomarkers, as determined by m-ddPCR in the Canada cohort (left; 100 CD, 53 controls) and Taiwan cohort (right; 40 CD, 40 controls). g, Comparison of the probability of disease (POD), calculated by the UC/CD model using m-ddPCR data and fecal calprotectin, between patients with inactive or active UC/CD and controls in the Canada and Taiwan cohorts. h, Diagnostic performance of fecal calprotectin and the UC model with ten bacterial species biomarkers determined by m-ddPCR in distinguishing patients with inactive UC (n = 81) and controls (n = 93). Shaded areas of ROC curves represent 95% CI of the AUC for each cohort. Data in boxplots show the median (center line), 25th and 75th percentiles (box limits) and 5th and 95th percentiles (whiskers). P values were calculated using the two-sided Wilcoxon rank-sum test.
Comparison between m-ddPCR panel and fecal calprotectin
For comparison of the diagnostic performance of our multibacteria biomarker panel with that of fecal calprotectin, we performed both tests on fecal samples from two independent cohorts from Canada (100 UC, 100 CD, 53 controls) and Taiwan (40 UC, 40 CD, 40 controls), with analysis blinded relative to each test. The multibacteria biomarker panel showed better performance than that of fecal calprotectin in UC (Fig. 6e; Canada, AUC = 0.74 versus 0.63; Taiwan, 0.79 versus 0.57), and performed slightly better than, or was comparable to, fecal calprotectin in the CD model (Fig. 6f; Canada, AUC = 0.77 versus 0.75; Taiwan, 0.71 versus 0.71). In the subgroup analysis, the CD multibacteria biomarker panel could differentiate patients with active and inactive CD from controls. In addition, the UC multibacteria biomarker panel showed higher performance than fecal calprotectin in discriminating inactive UC from controls (AUC = 0.78 versus 0.56) (Fig. 6g,h).
Discussion
In the present study, we comprehensively assessed the IBD-associated gut microbiome and its ability to distinguish IBD from non-IBD subjects. Through extensive and rigorous validation, whereby data were generated from eight countries and regions and data used for training were separated from those for testing, we identified disease-specific bacterial species and developed a noninvasive microbiome-based tool for IBD diagnosis. In particular, the metagenomics-based model trained on selected bacterial species from multiple studies maintained an AUC of 0.81 (95% CI: 0.80–0.82) in distinguishing patients with IBD from controls, which is above the threshold (AUC = 0.80) that is generally considered clinically useful25. Our multibacteria biomarker panel also showed numerically higher diagnostic performance than fecal calprotectin, a standard noninvasive clinical test for inflammation commonly used in IBD.
To date, microbial identification and analysis using mass spectrometry26,27 and 16S amplicon and metagenomics sequencing28,29 face challenges including high cost, complex operations and interpretation procedures. Targeted detection technologies, such as fluorescent quantitative PCR, nucleic acid hybridization30, fluorescent probe labeling31 and digital PCR32, have been applied in pathogen detection, environmental monitoring and liquid biopsy33,34, but there is limited research in applying these techniques for disease diagnosis. Herein, we have taken existing IBD microbiome research a step further by translating metagenomic-generated data to bacterial detection based on m-ddPCR, which is more user friendly and less operator dependent. We developed specific primers and probes for bacterial species identified from metagenomics and designed m-ddPCR assays for quantification. M-ddPCR-based results replicated performance from metagenomics with moderately higher accuracy than fecal calprotectin. Importantly, the potential cost of m-ddPCR is substantially lower and the turnaround time shorter than those of metagenomic-based tools. For clinical implementation, further studies in different populations to confirm the analytical and clinical performance of the multibacteria biomarker panel, in comparison with existing noninvasive testing or colonoscopy according to the regulatory requirements for in vitro diagnostics, will be helpful35.
Our metagenomic findings confirmed enrichments of E. coli and B. fragilis in the gut of patients with CD22,36. Specifically, adherent-invasive E. coli was present in more than half of patients with CD12 and has been linked to mucosal dysbiosis and functional alteration, associated with disease activity and endoscopic recurrence following surgery37,38, and B. fragilis may induce intestinal inflammation through toxin production39,40,41,42. In addition, we identified a new oral bacterium, Actinomyces sp. oral taxon 181, that was significantly enriched in stool samples of patients with CD and UC. It is possible that these resident oral bacteria translocate to the GI tract through the bloodstream or the digestive system and colonize and induce inflammation in the gut by activation of the intestinal immune system16. We also observed an increased abundance of G. morbillorum in patients with active UC compared with those in remission, suggesting its potential role in the inflammatory process10,43.
We also explored the underlying mechanisms to further understand the role of selected bacteria in IBD pathogenesis. The functional microbiome has emerged as a prerequisite for host phenotype and physiology, and increasing efforts have been made to link functional traits and mechanisms of organisms to their environments for prediction of survival and community structure44,45. In this study, functional metabolic perturbations were observed in patients with IBD, and the probability of disease obtained from our multibacteria biomarker panel could reflect these metabolic dysregulations. Notably, some of bacterial species drove the functional alterations. For example, reduction in bacterial species with putative anti-inflammatory properties may lead to a reduced capacity for fermentation of dietary fiber and/or production of short-chain fatty acid39,46,47, whereas their deficiency is often connected with impaired intestinal mucosal barrier function and induction of intestinal inflammation48. We also demonstrated that several bacteria depleted in IBD were major contributors to the amino acid biosynthesis pathway, and that impairment of these pathways may impact intestinal tissue repair and immune regulation in IBD47,49. In vivo and in vitro experiments can provide further mechanistic insights into the causal role of these bacteria and metabolic dysregulations in the pathogenesis of IBD, as well as their future clinical utility in disease monitoring or prediction of treatment response.
