
Abstract
In contrast to chimeric antigen receptor T cells, T cell receptor (TCR)-engineered T cells can target intracellular tumor-associated antigens crucial for treating solid tumors. However, most trials published so far show limited clinical activity. Here we report interim data from a first-in-human, multicenter, open-label, 3 + 3 dose-escalation/de-escalation phase 1 trial studying IMA203, an autologous preferentially expressed antigen in melanoma (PRAME)-directed TCR T cell therapy in HLA-A*02+ patients with PRAME+ recurrent and/or refractory solid tumors, including melanoma and sarcoma. Primary objectives include the evaluation of safety and tolerability and the determination of the maximum tolerated dose (MTD) and/or recommended dose for extension. Secondary objectives include the evaluation of IMA203 TCR-engineered T cell persistence in peripheral blood, tumor response as well as duration of response. A total of 27 patients were enrolled in the phase 1a dose escalation and 13 patients in the phase 1b dose extension. IMA203 T cells were safe, and the MTD was not reached. Of the 41 patients receiving treatment (that is, who started lymphodepletion), severe cytokine release syndrome was observed in 4.9% (2/41), and severe neurotoxicity did not occur. In the 40 patients treated with IMA203, an overall response rate consisting of patients with unconfirmed or confirmed response (u/cORR) of 52.5% (21/40) and a cORR of 28.9% (11/38) was observed with a median duration of response of 4.4 months (range, 2.4–23.0, 95% confidence interval: 2.6–not reached) across multiple indications. Rapid T cell engraftment and long-term persistence of IMA203 T cells were observed. IMA203 T cells trafficked to all organs, and confirmed responses were more frequent in patients with higher dose. T cell exhaustion was not observed in the periphery; deep responses were enriched at higher PRAME expression; and higher T cell infiltration resulted in longer progression-free survival. Overall, IMA203 showed promising anti-tumor activity in multiple solid tumors, including refractory melanoma. ClinicalTrials.gov identifier: NCT03686124.
Similar content being viewed by others

Main
Due to the limited success of chimeric antigen receptor T (CAR T) cells in solid cancers, T cell receptor T cell (TCR T) therapies have emerged to target intracellular tumor-associated antigens, presented on the surface of tumor cells by human leukocyte antigens (HLAs)1,2,3. Initial clinical studies have revealed that TCR T cells are effectively trafficked to solid tumor sites and can demonstrate anti-tumor activity4,5,6,7. However, most trials published so far included limited patient numbers and were restricted to rare disease entities based on the expression profile of the target antigen8,9.
Preferentially expressed antigen in melanoma (PRAME) is a cancer/testis antigen that has been detected not only in malignant melanoma but also in multiple other solid tumors10,11,12,13. Its expression in healthy tissues is limited to testis and at low RNA levels in the endometrium, ovaries, adrenal glands and a few other tissues10. HLA-complexed peptides derived from PRAME were first identified as targets for cytotoxic T cells presented by HLA-A*24 on melanoma cells10 and later described to be also potentially presented by HLA-A*02:01 (ref. 14).
Here we report a non-prespecified interim analysis of IMA203, an autologous TCR T product targeting a PRAME-derived peptide presented by HLA-A*02:01, in a first-in-human dose-escalation trial (NCT03686124). This analysis includes results from patients treated in the dose escalation (phase 1a) and in the dose extension (phase 1b), demonstrating safety, tolerability and initial anti-tumor activity. Data from 40 infused patients with a diverse set of advanced solid cancers are described. The trial is ongoing.
Results
Target identification and expression in solid tumors
The PRAME-derived HLA-A*02:01-presented target peptide PRAME-004 with the peptide sequence SLLQHLIGL was identified using a mass spectrometry (MS)-based antigen discovery platform15. Abundant PRAME-004 presentation could be demonstrated on patient-derived native (that is, unmodified, shock-frozen) primary tumor tissue and metastases while not quantifiable on 41 different normal tissue types (Supplementary Fig. 1). The mRNA threshold (set as described previously16) equals to PRAME target peptide copy numbers of approximately 40–50 copies per cell according to quantitative peptide presentation analysis. Applying this cutoff to The Cancer Genome Atlas (TCGA) RNA sequencing (RNA-seq) and own quantitative real-time (qRT) polymerase chain reaction (PCR) expression data, a high prevalence of PRAME expression across various tumor types could be confirmed (Supplementary Table 1). In indications such as cutaneous melanoma, synovial sarcoma, uterine carcinoma and ovarian carcinoma, expression of the target antigen was particularly strong and could be demonstrated in most patients (Extended Data Fig. 1). PRAME expression was found to be homogeneous applying mRNA in situ hybridization (Extended Data Fig. 2).
Derivation and optimization of the IMA203 TCR
The IMA203 TCR was derived from a natural TCR targeting PRAME-004, identified by screening of more than 20 healthy donors using in vitro priming of T cells followed by single-cell sorting and rapid amplification of complementary DNA (cDNA) ends (RACE) for individual TCR α-chains and β-chains. Fifty-two TCRs were selected for characterization after re-expression in healthy donors. Among those, the parental TCR for IMA203 recognized PRAME-004 peptide-loaded T2 cells with relatively high avidity (half-maximal effective concentration (EC50) of 0.8 nM; Supplementary Fig. 2a, left panel). The engineered TCR used for IMA203 is a pairing-optimized and affinity-enhanced variant of the parental TCR, with approximately fourfold enhanced avidity toward peptide-loaded T2 cells (EC50 = 0.2 nM; Supplementary Fig. 2a, right panel). The engineering comprised only two charge-complementary single amino acid substitutions at the α-chain (W44K) and β-chain (Q44E) interface for promoting pairing of both chains through electrostatic interactions. We used rational design for identifying these interface substitutions, and, despite their locations being distant from the TCRʼs complementarity-determining regions (CDRs), they enhanced the binding properties of the TCR, potentially by improving overall TCR stability. As a result, the engineered IMA203 TCR exhibited an improved binding affinity for PRAME-004 (5 µM by biolayer interferometry analysis compared to 18 µM for the parental TCR; Supplementary Fig. 2b,c).
The IMA203 TCR has a broad binding motif, recognizing six out of the nine target peptide residues, as determined by positional scanning. The TCR did not react against a set of 10 naturally expressed, MS-detected normal tissue peptides with high sequence similarity to the target peptide (Supplementary Fig. 3a). For more in-depth analysis of specificity, the human proteome and immunopeptidome were queried for peptides with amino acids matching the TCR binding motif, resulting in 57 predicted HLA-A*02 ligands. The IMA203 TCR only reacted weakly against two of those peptides (RASEF-001 and ATP-009; Supplementary Fig. 3b). Subsequent de-risking revealed that both hits were not MS detected on normal HLA-A*02 tissue. RASEF-001 corresponds to a minor single-nucleotide variant rarely expressed, and titration experiments with ATP-009 showed for the IMA203 TCR a more than 10,000-fold difference in response compared to the PRAME-004 target (Supplementary Fig. 3c). Peptide characteristics and weak recognition argued against a physiological relevance for the observed cross-reactivities. The engineered IMA203 TCR showed improved recognition of tumor cell lines with low endogenous PRAME-004 target expression (Supplementary Fig. 3d) but no reactivity toward non-malignant human primary cells (Supplementary Fig. 3e), except for a very low signal on human cortical renal epithelial cells with one of two donors. It is noteworthy that, so far, no specific renal toxicity has been observed in patients of the ongoing clinical trial.