There were limitations in this study. First, as a cross-sectional study, it is difficult to completely eliminate biases in cohort selection and other unmeasured environmental influences. Although we did not specifically collect dietary data, the consistent diagnostic performance achieved when using data from transethnic public datasets, whereby dietary habits vary, suggests that bacterial biomarkers are unlikely to be influenced by diet. Performance of the biomarker panels also remained unaffected regardless of different medications received by patients with IBD. Second, the distribution of disease and controls was balanced in this study, which may not reflect the true real-life prevalence of IBD, which is lower. Hence, future prospective studies consisting of a larger and more diverse group of patients with various GI symptoms would allow for a more comprehensive evaluation of bacterial biomarkers in a real-world population with GI problems. Furthermore, investigation into the microbiome characteristics of patients with different IBD phenotypes will provide insights into the potential of developing microbiome-based precision diagnostic tools. Lastly, the requirement of specialized equipment, such as m-ddPCR instruments and personnel trained in m-ddPCR techniques, may limit the implementation of a multibacterial diagnostic panel in certain healthcare settings. However, because m-ddPCR technology continues to advance and become more accessible, along with the introduction of training programs, these challenges can be addressed to enable successful worldwide implementation.
In conclusion, we have uncovered altered gut microbiome signatures and metabolic pathways associated with UC and CD. Our targeted ddPCR-based quantification of bacterial species, consistent with metagenomics data from different populations, serves as the foundation for diagnostic assays that are sufficiently robust, sensitive and cost effective for clinical application. The identification of reproducible bacterial biomarkers for IBD helps enable the design of noninvasive diagnostic tools for more precise and personalized approaches in IBD detection and management.
Methods
Ethics statement
This study has been approved by the Joint Chinese University of Hong Kong–New Territories East Cluster Clinical Research Ethics Committee (The Joint CUHK–NTEC CREC, CRE ref. nos. 2013.093, 2017.495 and 2022.308), the ethics committee of St Vincent’s Hospital (Melbourne, Australia), research ethics boards for each center involved in the research in Canada and National Taiwan University Hospital. All subjects provided written informed consent.
Subject recruitment
We recruited two IBD cohorts consisting of patients with CD and UC, and non-IBD controls. The first cohort consisted of 344 patients with UC, 266 with CD and 365 controls from the Prince of Wales Hospital and other hospitals in Hong Kong, which formed the basis for biomarker discovery (205 UC, 174 CD and 118 controls; Supplementary Table 1) and validation (139 UC and 139 controls; 92 CD and 108 controls; Supplementary Table 2). A second group of patients, recruited from St Vincent’s Hospital, Melbourne, Australia, was used as a validation cohort (98 CD and 81 controls; Supplementary Table 2). Information on gender was collected based on self-reporting. Patients with CD and UC were diagnosed according to the standard criteria of endoscopy, radiology and histology. CDAI scores of ≤150 (inactive) and >150 (active) for patients with CD, and Mayo scores of ≤2 (inactive) and >2 (active) for patients with UC, were collected. Individuals with no existing gut disorder, including IBD, cancer, advanced adenoma, IBS or other GI symptoms, were recruited as controls. Patients and control subjects were excluded if they had used antibiotics in the previous month or had known current sepsis (excluding uncomplicated infections such as influenza) and a known history of severe organ failure (including decompensated cirrhosis, malignant disease, kidney failure, epilepsy, active serious infection or acquired immunodeficiency syndrome), major bowel surgery in the past 6 months (excluding colonoscopy/procedure related to perianal disease), presence of an ileostomy or stoma or current pregnancy.
In addition, five in-house metagenomic cohorts of patients with GI disorders other than IBD, including CA (n = 162), CRC (n = 160) and IBS-D (n = 117), and patients not diagnosed with IBD but with non-GI disorders, including obesity (body mass index >28; n = 148) and CVD (n = 143), were also included for validation50 (Supplementary Table 3). Subjects with CRC and CA were diagnosed by colonoscopy and confirmed on histologic examinations. Subjects with IBS were diagnosed according to the ROME III criteria, and endoscopy and enteroscopy were performed to exclude other GI disorders including IBD, celiac disease, parasitic infestation or other organic disorders. Subjects with CVD were recruited from the public as part of a survey of cardiovascular health in the Hong Kong general population. Subjects underwent carotid ultrasound for measurement of intima/media thickness of the common, internal and external carotid arteries and carotid bulbs; those subjects with ≥50% stenosis in either single or multiple vessels were regarded as having risk of CVD.
Two independent international cohorts from Canada (100 UC, 100 CD, 53 controls) and Taiwan (40 UC, 40 CD, 40 controls) were recruited from several centers in Canada and from the National Taiwan University Hospital, respectively, according to the same inclusion and exclusion criteria previously mentioned. Information on gender was collected based on self-reporting. CDAI scores (≤150 or under, inactive; >150, active) or Harvey–Bradshaw index scores (≤4, inactive; >4, active) for patients with CD, and Mayo scores (≤2, inactive; >2, active) for patients with UC, were collected.
Sample collection
All participants were required to provide at least one spoonful of stool sample using a stool collection tube provided by the investigator in advance. Following collection, stool samples were divided into 2-ml tubes and promptly transferred to a −80 °C, ultralow-temperature freezer for storage until further processing. Aliquot tubes were used for different tests to avoid repeated freezing and thawing. Samples from other centers were processed using the same procedure, and shipped by courier to Hong Kong at low temperatures using dry ice.