Trial design and patient characteristics
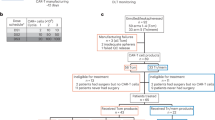
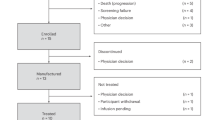
Between 17 April 2019 and 7 December 2022, a total of 399 patients were enrolled, of whom 85 HLA-A*02:01+ patients with advanced PRAME+ solid tumors underwent leukapheresis. Of these, 41 patients started treatment—that is, lymphodepletion—and 40 patients received IMA203. A total of 27 patients were treated in phase 1a dose escalation across four dose levels (DLs), ranging from 0.04 × 109 to 1.2 × 109 per m2 TCR T cells. Patients dosed between two DLs were defined as enrichment cohorts (ECs). Because we observed manageable toxicity until DL4 after clearance of this level, a protocol amendment was introduced to expand to higher doses. An additional 13 patients were treated in phase 1b dose extension, aimed to further evaluate DL4 and a higher DL5 (doses of 1.2 × 109 to 4.7 × 109 per m2 TCR T cells). A comprehensive overview on patient allocation can be found in Fig. 1.

CONSORT diagram indicating the number of patients screened and enrolled in the IMA203-101 trial and reasons for ineligibility. Diagram includes patients with first screening until 7 December 2022. *Patient was included into Supplementary Table 4 (safety data of IMA203) as the patient had treatment-related AEs due to lymphodepletion. ICF, informed consent form.
Patients with melanoma (n = 16, including cutaneous melanoma, n = 11; uveal melanoma, n = 3; unknown primary, n = 1; and mucosal melanoma, n = 1) were most frequent, followed by synovial sarcoma (n = 8), head and neck squamous cell carcinoma (HNSCC, n = 5) and ovarian carcinoma (n = 4). The median age was 53.5 years (range, 18–79). Patients were heavily pre-treated with a median of four prior lines of systemic therapies. Median tumor burden (sum of diameters of target lesions) was assessed at 108.9 mm, and 60% of patients had serum lactate dehydrogenase (LDH) levels above the upper limit of normal (ULN) at baseline. Of note, 42.5% of patients had liver lesions, and 20% had pre-existing brain lesions, which were required to be asymptomatic and stable before IMA203 treatment (Table 1).
Patients treated in phase 1a dose escalation received a median total dose of 0.409 × 109 TCR T (range, 0.078 × 109 to 2.090 × 109) across DL1–DL4, whereas the median dose applied in phase 1b dose extension was considerably higher (median, 4.16 × 109 TCR T cells; range, 1.3 × 109 to 8.84 × 109). Due to modification of the eligibility criteria for dose extension, patients in phase 1b showed slightly more favorable characteristics with a lower median number of prior treatment lines, lower LDH levels and lower tumor burden. In combination with the higher TCR T cell doses applied, this resulted in a higher TCR T cell dose to tumor burden ratio (Table 1). The respective information for patients with melanoma treated at DL4 and DL5 can be found in Extended Data Table 1 and, for individual patients, in Supplementary Table 2.
Manufacturing characteristics of TCR T drug product
IMA203 was manufactured (Supplementary Fig. 4) from autologous peripheral blood mononuclear cells (PBMCs; older process) in 7 d with an overall success rate of 93%. Four product failures occurred due to technical errors but were successfully re-manufactured and released. The success rate increased to 98% in phase 1b dose extension through modifications of the manufacturing protocol, primarily co-enrichment of CD4+ and CD8+ T cells from frozen leukapheresis. Heterogeneous source material contained a median 35.3% of CD3+ T cells (range, 7–54); the final product was highly enriched in T cells (median, 97.4% viable CD3+ cells) and nearly depleted of B cells, natural killer (NK) cells, NK T cells and γδ T cells as well as monocytes (grouped median, 2.2%) (Supplementary Fig. 5a). The final drug products had median 47.2% dextramer-positive (Dex+) out of CD3+ CD8+ T cells, median 5 vector copies per transduced cell and median 89.9% post-thaw viability (Supplementary Fig. 5b). A summary of individual patient product characteristics can be found in Supplementary Table 3.
The manufacturing process did not significantly alter the percentages of naive (TN) or central memory (CM) phenotype CD8+ T cells but led to a significant reduction of effector memory T cells re-expressing CD45RA (TEMRA), and T effector memory (TEM) cells increased from starting material to final product (Extended Data Fig. 3a–d). The percentages of CD62L-expressing, CD28-expressing and CD45RO-expressing CD8+ T cells increased, and CD57+ cells were significantly less abundant in the final product. Compared to apheresis, no significant change was observed in the ratio of CD27+ among CD8+ T cells; drug products had, on average, fewer programmed cell death protein (PD)-1+ cells but highly elevated T cell immunoglobulin and mucin ___domain (TIM)-3 and lymphocyte activation gene (LAG)-3 levels, whereas T cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibition motif ___domain (TIGIT) expression remained relatively the same (Extended Data Fig. 3e–m).
Drug products demonstrated in vitro cytotoxicity against high, medium or low PRAME-expressing tumor cell lines (Supplementary Fig. 6). However, a higher effector-to-target ratio was required to eliminate PRAMElow tumor cell lines.
Primary endpoint: safety
Safety analysis comprised 41 patients who received at least one dose of lymphodepletion, including one patient not receiving IMA203 due to an intercurrent infection. All patients experienced at least one treatment-emergent adverse event (TEAE) (Table 2 and Supplementary Table 4), predominantly of mild to moderate intensity. The most common TEAEs with higher severity (≥grade 3) were cytopenias associated with lymphodepletion (neutropenia (82.9%), lymphopenia (61%) and leukopenia (56.1%)). Almost all patients (92.7%) experienced cytokine release syndrome (CRS), mostly of grade 1 (46.3%) or grade 2 (41.5%). Only two patients (4.9%) had grade 3 CRS (one occurred approximately 6 weeks after an experimental second IMA203 infusion and several months after the initial infusion). Both events improved to grade 2 or lower within 3 d and 5 d. These two patients did not require vasopressor or ventilatory support, with grade 3 CRS being defined by grade 3 rash and grade 4 transaminitis, respectively, according to Neelapu et al.17. No grade 4 or grade 5 CRS events were observed within this trial, and no treatment-related patient deaths were reported. The median time to onset of CRS was 1 d (range, 0–6) with CRS occurring after the first administration of interleukin (IL)-2 in most patients. In seven patients, CRS was observed already at day 0 before the first application of IL-2. The median duration of CRS was 9 d (range, 3–51; Supplementary Fig. 7). In patients with CRS, the IL-2 dose was reduced in two patients (5%), temporarily interrupted in 10 patients (26.3%) and discontinued early in seven patients (18.4%). Tocilizumab was used for management of CRS in 36.8% (14/38) of patients who experienced any grade of CRS. The duration of these CRS cases was 9.5 d (range, 3–36), and CRS was fully resolved after a median of 4.0 d after first tocilizumab administration (range, 0–30). Overall, CRS events were transient and did not reveal a clear dose dependency or response dependency (Supplementary Fig. 8).