Fecal DNA extraction and metagenomic sequencing
Fecal bacterial DNA was extracted using the Maxwell RSC PureFood GMO and Authentication Kit (Promega), with modifications to the standard protocol to increase the yield of DNA. Briefly, approximately 100 mg of stool from each subject was suspended in 1 ml of double-distilled H2O and pelleted by centrifugation at 13,000g for 1 min. To washed samples were added 800 µl of TE buffer (pH 7.5), 16 µl of beta-mercaptoethanol and 250 U lyticase, with sufficient mixing and digestion at 37 °C for 90 min, followed by pelleting by centrifugation at 13,000g for 3 min. Following pretreatment, the precipitate was resuspended in 800 µl of CTAB buffer and mixed well. Following heating of samples at 95 °C for 5 min and cooling for 2 min, nucleic acid was released by vortexing with 0.5- and 0.10-mm beads at 4,500 rpm for 5 min. Following this, 40 µl of Proteinase K and 20 µl of RNase A were added and nucleic acid was digested at 70 °C for 10 min. Finally, the supernatant was obtained following centrifugation at 13,000g for 5 min and placed in a Maxwell RSC instrument for DNA extraction. Following quality control procedures by Qubit 2.0, agarose gel electrophoresis and an Agilent 2100, metagenomic libraries were prepared using the Nextera DNA Flex Library Preparation kit (Illumina), pooled and sequenced at the Microbiota-I Centre (MagIC) or outsourced (Novogene) using the Illumina NovaSeq 6000 targeting 6 Gb of sequence per sample with 150 base pairs of paired-end reads.
Public datasets of IBD and controls
Available public datasets were systematically searched from all published papers and NCBI with metagenomic datasets using the keywords ‘inflammatory bowel disease’, 'ulcerative colitis’, 'Crohn’s disease’, 'metagenomics’ and ‘gut microbiome’. Raw fecal metagenomic sequencing data and clinical metadata of the three populations from the United States, Europe and mainland China (101 UC, 136 CD, 125 controls) were downloaded from the European Nucleotide Archive (ENA) using the following ENA identifiers: PRJNA400072 (ref. 21) for cohorts from the United States and the Netherlands; and PRJNA429990 (ref. 51) and PRJEB15371 (ref. 52) for the two cohorts from mainland China (Supplementary Table 2). Information on treatment and fecal calprotectin data from cohorts in the United States and the Netherlands (PRJNA400072) were extracted for subgroup analysis. The IBS cohort from Mars RAT 2020 (ref. 53) was downloaded using the ENA identifier PRJEB37924. Metagenomic taxonomy profiling data from cohorts Hall AB 2017 (PRJNA385949)54, Nielsen HB 2014 (PRJEB1220)55, HMP 2019 IBDMDB (PRJNA398089)56, LifeLD VilaAV 2018 (EGAS00001001704, EGAD00001004194)39,57 and Ijaz UZ 2017 (PRJEB18780)58, from CRC and CA cohorts (refs. 59,60,61,62) and from the obesity cohort (LeChatelierE 2013 (ref. 63)) were acquired from curatedMetagenomicData (v.3.10.0)64.
Sequence data preprocessing and microbiome profiling
Adapters were removed using trimmomatic-0.39 in paired-end mode. Sequencing reads were trimmed when the average quality per base fell below 20 with a four-base-wide sliding window. Sequencing reads <50 bases in length were discarded. Host-contaminating reads were filtered using Kneaddata (v.0.10.0) with the human reference genome (hg37decv0.1, https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/001/405/GCF_000001405.25_GRCh37.p13/). Samples of low read count following quality filtration (<1 million reads) were excluded from our analysis. Furthermore, microbiome profiling was performed using MetaPhlAn3 (v.3.0.13). Sequences alignment of clade-specific markers gene and annotation of species pangenomes were performed with Bowtie2 (v.2.4.2). Relative abundance tables of taxa were acquired for further analysis. Microbiome functional pathways were profiled using HUMAnN (v.3.0).
Metabolic pathway analysis
To determine the dysbiotic metabolic functions in the discovery cohort, we used a functional dysbiosis score from Bray–Curtis dissimilarities based on the signature of metabolic functional pathways, as described in a previous study37. Briefly, a ‘reference set’ of samples was constructed from controls; the functional dysbiosis score of a given sample was then defined as the median Bray–Curtis dissimilarity to this reference sample set. To assess the metabolic dysbiosis in different groups, the 90th percentile of the functional dysbiosis scores for control samples was set as the threshold.
Microbiome analysis
Alpha-diversity, such as Shannon index and observed species, was calculated for each sample using the R package phyloseq (v.1.34.0). Beta-diversity analysis based on Bray–Curtis distances was calculated ith the R package vegan (v.2.6.4). Microbiome composition differences among different phenotypes (for example, disease subtype, age and gender) were calculated by permutational multivariate analysis of variance using Bray–Curtis distances with the adonis2 function in the R vegan package (v.2.6.4), with 999 permutations. MaAsLin2 were performed to determine the multivariable association between bacterial species and functional pathways and clinical data (for example, disease, age and gender), with controls as reference, using the R package MaAsLin2 (v.1.4.0). Heat maps were generated using pheatmap (v.1.0.12). Boxplots and violin plots were plotted using the R packages ggpubr (v.0.6.0) and ggplot2 (v.3.4.4). Correlation among disease-associated bacterial species was calculated by Spearman correlation and visualized by pheatmap (v.1.0.12).
Construction of random forest-based diagnostic models
The random forest algorithm was used to build the diagnostic models. The discovery cohort was randomly divided into 70% as the training set and 30% as the testing set. Preselected bacterial species candidates were input to construct the model with fivefold cross-validation. Each time, the weakest feature was recursively removed according to the ‘mean decrease accuracy’ value generated by the random forest algorithm. The ROC AUC value for different feature numbers was then obtained. Following selection of the best-performing features, we trained a model with well optimized hyperparameters (for example, mtry, maxnodes, nodesize, ntree) and fivefold cross-validation to avoid overfitting issues. Model performance was evaluated on a battery of assessment metrics, including AUC, accuracy, sensitivity, specificity and positive and negative predictive values. Independent validation cohorts, public datasets and other non-IBD disease datasets were input to the well-trained model. Results were evaluated by the assessment metrics mentioned above. The probability of disease was generated in the random forest model based on votes by all trees (R packages RandomForest (v.4.6-14), caret (v.6.0-94) and pROC (v.1.18.2)) in this analysis. The Youden index (Y = sensitivity + specificity − 1) was used to determine the optimal cutoff value for the probability of disease in the test set. Kernel Shapley additive explanation (SHAP) values were calculated and visualized using the R packages kernelshap (v.0.3-7) and shapviz (v.0.8-0).