Neurotoxicity in the form of mild to moderate immune effector cell-associated neurotoxicity syndrome (ICANS) was reported in five patients (12.2%), all treated during dose escalation (DL1 n = 1, EC1 n = 1, DL2 n = 1, DL3 n = 2). ICANS occurred at a median of 6 d (range, 5–7) after IMA203 infusion and was preceded by CRS in all five patients. ICANS was managed with short courses of steroids (9 d and 5 d) in two of the patients. All five events resolved without sequelae within a median of 3 d (range, 1–11). No grade 3 or higher neurotoxicities occurred. Analyses of serum cytokines revealed trends such as higher peak serum levels of tumor necrosis factor (TNF) and monocyte chemoattractant protein (MCP)-1 in patients with CRS grade 2 and grade 3. Furthermore, higher peak levels of serum IL-18 were observed in patients with ICANS (Supplementary Fig. 9).
A single dose-limiting toxicity (DLT) was observed with a patient treated at DL2 experiencing grade 3 atrial fibrillation, hypotension with hemodynamic instability and transfer to intensive care unit. The event resolved in 3 d after treatment with intravenous fluids, low-dose phenylephrine, amiodarone and tocilizumab. A formal maximum tolerated dose (MTD) was not reached for IMA203 and the highest dose levels (DL4/DL5), and an equivalent range of 1 × 109 to 10 × 109 total transduced TCR T cells was selected for further clinical development.
Secondary endpoint: clinical efficacy
Until the data cutoff on 4 July 2023, all 40 patients treated with IMA203 TCR T cells had at least one tumor response assessment after T cell infusion. Target lesions were evaluable for 39 patients (Fig. 2a). For patients treated during the phase 1a dose-escalation part (n = 27), the overall response rate (ORR) consisting of patients with unconfirmed and confirmed response (u/cORR) according to Response Evaluation Criteria in Solid Tumors (RECIST) 1.1 was 48.1% (13/27; 95% confidence interval (CI): 28.7–68.1), and the cORR equaled 18.5% (5/27; 95% CI: 6.3–38.1). The median duration of response (mDOR) in this cohort was 4.4 months (range, 2.4–23.0; 95% CI: 2.4–not reached); the median progression-free survival (PFS) was 2.8 months (range, 1.3–24.4; 95% CI: 2.4–3.1); and the median overall survival (OS) was 7.5 months (range, 1.9–34.2+; 95% CI: 4.0–10.9). For patients treated in the phase 1b dose extension (n = 13), the u/cORR increased to 61.5% (8/13; 95% CI: 31.6–86.1) with a cORR of 54.5% (6/11; 95% CI: 23.4–83.3). At the time of data cutoff, four of eight responses remained ongoing, with two patients being in partial response (PR) more than a year after T cell infusion (Fig. 2b). With a median follow-up of 11.5 months, the mDOR was 8.1 months (range, 2.6–11.8+; 95% CI: 2.6–not reached), and the median PFS was 5.7 months (range, 1.2–13.1+; 95% CI: 2.4–not reached). The median OS was not reached at data cutoff (Table 3).

a, Best percent change in sum of diameter of target lesions from baseline and BOR by RECIST 1.1 in IMA203 monotherapy population. Each bar represents an individual patient. Left, BOR of patients treated in the dose escalation (DL1–DL4; n = 27). #Synovial sarcoma patient 19 (DL3) PD at week 6 is not shown as target lesions were not evaluable. Right, BOR of patients treated in the dose extension (DL4 and DL5; n = 13). Ovarian cancer patient 35 (DL5) erroneously received one dose of nivolumab. *Maximum change of target lesions and RECIST 1.1 response at different timepoints. b, Percent change in sum of diameter of target lesions from baseline over time. Left, response over time in IMA203 monotherapy dose-escalation population (n = 27). #Synovial sarcoma patient 19 (DL3) PD at week 6 is not shown as target lesions were not evaluable. Right, response over time in IMA203 monotherapy dose-extension population (n = 13). Ovarian cancer patient 35 (DL5) erroneously received one dose of nivolumab. *Response of patient 30 (DL4) until 5.7 months after infusion; target lesion response assessment is not available (external assessment). Each line represents one patient, with the dots representing tumor assessments. The arrow indicates ongoing response at data cutoff. Colors indicate BOR according to RECIST1.1. The data show deeper and more durable responses in the dose-extension cohort compared to dose escalation. BL, baseline; BOR, best overall response; cPR, confirmed partial response; Cut, cutaneous; MPNST, malignant peripheral nerve sheath tumor; NET, neuroendocrine tumor; NSCLC, non-small cell lung cancer; PD, progressive disease; SCN, small cell neuroendocrine; SD, stable disease; Unk, unknown.
A post hoc analysis of patients with melanoma (n = 10) treated at DL4/DL5 across phase 1a and phase 1b cohorts revealed a u/cORR of 70% (7/10; 95% CI: 34.8–93.3) with a cORR of 50% (5/10; 95% CI 18.7–81.3). The mDOR for this cohort has not been reached at data cutoff, with three of seven responses ongoing at 9+ months (one patient) and 12+ months (two patients) after T cell infusion as of data cutoff (Fig. 2b and Table 3).
Clinical predictors of response to IMA203
Response to IMA203 was not limited to certain body organs, suggesting successful trafficking of TCR T cells to lung, liver, pleura, peritoneum, skin, lymph node, adrenal gland, bladder, kidney, spleen and muscle (Extended Data Fig. 4a). Representative durable responses are shown in Extended Data Fig. 4b.
No significant association between number of prior lines of treatment or baseline LDH with response was observed at data cutoff (Supplementary Fig. 10). However, confirmed responses were significantly more frequent in patients with higher administered dose, lower baseline tumor burden and, therefore, a higher TCR T dose to tumor burden ratio (Extended Data Fig. 5a).
Even though confirmed responses were observed across a wide range of PRAME expression levels above the prospectively defined expression threshold, higher PRAME expression was seen in confirmed responders (Extended Data Fig. 5b). When all responders with confirmed and unconfirmed PR were compared to non-responders, the difference was statistically significant (Supplementary Fig. 11). A significant correlation was also demonstrated between PRAME expression and depth of response, and a trend toward longer PFS was noted in patients with higher PRAME expression (Extended Data Fig. 5c).
Secondary endpoint: IMA203 pharmacokinetics
Pharmacokinetics characterization (Supplementary Table 5) of IMA203 revealed rapid IMA203 T cell engraftment in all patients (median peak day, Tmax: 3.5; range, day 1 to day 15) irrespective of tumor indication (Extended Data Fig. 6). Higher T cell dose led to higher maximal IMA203 transgene levels in blood (Cmax), and higher Cmax was significantly associated with a higher percentage of TCR+ T cells in the drug product (Supplementary Fig. 12). IMA203 in peripheral blood declined over time, but no patient showed complete loss during the period of assessment. The persistence of IMA203 T cells was observed for more than 2 years in two patients who underwent long-term follow-up. No correlation was observed between response and Cmax or integrated transgene levels over the first 4 weeks (AUC0–28d) at data cutoff. However, Cmax and AUC0–28 normalized to tumor burden were higher in responders compared to non-responders (Supplementary Table 5 and Supplementary Fig. 13).
Dynamics of IMA203 immunophenotype (exploratory endpoint)
In the IMA203 T cell product, with the exception of TEMRA, the relative abundance of TN, CM and TEM within the transduced CD8+ compartment was not associated with response and neither was the expression of activation/differentiation markers (CD62L, CD27, CD28, CD45RO, CD57) or activation/exhaustion markers (PD-1, TIM-3, LAG-3, TIGIT) (Supplementary Fig. 14).