Fecal calprotectin test
Fecal calprotectin levels were measured in samples from 72 patients with IBD (36 UC and 36 CD) and 36 with IBS from in-house sequencing cohorts, as well as from independent validation cohorts from Canada (100 UC, 100 CD, 53 controls) and Taiwan (40 UC, 40 CD, 40 controls), using BÜHLMANN fCAL ELISA assays according to the manufacturer’s protocol. All fecal calprotectin levels measured are represented as micrograms of of calprotectin detected per gram of fecal matter (µg g−1).
m-ddPCR
The abundance of bacterial species biomarkers was determined by newly designed m-ddPCR assays. The sequences of primers and probes used for detectiion of the ten UC bacterial species biomarkers, nine CD bacterial species biomarkers and the reference gene are listed in Supplementary Table 4. The ddPCR mixture consisted of 10 μl of ddPCR Supermix for Probes (no dUTP, Bio-Rad, cat. no. 1863024), primers, probes (allocated combination and probe concentration are listed in Supplementary Table 4), 2 μl of the above-mentioned DNA extracted from fecal sample (diluted to 5 ng μl−1 in reactions 1 and 2, diluted to 0.05 ng μl−1 in reaction 3) and nuclease-free water (to 20 μl). The 20-μl ddPCR mixture and 70-μl Droplet Generation Oil for Probes (Bio-Rad, cat. no. 1863005) were then loaded into DG8 Cartridges (Bio-Rad, cat. no. 1864008). DG8 Gaskets (Bio-Rad, cat. no. 1863009) were then hooked over the cartridge holder. The droplets for each sample were generated using the QX200 Droplet Generator (Bio-Rad) and then transferred to ddPCR 96-well plates (Bio-Rad, cat. no. 12001925). After covering the plate with foil seal (Bio-Rad, cat. no. 1814040) and sealing in PX1 PCR Plate Sealer (Bio-Rad), the 96-well plates were run on the Bio-Rad T100 PCR System. The PCR program was: (1) initial denaturation at 95 °C for 10 min; (2) 40 cycles of denaturation at 94 °C for 30 s, and annealing and extension at 59 °C for 1 min; and (3) enzyme deactivation at 98 °C for 10 min. Following PCR, droplet fluorescence was detected in a QX200 Droplet Reader (Bio-Rad) and data were analyzed by QuantaSoft Analysis Pro (v.1.0.596). The concentration of each bacterial species biomarker was determined and then normalized by the concentration of the 16S reference gene (normalized abundance of target = concentration of the target/concentration of 16S). The random forest models for UC and CD based on ddPCR data were subsequently established for IBD diagnosis, forming the stool microbial biomarker test.
Statistical analyses
Comparison of alpha-diversity and the relative abundance of bacterial species biomarkers between disease and control groups were performed with a nonparametric test such as the Wilcoxon signed-rank test. Correlations between the abundance of bacterial species biomarkers, as determined by metagenomics and m-ddPCR, were given by Spearman correlation. Correlations among selected bacterial species biomarkers and pathways, and correlation between functional dysbiosis scores and probability of disease, were calculated using Spearman correlation. AUC values were determined by random forest models.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The metagenomic sequencing data generated in this study have been deposited in the NCBI Sequence Read Archive database with BioProject no. PRJNA1086048. Other raw metagenomic data are available in the SRA and European Nucleotide Archive under accession nos. PRJNA400072, PRJNA429990 and PRJEB15371. Source Data are provided with this paper.
Code availability
The software packages used in this study are free and open source. The bioBakery tools (including KneadData, MetaPhlAn3 and HUMAnN3) are available via http://huttenhower.sph.harvard.edu/biobakery as source code and installable packages. The code and scripts used for microbiome analyses or figure visualization are available via GitHub at https://github.com/WooJenny27/IBD-project/.
References
GBD 2017 Inflammatory Bowel Disease Collaborators. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 5, 17–30 (2020).
Ng, S. C. Emerging trends of inflammatory bowel disease in Asia. Gastroenterol. Hepatol. (N. Y.) 12, 193–196 (2016).
Ng, S. C. et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet 390, 2769–2778 (2017).
Noble, A. J., Nowak, J. K., Adams, A. T., Uhlig, H. H. & Satsangi, J. Defining interactions between the genome, epigenome, and the environment in inflammatory bowel disease: progress and prospects. Gastroenterology 165, 44–60 (2023).
Ananthakrishnan, A. N. et al. Environmental triggers in IBD: a review of progress and evidence. Nat. Rev. Gastroenterol. Hepatol. 15, 39–49 (2018).
Danese, S., Fiorino, G. & Peyrin-Biroulet, L. Early intervention in Crohn’s disease: towards disease modification trials. Gut 66, 2179–2187 (2017).
D’Haens, G. et al. Early combined immunosuppression or conventional management in patients with newly diagnosed Crohn’s disease: an open randomised trial. Lancet 371, 660–667 (2008).
Jayasooriya, N. et al. Systematic review with meta-analysis: time to diagnosis and the impact of delayed diagnosis on clinical outcomes in inflammatory bowel disease. Aliment. Pharm. Ther. 57, 635–652 (2023).
Zheng, J., Sun, Q., Zhang, J. & Ng, S. C. The role of gut microbiome in inflammatory bowel disease diagnosis and prognosis. United European Gastroenterol. J. 10, 1091–1102 (2022).
Gevers, D. et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 15, 382–392 (2014).
Moayyedi, P. et al. Fecal microbiota transplantation induces remission in patients with active ulcerative colitis in a randomized controlled trial. Gastroenterology 149, 102–109 (2015).
Palmela, C. et al. Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut 67, 574–587 (2018).
Zhang, J. et al. Elucidation of Proteus mirabilis as a key bacterium in Crohn’s disease inflammation. Gastroenterology 160, 317–330 (2021).
Federici, S. et al. Targeted suppression of human IBD-associated gut microbiota commensals by phage consortia for treatment of intestinal inflammation. Cell 185, 2879–2898 (2022).
Zhang, Q. et al. Klebsiella pneumoniae induces inflammatory bowel disease through Caspase-11-mediated IL18 in the gut epithelial cells. Cell. Mol. Gastroenterol. Hepatol. 15, 613–632 (2023).
Kitamoto, S. et al. The intermucosal connection between the mouth and gut in commensal pathobiont-driven colitis. Cell 182, 447–462 (2020).
Fujimoto, T. et al. Decreased abundance of Faecalibacterium prausnitzii in the gut microbiota of Crohn’s disease. J. Gastroenterol. Hepatol. 28, 613–619 (2013).
Takahashi, K. et al. Reduced abundance of butyrate-producing bacteria species in the fecal microbial community in Crohn’s disease. Digestion 93, 59–65 (2016).
Glassner, K. L., Abraham, B. P. & Quigley, E. M. M. The microbiome and inflammatory bowel disease. J. Allergy Clin. Immunol. 145, 16–27 (2020).
Veltkamp, C. et al. Continuous stimulation by normal luminal bacteria is essential for the development and perpetuation of colitis in Tgϵ26 mice. Gastroenterology 120, 900–913 (2001).
Franzosa, E. A. et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol 4, 293–305 (2019).
Ning, L. et al. Microbiome and metabolome features in inflammatory bowel disease via multi-omics integration analyses across cohorts. Nat. Commun. 14, 7135 (2023).
Pratt, M., Forbes, J. D., Knox, N. C., Bernstein, C. N. & Van Domselaar, G. Microbiome-mediated immune signaling in inflammatory bowel disease and colorectal cancer: support from meta-omics data. Front. Cell Dev. Biol. 9, 716604 (2021).
Duvallet, C., Gibbons, S. M., Gurry, T., Irizarry, R. A. & Alm, E. J. Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat. Commun. 8, 1784 (2017).
Çorbacıoğlu Ş, K. & Aksel, G. Receiver operating characteristic curve analysis in diagnostic accuracy studies: a guide to interpreting the area under the curve value. Turk. J. Emerg. Med. 23, 195–198 (2023).
Sauer, S. & Kliem, M. Mass spectrometry tools for the classification and identification of bacteria. Nat. Rev. Microbiol. 8, 74–82 (2010).
Patel, R. MALDI-TOF MS for the diagnosis of infectious diseases. Clin. Chem. 61, 100–111 (2015).
Turnbaugh, P. J. et al. A core gut microbiome in obese and lean twins. Nature 457, 480–484 (2009).
Schloissnig, S. et al. Genomic variation landscape of the human gut microbiome. Nature 493, 45–50 (2013).
Zhang, J., Wang, J., Zhang, X. & He, F. Rapid detection of Escherichia coli based on 16S rDNA nanogap network electrochemical biosensor. Biosens. Bioelectron. 118, 9–15 (2018).
Chen, W. et al. Identification of bacteria in water by a fluorescent array. Angew. Chem. Int. Ed. Engl. 53, 13734–13739 (2014).
Chen, W. et al. Breast cancer subtype classification using 4-plex droplet digital PCR. Clin. Chem. 65, 1051–1059 (2019).
Lewin, A. S. et al. Multiplex droplet digital PCR assay for detection of Flavobacterium psychrophilum and Yersinia ruckeri in Norwegian aquaculture. J. Microbiol. Methods 177, 106044 (2020).
Taly, V. et al. Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients. Clin. Chem. 59, 1722–1731 (2013).
Statistical Guidance on Reporting Results from Studies Evaluating Diagnostic Tests - Guidance for Industry and FDA Staff (Center for Devices and Radiological Health, 2007); https://www.fda.gov/regulatory-information/search-fda-guidance-documents/statistical-guidance-reporting-results-studies-evaluating-diagnostic-tests-guidance-industry-and-fda
Ma, Y. et al. Metagenome analysis of intestinal bacteria in healthy people, patients with inflammatory bowel disease and colorectal cancer. Front. Cell Infect. Microbiol 11, 599734 (2021).
Lloyd-Price, J. et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662 (2019).
Darfeuille-Michaud, A. et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 127, 412–421 (2004).
Vich Vila, A. et al. Gut microbiota composition and functional changes in inflammatory bowel disease and irritable bowel syndrome. Sci. Transl. Med. 10, eaap8914 (2018).
Chung, L. et al. Bacteroides fragilis toxin coordinates a pro-carcinogenic inflammatory cascade via targeting of colonic epithelial cells. Cell Host Microbe 23, 203–214 (2018).
Prindiville, T. P. et al. Bacteroides fragilis enterotoxin gene sequences in patients with inflammatory bowel disease. Emerg. Infect. Dis. 6, 171–174 (2000).
Toprak, N. U. et al. A possible role of Bacteroides fragilis enterotoxin in the aetiology of colorectal cancer. Clin. Microbiol Infect. 12, 782–786 (2006).
Pisani, A. et al. Dysbiosis in the gut microbiota in patients with inflammatory bowel disease during remission. Microbiol. Spectr. 10, e0061622 (2022).
Tremaroli, V. & Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 489, 242–249 (2012).
Brown, E. M., Clardy, J. & Xavier, R. J. Gut microbiome lipid metabolism and its impact on host physiology. Cell Host Microbe 31, 173–186 (2023).
Hiippala, K. et al. Novel Odoribacter splanchnicus strain and its outer membrane vesicles exert immunoregulatory effects in vitro. Front. Microbiol. 11, 575455 (2020).