In patients’ blood, a gradual increase in TEMRA associated with a decrease in TN, CM and TEM subsets after infusion was observed, suggesting in vivo activation and differentiation of IMA203 cells (Supplementary Fig. 15). Two weeks after infusion, responders trended to retain a higher degree of CM phenotype as compared to non-responders (Extended Data Fig. 7a). After infusion, in vivo activation of transduced CD8+ T cells could be followed by loss of CD62L, CD27 and CD28 expression over time, whereas temporal upregulation of CD45RO and gradual increase of CD57 were seen (Supplementary Fig. 16). Two weeks after infusion, responders trended to have more CD27-expressing and CD45RO-expressing transduced TCR T cells, and depth of response trended to correlate with higher CD27 and CD45RO expression at week 2 (Extended Data Fig. 7b).
A transient upregulation of PD-1 and TIGIT and a gradual downregulation of TIM-3 and LAG-3 expression in peripheral blood T cells after IMA203 infusion were observed (Supplementary Fig. 17). Two weeks after infusion, upregulation of PD-1 was more prominent in responders together with upregulation of TIGIT by trend compared to non-responders, and depth of response correlated with ratio of upregulation of PD-1 and TIGIT expression at week 2 compared to final product (Extended Data Fig. 7c). None of these activation/exhaustion-related markers (PD-1, TIM-3, LAG-3, TIGIT) was upregulated 8 weeks after infusion, and, thus, no evidence of IMA203 exhaustion was observed in the periphery.
IMA203 in tumor microenvironment (exploratory endpoint)
T cell infiltration into tumor tissues was tested through preplanned post-infusion biopsies available from 21 treated patients. TCR-transgenic T cells could be detected in all evaluable post-infusion biopsies except one independent of the cancer type. Responders were shown to have a significantly higher degree of IMA203 infiltration than non-responders, and a higher degree of tumor infiltration correlated with deeper response and longer PFS (Fig. 3).

T cell infiltration correlates with clinical efficacy. a, Bar graph (mean ± s.d.) showing IMA203 TCR T cell infiltration into tumors in patients at day 42 post-infusion biopsies (n = 22). Genomic DNA (gDNA) isolated from pre-infusion and post-infusion biopsies was analyzed by qPCR using lentiviral Psi sequence-specific primers, and the results are expressed as vector copies detected per microgram (μg) of gDNA. Two-tailed Mann–Whitney statistical test was used. The P values are depicted in the respective graphs. b, IMA203 T cell infiltration (vector copies/μg gDNA) values for each patient with post-infusion biopsies plotted against depth of response max % change in sum of the longest diameter of target tumor lesions from baseline (n = 22). c, Best % change in sum of diameter of target lesions compared to baseline according to RECIST 1.1 response plotted against PFS. The P value was determined by two-sided Spearman correlation. The P values and correlation coefficients are depicted in the respective graphs. Each point represents one patient and the respective BOR (color-coded) according to RECIST 1.1. Triangles indicate censored patients (PFS; n = 39). BOR, best overall response; cPR, confirmed partial response; NR, non-responder; PD, progressive disease; R, responder; SD, stable disease.
Serial—that is, paired pre-treatment and post-treatment—IMA203 biopsies were available in 13 patients, and, although the data are limited, no evidence of general PRAME downregulation was found. Numerically, PRAME expression values were lower in the post-treatment biopsies in four out of 13 patients (two responders and two non-responders) but higher in biopsies in five out of 13 patients (two responders and three non-responders) (Supplementary Fig. 18).
An altered expression profile for gene products modulating antigen presentation was observed in responders (four out of five) with at least twofold higher expression noted in post-treatment biopsies compared to pre-treatment biopsies, whereas most of the gene products remained unchanged in non-responders without any apparent loss nor significant downregulation of HLA-A or β2-microglobulin (β2M) expression (Extended Data Fig. 8). Because many of these genes (ERAP1, ERAP2, FCGR1A, IFI30, TAPBP) are interferon (IFN)-γ inducible, data suggest active IFN-γ signaling in the tumor microenvironment of responders. Similarly, genes that are responsible for negative regulation of T-cell-mediated immunity (LILRB, VISTA) and T cell exhaustion (FCGR2B, CD244, HAVCR2, KLRC1, LAG3, TIGIT) were also upregulated in the responders along with immune checkpoint molecules with IFN-γ-dependent regulation (CD274, PDCD1LG2) (Extended Data Fig. 9).
Discussion
The results of this phase 1 trial provide a strong rationale to further evaluate IMA203 TCR T cells targeting PRAME-004 with the pairing-optimized, affinity-enhanced and highly selective IMA203 TCR in an autologous setting across multiple solid tumors. IMA203 was found to be safe and well tolerable, and no treatment-related fatalities occurred in 40 patients receiving up to 8.8 × 109 TCR T cells. Consistent with other autologous T cell therapies, most patients experienced CRS, but higher-grade CRS (≥grade 3) was uncommon and occurred in only 5% of patients. This is in line with other TCR T trials7,18 but lower than with CAR T cell therapies19. Neurotoxicity was rare (12% of the patients), with no patient experiencing grade 3 or higher ICANS. Again, this is similar to other TCR T cell products targeting melanoma-associated antigen (MAGE)-A4, MAGE-A10 and NY-ESO1 describing ICANS in 0–20% of patients with infrequent or no grade 3 or higher events9,20,21,22,23,24. In contrast, neurotoxicity of grade 3 or higher was reported in 10–22% of patients treated with approved CD19 CAR T cell products25,26,27.
Within phase 1a dose escalation, we observed a u/cORR of 48.1%, whereas the cORR was only 18.5%. One possible explanation for this observation is a transient response of the tumor to the chemotherapeutics used for lymphodepletion. There is, however, a documented lack of activity of fludarabine in the disease entities that responded to IMA203 (refs. 28,29). Data on single-agent cyclophosphamide are limited, but some evidence suggests that it may induce responses in ovarian cancer30 and soft tissue sarcoma31. There are no data supporting a role for cyclophosphamide in the treatment of melanoma; in fact, this entity is perceived to be largely chemotherapy resistent32. In addition, we used a similar lymphodepleting regimen in the IMA202-101 trial, testing a MAGE-A1-directed TCR T product, and did not see a single response in a cohort of 16 patients, including melanoma, sarcoma and HNSCC33. Of note, the gap between initial and cORR was less pronounced in phase 1b treating all patients at DL4/DL5 (u/cORR 61.5% versus cORR 54.5%), which indicates that higher doses are needed to achieve higher response rates.
The mDOR was 4.4 months in phase 1a dose escalation and 8.1 months in phase 1b dose extension with responses exceeding 12 months, suggesting a clear dose–response relationship. Clinical responses were observed in heavily pre-treated cutaneous, mucosal and uveal melanoma, sarcoma, HNSCC and ovarian cancer, suggesting that IMA203 has therapeutic potential across immunologically hot tumors (melanoma and head and neck) and cold tumors (ovarian and synovial sarcoma)34.
In a previous phase 1, multi-indication, solid tumor basket trial, a MAGE-A1-targeting TCR T cell product (called IMA202) that shared many features with IMA203 was administered to a similar patient population. The therapy demonstrated manageable safety but only limited clinical activity33. Apart from targeting a different cancer/testis antigen, the enhanced clinical efficacy seen here for IMA203 as compared to the previous trial may be attributed to (1) the higher affinity of the TCR (5 μM for IMA203 versus 8.7 μM for IMA202) with higher functional avidity (IFN-γ EC50: 0.81 nM versus 11 nM); (2) the higher cell doses investigated (up to 4.7 × 109 cells per m2 body surface area (BSA) ± 20% versus 1 × 109 cells per m2 BSA ± 20%); and (3) the shorter manufacturing (median 7 d versus 10 d), which could have contributed to a more favorable co-stimulatory phenotype. These results highlight the importance of optimizing TCR affinity, achieving suitable doses and designing manufacturing process for favorable T cell fitness.
There was no obvious organ restriction to IMA203, and infiltration of transduced cells into tumor tissue was observed in all post-treatment biopsies assessed. This suggests that IMA203 cells were activated and expressed the chemokine receptor signature required to overcome stromal or vasculature barriers and effectively engage with PRAME target35. Consistent with other reports36,37,38, we observed a significant correlation between tumor T cell infiltration and response as well as depth of response. Deeper responses to IMA203 predicted for longer duration of response, which is concordant with other immunotherapy trials in hematological malignancies and solid tumors39,40,41,42.
In contrast to other trials using TCR therapies in solid tumors43, no correlation was observed between the number of prior treatments received and response to IMA203. So far, we have not observed a correlation between elevated baseline LDH leading to poor response, as was reported in other adoptive T cell44 and checkpoint inhibitor45,46 trials. However, IMA203 responders were more likely to have a lower tumor burden, which fits well with other data suggesting similar associations in lymphoma26 and sarcoma43 treated with CAR and TCR T cells, respectively. Of note, a higher dose of IMA203 was clearly correlated with higher likelihood of durable response, and patients with a low tumor burden receiving a high TCR T cell dose were the most likely to derive long-term benefit. A similar association was described for CAR T cell therapy in lymphoma, where a higher effector to tumor burden ratio was a key driver of durable response47. In contrast to most CAR T cell trials48 and a TCR T trial targeting NY-ESO1 (ref. 18), we have not so far observed any differences in Cmax, Tmax and AUC0–28 between non-responders and responders. The fact that we observed a correlation between TCR T cells in the tumor and depth of response, but no correlation for classical predictive pharmacokinetics parameters measured in the peripheral blood that are well described for CAR T cells, may be driven by the biological differences of solid versus liquid cancers and may highlight the importance of monitoring T cell infiltration as an additional key parameter in solid cancer trials.
Several trials18,47 emphasized the role of CM and TEM T cell phenotype content in the drug product as drivers of response; however, such differences were not observed in IMA203 drug products between responders and non-responders.
Although responses were seen across all PRAME expressions levels above the threshold, IMA203 responders had a higher median target antigen expression, paralleling observations with MAGE-A4-targeting TCR T cells7 and PRAME-directed T-cell-engaging bispecifics49. We also observed a trend for deeper responses and longer PFS in patients with higher PRAME expression. To our knowledge, antigen density has so far not been described as one of the known factors predicting durable remissions for CAR T48. This may be because data generated so far are dominated by CD19 and BCMA target antigens, which show very high and homogenous expression, a rare feature for tumor-associated antigens in solid tumors. Our data indicate that understanding antigen expression levels is key to designing drug products with defined activation thresholds tailored to the targeted disease and patient. This principle is potentially applicable for both liquid and solid cancers whenever targeted antigens do not show the properties of BCMA and CD19.
Given the limited availability of post-progression biopsies, it is difficult to speculate on potential mechanisms of resistance to IMA203. In contrast to observations in CD19-targeting50 and BCMA-targeting51 CAR T cells, there were no obvious signs of loss or significant downregulation of the target antigen PRAME nor of HLA-A or β2M in IMA203-treated patients. In contrast to observations in solid tumors treated with checkpoint inhibitors52 or TCR T cells8,18, we did not detect deficiencies in antigen-processing machinery or HLA expression in the post-treatment biopsies assessed. On the contrary, upregulation of IFN-γ-inducible genes in responders indicates active IFN-γ signaling in the tumors of these patients53.
The limitations of our trial include testing multiple DLs across many solid tumors in a heterogenous patient population. The results should be treated with caution, and extension cohorts in homogenous patients with a larger sample size are required to establish robustness of our conclusions. Nevertheless, treatment with IMA203 PRAME-targeting TCR T cells was well tolerated and demonstrated meaningful clinical efficacy in a diverse set of heavily pre-treated patients with solid tumors. Patients with high target antigen expression, relatively low tumor burden and high TCR T cell dose might have more robust and long-lasting TCR T cell activity. This work clinically validates PRAME as a target for treatment of multiple solid cancers and points to a new benchmark for a high rate of durable response in patients with last-line melanoma. Further studies are planned to evaluate the benefit of IMA203 in larger patient populations across multiple solid tumors.
Methods
This trial was approved by the US Food and Drug Administration and the German Paul Ehrlich Institut and was conducted in compliance with international guidelines, including the Declaration of Helsinki and current Good Clinical Practice standards as defined by the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use Guideline for Good Clinical Practice; all applicable national, state and local laws and regulations; and the applicable institutional review boards (IRBs)/independent ethics committees and other institutional requirements. This trial was approved by the Western IRB (reference number IRB00000533), the University of Pittsburgh IRB (reference number IRB00001476), the Columbia Research Human Research Protection Office IRB (reference number IRB00006882), the Fox Chase Cancer Center (Temple Health) IRB/Federalwide Assurance (reference number 00003846), the University of Miami IRB – Human Subject Research Office IRB/Federalwide Assurance (reference number 00002247) and the Ethikkommission an der Technischen Universität Dresden (reference number EK 537122019). This trial is registered with ClinicalTrials.gov (NCT03686124) and the European Union Drug Regulating Authorities Clinical Trials Database (2024-515114-41-00).
Trial design and statistical analysis
IMA203-101 is a first-in-human, multicenter, open-label, 3 + 3 dose-escalation/de-escalation phase 1/2 trial in patients with PRAME+ recurrent and/or refractory solid tumors. The trial is composed of a phase 1a dose escalation and a phase 1b dose extension (including a monotherapy cohort and a PD-1 inhibitor combination cohort). Here we report results for IMA203 monotherapy. Primary objectives are the evaluation of safety and tolerability and the determination of the MTD and/or recommended phase 2 dose (RP2D) of IMA203, and primary endpoints are the incidence and nature of TEAEs, adverse events (AEs) of special interest, treatment-emergent serious AEs and DLTs. Secondary endpoints include the evaluation of IMA203 TCR-engineered T cell persistence in peripheral blood, tumor response measured according to RECIST 1.1 and duration of response.
Since 17 April 2019, patients were recruited, and the trial is conducted at different locations in the United States (University of Miami Hospital and Clinics, Miami, Florida; Columbia University Medical Center, New York, New York; University of Pittsburgh Medical Center, Pittsburgh, Pennsylvania; The University of Texas MD Anderson Cancer Center, Houston, Texas) and Germany (University Hospital Würzburg, Würzburg; University Hospital Bonn, Bonn; University Hospital Carl Gustav Carus, Dresden; University Hospital Hamburg-Eppendorf, Hamburg).