Schirmer, M., Garner, A., Vlamakis, H. & Xavier, R. J. Microbial genes and pathways in inflammatory bowel disease. Nat. Rev. Microbiol. 17, 497–511 (2019).
Morrison, D. J. & Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 7, 189–200 (2016).
Ji, Y. et al. Insights into diet-associated oxidative pathomechanisms in inflammatory bowel disease and protective effects of functional amino acids. Nutr. Rev. 81, 95–113 (2022).
Su, Q. et al. Faecal microbiome-based machine learning for multi-class disease diagnosis. Nat. Commun. 13, 6818 (2022).
Weng, Y. J. et al. Correlation of diet, microbiota and metabolite networks in inflammatory bowel disease. J. Dig. Dis. 20, 447–459 (2019).
He, Q. et al. Two distinct metacommunities characterize the gut microbiota in Crohn’s disease patients. Gigascience 6, gix050 (2017).
Mars, R. A. T. et al. Longitudinal multi-omics reveals subset-specific mechanisms underlying irritable bowel syndrome. Cell 182, 1460–1473 (2020).
Hall, A. B. et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med. 9, 103 (2017).
Nielsen, H. B. et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 32, 822–828 (2014).
Schirmer, M. et al. Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat. Microbiol 3, 337–346 (2018).
Zhernakova, A. et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 352, 565–569 (2016).
Ijaz, U. Z. et al. The distinct features of microbial ‘dysbiosis’ of Crohn’s disease do not occur to the same extent in their unaffected, genetically-linked kindred. PLoS ONE 12, e0172605 (2017).
Feng, Q. et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat. Commun. 6, 6528 (2015).
Zeller, G. et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol. 10, 766 (2014).
Wirbel, J. et al. Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat. Med. 25, 679–689 (2019).
Yachida, S. et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 25, 968–976 (2019).
Le Chatelier, E. et al. Richness of human gut microbiome correlates with metabolic markers. Nature 500, 541–546 (2013).
Pasolli, E. et al. Accessible, curated metagenomic data through ExperimentHub. Nat. Methods 14, 1023–1024 (2017).
Acknowledgements
We thank J. Mak and A. Chan (Prince of Wales Hospital), C. Ng (Princess Margaret Hospital), M. Li (Tuen Mun Hospital), C. W. Chow (Tseung Kwan O Hospital), E. Shan (Caritas Medical Centre), C. Y. Lam (Kwong Wah Hospital), K. H. Chan (North District Hospital), F. H. Lo (United Christian Hospital), S. F. Sze (Queen Elizabeth Hospital), C. M. Leung (Pamela Youde Nethersole Eastern Hospital), A. J. Hui (Alice Ho Miu Ling Nethersole Hospital) and W. H. Chow (Yan Chai Hospital) for IBD patient recruitment and sample collection. We thank clinical research staff and students for their technical contribution to this study, including clinical data and sample collection, inventory and processing. We thank S. Lin for her assistance in coordinating the shipment of samples from other countries/regions and for helping with sample processing and testing. We thank H. Min Tun and J. Zhu for their assistance in bioinformatic analysis. This research has been conducted using the CU-Med Biobank Resource under Request ID R20221009 and R20231025. S.C.N. has received research funding from the Research Grants Council–Research Impact Fund (RGC-RIF, grant no. R4030-22), New Cornerstone Science Foundation (grant no. NCI202346), InnoHK, The Government of Hong Kong, Special Administrative Region of the People’s Republic of China and the Leona M. and Harry B. Helmsley Charitable Trust (grant no. 2017PG-IBD003). J. Zhang has received research funding from the Health and Medical Research Fund (grant no. 10210816), Research Grants Council-General Research Fund (RGC-GRF, grant no. 14121322) and the National Natural Science Foundation of China (grant no. 82100573). Those authors affiliated with MagIC are partially supported by InnoHK, The Government of Hong Kong, Special Administrative Region of the People’s Republic of China. The funder had no role in design of the study, data collection, data interpretation and analysis or writing of the manuscript.
Author information
Authors and Affiliations
Contributions
S.C.N. and J. Zhang conceived and supervised the study. J.C., C.P.C., A.L.H. and A.L.W.O. contributed to participant recruitment, sample collection and biobank management. S.C.W. and C.N.B. contributed sample materials for validation. J. Zheng, Q. Sun, M.Z., C.L., Q. Su, L.Z., Z.X. and W.L. generated metagenomic data. J.Y.Z., Q. Su, Z.X. and W.T. developed the metagenomic profiling workflows and/or performed taxonomic and functional profiling. J. Zheng performed statistical analysis and/or developed statistical analysis workflows. J. Zheng and C.L. designed and performed validation experiments. J. Zheng and J. Zhang designed and drew the figures. J. Zheng, J. Zhang and S.C.N. wrote the manuscript. C.N.B., D.T.R., E.B.C., M.M., M.A.K. and F.K.L.C. provided critical comments on the manuscript. All authors discussed and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
S.C.N. has served as an advisory board member for Pfizer, Ferring, Janssen and Abbvie and received honoraria as a speaker for Ferring, Tillotts, Menarini, Janssen, Abbvie and Takeda; has received research grants through her affiliated institutions from Olympus, Ferring and Abbvie; is a founder member, nonexecutive director, nonexecutive scientific advisor and shareholder of GenieBiome Ltd; and receives patent royalties through her affiliated institutions. F.K.L.C. is a Board Member of CUHK Medical Centre; is a cofounder, nonexecutive Board Chairman, nonexecutive scientific advisor, Honorary Chief Medical Officer and shareholder of GenieBiome Ltd; receives patent royalties through his affiliated institutions; and has received fees as an advisor and honoraria as a speaker for Eisai Co. Ltd, AstraZeneca, Pfizer Inc., Takeda Pharmaceutical Co. and Takeda (China) Holdings Co. Ltd. Q. Su and Z.X. are