According to the most current clinical trial protocol, the following TEAEs occurring from day 0 (IMA203 infusion) through day 28 were defined as DLTs: any grade 3 or higher non-hematological AE (with exceptions of transient nausea, vomiting and diarrhea, responding to supportive care) that was at least possibly related to the trial drug; any grade 2 or higher bronchospasm requiring discontinuation of T cell infusion; all grade 2 or higher hypersensitivity reactions related to treatment; any grade 2 or higher autoimmune reactions (CRS was not considered an autoimmune reaction for the purpose of DLT definition); and any AE that led to a discontinuation of T cell infusion. Treatment-emergent grade 3 or higher toxicities unrelated to disease progression occurring beyond the DLT observation window (that is, after 28 d) may have been considered a DLT by the investigator after consultation with the sponsor and the Data Safety Monitoring Board. Patients experiencing an IMA203 T cell product DLT were counted as a DLT and removed from trial treatment without being replaced.
We tested a total of five DLs starting with 40 × 106 to 60 × 106 transduced T cells (CD3+CD8+Dex+ T cells) per m2 BSA (DL1) and then escalating to 120 × 106 to 180 × 106 cells per m2 BSA (DL2), 200 × 106 to 480 × 106 cells per m2 BSA (DL3), 200 × 106 to 1,200 × 106 cells per m2 BSA (DL4) and 1,201 × 106 to 4,700 × 106 cells per m2 BSA (DL5). Additionally, patients were allowed to be enrolled at DLs already cleared for safety or at any intermediate DLs (ECs) to better understand the safety and tolerability of IMA203 and to provide a T cell product to patients in need.
The phase 1 part of the trial used a standard 3 + 3 trial design with a maximum of six investigated DLs (including DL1) and with up to 36 patients. For the phase 1b extension phase, up to 20 additional patients can be enrolled for IMA203 monotherapy and up to 18 for the PD-1 inhibitor combination cohort. No statistical method was used to predetermine sample size; the experiments were not randomized; and investigators were not blinded to allocation during experiments and outcome assessment. Analyses were based on data cutoff on 4 July 2023. The safety analysis set comprised 41 patients who received at least one dose of lymphodepletion, including one patient who started lymphodepletion but did not receive IMA203 due to intercurrent, fatal sepsis. Efficacy analyses were based on all 40 patients in the safety analysis set who received IMA203 (that is, excluding one patient). Although dose escalation was completed with the reported patients, the phase 1b part of the trial is still ongoing to expand the dataset at DL4 and DL5.
ORR (for patients with at least one scan after infusion or clinical progression) and cORR (patients with at least two tumor assessments after infusion or with clinical/radiological progressions at any timepoint) were determined according to RECIST 1.1 and evaluated using a Clopper–Pearson 95% CI. DOR was defined as the time from first documented PR until first documented date of disease progression or death due to any cause, whichever occurred first in patients with confirmed response. PFS was defined as the time between T cell infusion and event (progression or death, whichever occured first). A patient who experiences any form of progression (radiological and clinical) as determined by the local investigator was evaluated as a patient with an event. Patients who did not record disease progression or death as of the cutoff date for the analysis were censored at the cutoff date. OS was defined as the time between T cell infusion and death of the patients. Patients who did not record death as of the cutoff date for the analysis were censored at the last date that they were known to be alive. Median follow-up of confirmed responses was defined as the time from first documented PR that patients with confirmed responses were followed-up on and was calculated using the reverse Kaplan–Meier method. Patients with disease progression were censored at the timepoint of progressive disease, and patients without documented progressive disease at the cutoff date for the analysis were counted as events at data cutoff. DOR, PFS and OS were summarized using the Kaplan–Meier method to estimate the median survival time, including minimum and maximum values and 95% CIs based on Brookmeyer and Crowley methodology. Formal statistical testing was not prespecified. In the post hoc analysis, differences between two independent proportions were analyzed using Fisher’s exact test; medians of two groups were compared using the Mann–Whitney U-test; multiple comparisons were performed using the Kruskal–Wallis test; and correlations were calculated using Spearman’s rank correlation. The P values were considered statistically significant for P < 0.05. Where appropriate, 95% exact CIs were provided.
Statistical analyses were conducted using GraphPad Prism version 9 and R version 4.3.0. No data were excluded from the analyses, but data points may be missing in cases where patient samples were not available. Sample numbers are mentioned in the respective figure legends.
An independent Data and Safety Monitoring Board closely monitored the safety of this trial.
Until the cutoff for this publication, the clinical trial underwent five noteworthy amendments related to IMA203. The first (7 June 2019; version 2.0) modified the cell doses in dose cohorts 2 (from 150 × 106 to 300 × 106 CD3+CD8+Dex+ cells per m2 BSA) and 3 (from 500 × 106 to 1,000 × 106 CD3+CD8+Dex+ cells per m2), increased the total maximum number of viable CD3+ cells that can be infused from 1 × 1010 to 2 × 1010 and split the protocol into a 1-year treatment and observation phase, a 2-year follow-up phase and a 12-year long-term follow-up, which will be covered in a separate protocol. The second amendment (2 December 2020; version 5.0) reflected specific risks and mitigation measures for coronavirus disease 2019 (COVID-19). It also reduced the lymphodepletion regimen and dosages for patients with hepatocellular carcinoma or renal impairment as a safety precaution, revised DLs, added DL4 (200 × 106 to 1,200 × 106 cells per m2 BSA) with the same maximal dose as the previous DL3 and shortened the DLT observation period from 30 d to 28 d. The third amendment (6 April 2021; version 6.0) increased the cell cap from 2 × 1010 to 5 × 1010 total viable CD3+ cells. Additionally, it introduced modifications to the IL-2 dose and schedule: low-dose IL-2 treatment now starts the day after IMA203 infusion (day 1), approximately 24 h after T cell infusion, instead of the previous start time of 6 h after IMA203 infusion. Furthermore, IL-2 treatment could be resumed until day 10 at the longest, if IL-2 administration had to be paused. Moreover, the administration period was reduced from twice daily for 14 d to once daily for 5 d, followed by twice daily for 5 d. The lymphodepletion regimen was revised to align with doses used by sponsors with similar cancer products, incorporating specific dose regimens based on renal function and bone marrow reserve. Additionally, the IMA203 DLT definition was revised. The fourth amendment (27 January 2022; version 7.0) expanded dose escalation to include DL5 (1,201 × 106 to 4,700 × 106 cells per m2 BSA), prolonged the follow-up phase from 2 years to 4 years and added tumor lysis syndrome as a risk. The fifth amendment (28 April 2023; version 9.0) implemented DL5 as the provisional RP2D and provided guidance for the clinical management of hemophagocytic lymphohistiocytosis. Additional major changes to the protocol included the extension from phase 1 to phase 1/2, the implementation of cohorts for the evaluation of combination therapy of IMA203 with a checkpoint inhibitor and the treatment with the IMA203 next-generation product (IMA203CD8). These cohorts are not relevant for this publication.
Patients
Patients voluntarily signed an informed consent form before initiation of any trial-specific screening procedures and before start of treatment, stating that they were aware of the investigational nature of the trial. Eligible patients were 18 years of age or older, were diagnosed with pathologically confirmed advanced and/or metastatic solid tumor with recurrent/progressing and/or refractory disease and received or were not eligible for all available indicated standard-of-care treatments known to confer clinical benefit. Patients had to have measurable disease according to RECIST 1.1 and be HLA-A*02:01+, and tumors had to express PRAME as assessed by RT–qPCR from a fresh biopsy. In addition, eligible patients had to have an Eastern Cooperative Oncology Group performance status (ECOG-PS) of 0–1, adequate organ/marrow function (absolute neutrophil count ≥1.0 × 109 per liter without granulocyte colony-stimulating factor support, platelets ≥75,000 per microliter, hemoglobin ≥8 g dl−1), adequate hepatic function (total bilirubin level ≤1.5× ULN, unless the patient has known Gilbert’s syndrome, and alanine aminotransferase/aspartate aminotransferase ≤2.5× ULN or ≤5× ULN for patients with liver metastases or tumor involving the liver) as well as pulmonary function, life expectancy more than 3 months, serum creatinine within 1.5× normal range for age or creatinine clearance with a recommended estimated glomerular filtration rate ≥50 ml min−1 1.73 m−2, acceptable coagulation status (international normalized ratio of prothrombin time of blood coagulation ≤2.0× ULN and partial thromboplastin time ≤2.0× ULN), received available standard-of-care treatments, recovered from any side effects of prior therapy to grade 1 or lower (except for non-clinically substantial toxicities) before lymphodepletion and confirmed and adequate production timelines and capacities for the patient’s IMA203 product. Patients with hepatocellular carcinoma had a Child–Pugh score of less than 6 and a total bilirubin level of ≤2.5× ULN. Male patients had to agree to use effective contraception or be abstinent while on trial for 6 months after IMA203 infusion. Female patients of childbearing potential had to use adequate contraception before trial entry until 12 months after IMA203 infusion.
Patients were excluded if they fulfilled one of the following criteria: history of other malignancies (except for adequately treated basal or squamous cell carcinoma or carcinoma in situ) within the last 3 years, pregnancy or breastfeeding, prior allogenic stem cell transplantation or solid organ transplantation, any condition contraindicating leukapheresis, HIV infection, active hepatitis B virus (HBV) and/or hepatitis C virus (HCV) infection, ongoing active anti-HCV treatment or detectable HBV or HCV viral load (except resolved or chronic stable HBV infection), receipt of systemic corticosteroids (≥10 mg d−1 prednisone or equivalent) within 2 weeks before leukapheresis, concurrent severe and/or uncontrolled medical disease (for example, uncontrolled diabetes, severe infection requiring active treatment, severe malnutrition, chronic severe liver or renal disease), history of, or current, immunodeficiency disease or prior treatment compromising immune function (inclusion at the discretion of the investigator), history of hypersensitivity (to cyclophosphamide, fludarabine, IL-2 or any rescue medication), solid tumor indications with low likelihood of PRAME expression (colon adenocarcinoma, glioblastoma multiforme, kidney chromophobe, lower-grade glioma, mesothelioma, pancreatic adenocarcinoma, pheochromocytoma, paraganglioma, prostate adenocarcinoma, rectum adenocarcinoma or thyroid carcinoma) and active brain metastases (patients with a history of brain metastases are eligible if imaging studies performed ≥4 weeks after treatment indicate stable disease, the patient is asymptomatic and steroid therapy has been discontinued for ≥2 weeks). Furthermore, patients with a history of serious autoimmune diseases were excluded, such as active serious inflammatory bowel disease (including Crohn’s disease and ulcerative colitis) and autoimmune disorders such as rheumatoid arthritis, multiple sclerosis, systemic progressive sclerosis (scleroderma), systemic lupus erythematosus or autoimmune vasculitis (for example, Wegener’s granulomatosis). Patients may be included if their disease is well controlled without the use of immunosuppressive agents. Other exclusion criteria included unfavorable cardiac conditions, such as uncontrolled hypertension despite optimal therapy, uncontrolled angina, ventricular arrhythmias, congestive heart failure (New York Heart Association Class 2 or higher), baseline left ventricular ejection fraction ≤50%, prior or current cardiomyopathy, atrial fibrillation with heart rate over 100 beats per minute and unstable ischemic heart disease (myocardial infarction within 6 months before leukapheresis or angina requiring use of nitrates more than once weekly). Patients with active viral infection (for example, COVID-19, influenza and severe acute respiratory syndrome (SARS)) during initial screening were excluded, but screening may have started/continued after recovery. For patients with confirmed or suspected COVID-19 infection, screening or lymphodepletion may have started or continued at the earliest 2 weeks after full recovery or a negative severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) test. For patients recovered from other viral infections, lymphodepletion may start after full recovery. No patients who received any anti-tumor therapy, such as chemotherapy, surgery, palliative radiotherapy, tyrosine kinase inhibitors (for example, erlotinib and gefitinib) and investigational therapies 7 d before leukapheresis were included. The same was true for patients receiving (1) chemotherapy, radiotherapy or investigational therapies within 1 week; (2) major surgery within 2 weeks; (3) extensive radiotherapy to the lung or liver within 4 months; (4) live vaccines within 6 weeks; or (5) inactivated vaccines within 2 weeks before lymphodepletion. Another exclusion criterion was the existence of any other condition that would, in the investigator’s judgment, contraindicate the patient’s participation in the clinical trial because of safety concerns or compliance with clinical trial procedures. Concurrent participation in an interventional part of another clinical trial was an exclusion criterion as well.
Gender was determined based on self-report. Due to the limited number of eligible patients, the sex and/or gender of the patients could not be specifically considered before enrollment. As a result of the small sample size, no disaggregated analysis for sex was conducted.
Patients did not receive compensation for taking part in this trial.
Trial procedures and treatment
After confirmation of eligibility, patients underwent leukapheresis, and IMA203 product was manufactured under current Good Manufacturing Practice–compliant conditions using a lentiviral vector to transduce a PRAME-specific TCR into autologous T cells. After expansion, T cells were harvested, washed and cryopreserved. Before infusion, release testing was performed to ensure sterility, safety and quality of the final product. Lymphodepletion with fludarabine and cyclophosphamide was applied from day −6 to day −3 before IMA203 infusion at day 0 (Supplementary Fig. 19). IL-2 dosing regimen consisted of 1 × 106 IU (approximately 5.5 × 105 IU m−2) and was administered subcutaneously approximately 24 h after IMA203 infusion, followed by subcutaneous injection every 24 h for 5 d and then approximately every 12 h for another 5 d. IL-2 administration was interrupted at the discretion of the investigator in case of toxicities. IL-2 treatment could be resumed until day 10 at the longest, if IL-2 administration had to be paused. Injection of IL-2 was paused if grade 2 or higher CRS was suspected or if it was decided to apply tocilizumab. Before continuing IL-2 administration, patient had to recover to at least grade 1 CRS. When resumed, IL-2 dose may have been adapted to any lower dose for safety reasons.
Prophylaxis for infections started on day −6, continuing as clinically indicated, and the prophylaxis for allergic reactions (acetaminophen (paracetamol) 500–650 mg and diphenhydramine hydrochloride 25–50 mg orally or intravenously, according to institutional guidelines) started on day 0, continuing as clinically indicated.
After IMA203 infusion, patients were closely observed during the treatment and observation phase until month 12, progressive disease, death or early discontinuation. Thereafter, the follow-up started, which ranged up to 25.6 months (median, 1.9 months). No patients were lost to follow-up, but three patients withdrew consent after progression. During follow-up, patients were evaluated for changes in health status, vital signs and physical examination, tumor assessment and OS. In addition, blood samples were collected to be tested for replication-competent lentivirus, and monitoring of T cell persistence was continued.