Scientists (Diagnostics) of GenieBiome Ltd. W.T. is Consultant (Regulatory Affairs) of GenieBiome Ltd. J. Zhang is Chief Scientist (Diagnostics) of GenieBiome Ltd. J. Zheng, W.T., J. Zhang, F.K.L.C. and S.C.N. are named inventors of patent applications held by MagIC that cover the therapeutic and diagnostic use of microbiome related to IBD (nos. 63/562,232; 63/675,266; 63/689,864 USA, 2024). C.N.B. is supported by the Bingham Chair in Gastroenterology; has served on advisory Boards for AbbVie Canada, Amgen Canada, Bristol Myers Squibb Canada, Ferring Canada, JAMP Pharmaceuticals, Lilly Canada, Janssen Canada, Pendopharm Canada, Sandoz Canada, Takeda Canada and Pfizer Canada; has received educational grants from Abbvie Canada, Bristol Myers Squibb Canada, Ferring Canada, organon Canada, Pfizer Canada, Takeda Canada, Boston Scientific and Janssen Canada; has served on a Speaker’s panel for Abbvie Canada, Janssen Canada, Pfizer Canada and Takeda Canada; and has received research funding from Abbvie Canada, Amgen Canada, Sandoz Canada, Takeda Canada and Pfizer Canada. D.T.R. has received grant support from Takeda and has served as a consultant for Abbvie, Altrubio, Amgen, Bausch Health, Bristol Myers Squibb, Connect BioPharma, Ferring Pharma, Image Analysis Group, Iterative Health, Janssen Pharmaceuticals, Lilly, Merck, Pfizer, Prometheus Biosciences (now Merck), Reistone Biopharma, USA, Takeda and Trellus Health. E.B.C. is Founder and Chief Scientific Officer of Gateway Biome, Inc. M.M. has received research grants from Soho Flordis International Australia Research, Bayer Steigerwald Arzneimittelwerk (Bayer Consumer Health) and Yakult-Nature Global Grant for Gut Health; Speaker’s honoraria and travel sponsorship from Janssen Australia; consultancy fees from Bayer Steigerwald Arzneimittelwerk (Bayer Consumer Health), Sanofi Australia and Danone-Nutricia Australia; Speaker honoraria and travel sponsorship from Perfect Company (China); and travel sponsorship from Yakult Inc. (Japan); is coinventor of PCT/AU2022/050556 ‘Diagnostic marker for functional gastrointestinal disorders’ from the University of Newcastle and UniQuest (University of Queensland), and US20110076356 A1 ‘Novel Fibro-biotic bacterium isolate’ via the US Department of Agriculture; acknowledges funding from NHMRC Australia, Australian Research Council, Princess Alexandra Hospital Research Foundation, Medical Research Futures Fund of Australia, Helmsley Charitable Trust via the Australasian Gastrointestinal Research Foundation and the United States Department of Defense; and serves on the science advisory board (nonremunerated) for GenieBiome, Hong Kong. The other authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Alison Farrell and Liam Messin, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Factors explaining microbiota variance.
Multivariate analysis showing the amount of explained variance and the respective P value determined by PERMANOVA based on Bray-Curtis dissimilarity at species level.
Extended Data Fig. 2 Comparisons of the relative abundance of six phyla among patients with UC (N = 205), CD (N = 174), and controls (N = 118).
Data were shown in boxplots as the median (centre line), 25th and 75th percentiles (box limits), and 5th and 95th percentiles (whiskers). P values were calculated using the two-sided Wilcoxon rank-sum test. CD: Crohn’s disease; UC: Ulcerative colitis.
Extended Data Fig. 3 Performance of model with different number of used features to discriminate patients with UC (N = 205) or CD (N = 174) from controls (N = 118).
a, A total of 125 species features were used in the UC diagnostic model. The vertical dotted line in x = 7 represented the minimum number of features to maintain a relatively stable performance of the model (horizontal dotted line, AUC = 0.8937). b, A total of 161 species features were used in the CD diagnostic model. The vertical dotted line in x = 8 represented the minimum number of features to maintain relatively stable performance of the model (horizontal dotted line, AUC = 0.9096). The AUC values were yield from 5-fold validation. The black dot indicates the mean, and the error bars indicate standard deviation. CD: Crohn’s disease; UC: Ulcerative colitis.
Extended Data Fig. 4 Differential functional pathway between UC/ CD patients and controls, and their correlation with bacterial species biomarkers.
a, Differential functional pathways between UC patients and controls determined by MaAsLin2 with age and gender adjusted. b, Differential functional pathways between CD patients and controls determined by MaAsLin2 with age and gender adjusted. The correlation coefficient and two-sided P value between ten UC or nine CD bacterial species biomarkers and differential functional pathways were given by Spearman correlation. *, p < 0.05; **, p < 0.01; ***, p < 0.001. CD: Crohn’s disease; UC: Ulcerative colitis.
Extended Data Fig. 5 Functional pathways in UC and CD patients compared with controls.
a-b, Contribution of bacterial species biomarkers in differential functional pathways in UC (N = 205) and CD patients (N = 174) compared with controls (N = 118). The stacked bar plot indicates the contribution of bacterial species biomarkers and other bacteria in each sample. Data were shown in boxplots as the median (centre line), 25th and 75th percentiles (box limits), and 5th and 95th percentiles (whiskers). The gray diamond represents the mean value. c, Distribution of functional dysbiosis scores determined by median Bray-Curtis dissimilarity between a sample and controls. The dash line indicates the 90th percentile of the functional dysbiosis scores for controls samples. d, Comparison of functional dysbiosis scores among UC (N = 205), CD (N = 174) and controls (N = 118). The dash lines in violin plot represent Q1, median, Q3. P values were calculated using the two-sided Wilcoxon rank-sum test. CD: Crohn’s disease; UC: Ulcerative colitis.
Extended Data Fig. 6 Relative abundance of bacterial species biomarkers in controls and patients at inactive and active status.
a, The relative abundance of ten UC bacterial species biomarkers in controls and UC patients at inactive and active status. b, The relative abundance of nine CD bacterial species biomarkers in controls and CD patients at inactive and active status. e, Comparison of the probability of disease calculated by the random forest model between CD patients at inactive (N = 69) and active (N = 9) status. c, Comparison of the probability of disease calculated by the random forest model between UC patients at inactive (N = 110) and active (N = 11) status. d, Model performance in distinguishing inactive UC patients (N = 40) and controls (N = 42). f, Model performance in distinguishing inactive CD patients (N = 69) and controls (N = 108). Shaded areas of the ROC curves represent the 95% confidence interval of the AUC for each cohort. Data were shown in boxplots as the median (centre line), 25th and 75th percentiles (box limits), and 5th and 95th percentiles (whiskers). The gray diamond represents the mean value. P values were given by the two-sided Wilcoxon rank sum test. CD: Crohn’s disease; UC: Ulcerative colitis; IBS, Irritable bowel syndrome.
Extended Data Fig. 7 Abundance and prevalence of bacterial species biomarkers and the performance of diagnostic models in cohorts from different ethnicities and regions.
a-b, Signature of bacterial species biomarkers for UC and CD diagnosis in patients and healthy individuals of discovery cohort, validation cohort, and three downloaded public datasets. The abundance of species was normalized to log2 fold change (log2FC) relative to the mean of control samples. P values were calculated using the two-sided Wilcoxon rank-sum test. P values were then converted to -log10(P-value) after using Benjamini–Hochberg correction to control for multiple testing. Prevalence indicates the proportion of bacterial presence in UC, CD, and healthy group of each cohort. c, The probability of disease calculated by the random forest model between UC/CD patients and controls in Hong Kong discovery cohort (205 UC,174 CD, 118 controls), validation cohort from Hong Kong (139 UC, 139 controls; 92 CD, 108 controls) and Australia (98 CD, 81 controls), and public datasets from the United States (53 UC, 68 CD, 34 controls), Netherlands (23 UC, 20 CD, 22 controls) and mainland China (25 UC, 15 controls; 48 CD, 54 controls). Data were shown in boxplots as the median (centre line), 25th and 75th percentiles (box limits), and 5th and 95th percentiles (whiskers). P values were calculated using the two-sided Wilcoxon rank-sum test. d, Correlation among the ten UC bacterial species biomarkers. UC-depleted bacteria were labelled with green color while the UC-enriched ones were labeled with yellow color. e, Correlation among the nine CD bacterial species biomarkers. CD-depleted bacteria were labelled with green color while the CD-enriched ones were labeled with orange color. Grids in red indicated positive correlation, while grids in blue indicated negative correlation. The correlation coefficient and two-sided P value were given by Spearman correlation. *p < 0.05, **p < 0.01, ***p < 0.001. CD: Crohn’s disease; UC: Ulcerative colitis.
Extended Data Fig. 8 Relative abundance of bacterial species biomarkers in UC/CD and other non-IBD disease groups in Hong Kong cohort.
a, Relative abundance of 10 UC bacterial species biomarkers in UC (N = 205) and other non-IBD disease group (162 CA, 160 CRC, 117 IBS-D, 148 obesity, 143 CVD, 118 Controls). b, Relative abundance of 9 CD bacterial species biomarkers in CD (N = 174) and other non-IBD disease group (162 CA, 160 CRC, 117 IBS-D, 148 obesity, 143 CVD, 118 Controls). Data were shown in boxplots as the median (centre line), 25th and 75th percentiles (box limits), and 5th and 95th percentiles (whiskers). The gray diamond represents the mean value. P values were calculated using the two-sided Wilcoxon rank-sum test. CD: Crohn’s disease; UC: Ulcerative colitis; IBS-D, Irritable bowel syndrome (diarrhea subtype); CA, Colorectal adenomas; CRC, Colorectal cancer; CVD, Cardiovascular disease.
Extended Data Fig. 9 Performance of general IBD model in classifying IBD from and non-IBD subjects.
a, ROC of general IBD model in classifying IBD from controls and non-IBD in test set, IBD validation cohort, and non-IBD cohort. b, Prototypical standards for reporting diagnostic accuracy studies (STARD) diagram reporting the flow of participants in independent international IBD cohort (IBD = 1882, Controls=2027). c, Comparison of diagnostic performance of general IBD model and fecal calprotectin in classifying IBD from and IBS subjects. Shaded areas of the ROC curves represent the 95% confidence interval of the AUC for each cohort. IBD, Inflammatory Bowel Disease; IBS, Irritable bowel syndrome.
Extended Data Fig. 10 Difference of probability of disease (POD) calculated by metagenomics-based model and m-ddPCR-based model for UC and CD.
CD: Crohn’s disease; UC: Ulcerative colitis.
Supplementary information
Supplementary Information
Supplementary Tables 1–4.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 7
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zheng, J., Sun, Q., Zhang, M. et al. Noninvasive, microbiome-based diagnosis of inflammatory bowel disease. Nat Med 30, 3555–3567 (2024). https://doi.org/10.1038/s41591-024-03280-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-024-03280-4
This article is cited by
-
Intestinal microbiota profiles of captive-bred cynomolgus macaques reveal influence of biogeography and age
Animal Microbiome (2025)
-
Deciphering microbial and metabolic influences in gastrointestinal diseases-unveiling their roles in gastric cancer, colorectal cancer, and inflammatory bowel disease
Journal of Translational Medicine (2025)
-
From gut inflammation to psychiatric comorbidity: mechanisms and therapies for anxiety and depression in inflammatory bowel disease
Journal of Neuroinflammation (2025)
-
Elevated drain fluid calprotectin as a predictor of early anastomotic leakage in colorectal surgery
World Journal of Surgical Oncology (2025)
-
[1]The human gut microbiota in IBD, characterizing hubs, the core microbiota and terminal nodes: a network-based approach
BMC Microbiology (2025)