Safety was assessed by physical examinations (for example, assessment of cardiovascular, respiratory, gastrointestinal and neurological systems), measurement of vital signs (that is, temperature, systolic and diastolic blood pressure, pulse rate, respiratory rate and oxygen saturation), electrocardiograms and transthoracic echocardiogram, pulmonary function tests, ECOG-PS and clinical safety laboratory assessments (among others, hematology, clinical chemistry, urinalysis, thyroid function and coagulation). Early assessment, grading and treatment of CRS followed the recommendations of Lee et al.54 and Neelapu et al.17. Early assessment and management of neurotoxicity followed published guidance from Neelapu et al.17 with more aggressive treatment being possible for patients with high fever (temperature ≥39.5 °C). Interruption of IL-2 application was recommended in case of CRS grade 2 or higher, or tocilizumab was applied.
Tumor response was assessed according to RECIST 1.1 (ref. 55).
pHLA isolation and relative quantitation of PRAME pHLA
Primary human tissue samples were extracted surgically or postmortem from HLA-A*02+ patients with cancer or HLA-A*02+ normal tissue donors. All specimens were procured with written informed consents and with appropriate ethics committee approvals. The resulting sample set covered 38 different organs and 24 tumor types. Tissue samples were snap frozen in liquid nitrogen after excision and stored until isolation at −80 °C for subsequent RNA and pHLA analyses. Tissue samples were fixed in formalin and embedded in paraffin (FFPE) for histological assessment.
After tissue homogenization and lysis, pHLA complexes were isolated by immunoprecipitation using BB7.2 (Department of Immunology, University of Tübingen, Germany) coupled to cyanogen bromide–activated sepharose resin (GE Healthcare Europe). Peptides were eluted from antibody resin by acid treatment and purified by ultrafiltration. HLA peptidomics was performed using an in-house target discovery platform as previously described16. In brief, peptidome samples were separated by reversed-phase ultra-performance liquid chromatography (UPLC) (Waters, nanoAcquity) using ACQUITY UPLC BEH C18 columns (75 μm × 250 mm; Waters) and a gradient ranging from 1% to 34.5% acetonitrile over the course of 70 min or 190 min. MS was performed on online coupled Orbitrap mass spectrometers Fusion, Velos and Linear trap quadrupole (Thermo Fisher Scientific) in data-dependent acquisition mode. Samples were analyzed in at least three replicate runs, acquiring MS/MS data in collision-induced dissociation and higher collisional energy dissociation mode. Data processing was performed using a proprietary pipeline, which combines database search, spectral clustering, feature detection, retention time alignment and global normalization for the generation of population-scale, relative-quantitative peptide presentation profiles.
TCR identification of the PRAME-specific TCR
The PRAME-specific TCR for IMA203 was identified from CD8+ healthy donor T cells after a 3-week in vitro priming using streptavidin-coated microspheres as artificial antigen-presenting cells (APCs) loaded with anti-CD28 antibody (clone 9.3, purified from mouse hybridoma supernatant, University of Tübingen, Germany) and target pHLA monomers56. Single cells of two-dimensional (2D) target tetramer-binding populations were sorted on a BD FACSAria III device (BD FACSDiva 8.0.1) into lysis buffer (64.9 mmol L−1 Tris, 810.8 mmol L−1 LiCl, 6.5 mmol L−1 ethylenediaminetetraacetic acid (EDTA), pH 7.5) for single-cell rapid amplification of cDNA 5′ ends (5′RACE). After cell lysis, mRNA was captured by paramagnetic oligo-deoxythymine beads, and cDNA was synthesized using TCR gene-specific primers (ATAGAGGATGGTGGCAGACAGG and ACCGAGGTAAAGCCACAGTC for the β-chain and AGCAGTGTTTGGCAGCTCTT for the α-chain). TCR transcripts were amplified using nested multiplex PCR57. The resulting PCR products were analyzed by Sanger sequencing. The sequencing data were used to assemble full-length coding DNA sequences in silico using BLAST, CDR3 determination and final chain assembly (MacVector 15.5.3). The TCRs were resynthesized by gene synthesis at GenScript.
TCR characterization
For TCR characterization, identified TCRs were re-expressed in healthy donor cells from in vitro transcribed TCR mRNA (mMESSAGE mMACHINE T7 Transcription Kit). As a template for in vitro transcription, individual TCR chains were PCR amplified with T7 and Kozak sequences at the 5′ end and a 64-adenine 3′ tail. Primary CD8+ T cells were isolated from leukaphereses from HLA-A*02+ donors by CD8+ magnetic-activated cell sorting (Miltenyi Biotec) and 3–4-d stimulation with plate-bound anti-CD3 (10 µg ml−1 coating concentration) and soluble anti-CD28 (0.1 µg ml−1) antibodies. Cells were electroporated with TCR mRNA in an ECM830 electroporator (BTX) at 500 V for 3 ms and rested for 20 h before use in functional experiments.
T cell functionality
Electroporated T cells were used for activation assay by IFN-γ release ELISA after co-culture with target cells loaded with peptides or target-expressing tumor cell lines as well as primary cells from healthy tissues. Released IFN-γ levels were determined after 20 h of co-culture with BD OptEIA Human IFN-γ ELISA or BioLegend Human IFN-γ ELISA MAX Deluxe Kits. Primary cells from healthy tissues were obtained from PromoCell, or induced pluripotent stem cell (iPSC)-derived cell types were obtained from FUJIFILM Cellular Dynamics. Tumor cell lines were obtained from the American Type Culture Collection (ATCC) or the German Collection of Microorganisms and Cell Cultures (DSMZ). All cells were cultured according to the manufacturer’s instructions and genotyped for HLA-A*02. Culture periods were kept short to maintain cellular characteristics. T cell activation assays were performed in T cell medium to enable optimal activity of the effector cells. T cell medium consists of RPMI 640 GlutaMAX supplemented with 10% heat-inactivated human serum, 1% penicillin–streptomycin, 0.2% gentamycin and 1% sodium pyruvate. The EC50 of the PRAME-specific TCR was determined using GraphPad Prism version 6 via nonlinear fit (sigmoidal, 4PL) of log dose (loaded peptide concentration) versus response (IFN-γ release).
Cytotoxicity assays
IMA203 patient drug products were thawed, washed and resuspended in culture medium and then treated with benzonase nuclease for 15 min. After a wash, cells were rested overnight in culture medium within a Grex vessel at 37 °C, 5% CO2. The next day, red fluorescent protein (RFP)-labeled tumor cell lines UACC257, hs695T and A375 were harvested using 0.05% trypsin, washed and then counted. These antigen-positive tumor cell lines were plated at 10,000 cells per well in a flat-bottomed 96-well ImageLock plate in 100 μl of culture medium. Plates were placed in an incubator at 37 °C, 5% CO2 until effector T cells were ready for plating. Overnight-rested effector T cells were removed from the incubator and counted. Effector numbers were normalized to TCR positivity and added to the 96-well ImageLock plate at specific effector-to-target ratios: 2:1 for UACC257, 4:1 for hs695T and 8:1 for A375. Then, 100 μl of effector cells were added to their respective wells. The 96-well ImageLock co-culture plates were then placed into an IncuCyte S3 imager at 37 °C, 5% CO2 and imaged every 4 h for the duration of the assay. Data were exported from IncuCyte S3 software (2022BRev2) into Microsoft Excel (version 2304, build 16327.20214) and GraphPad Prism (version 9.5.1) for further analysis. Fold tumor growth (RFP+ tumor cell count) was normalized to the 0-h timepoint.
TCR affinity determination
The PRAME-specific IMA203 TCR and its non-engineered parental TCR variant were expressed as soluble proteins and refolded according to a published protocol in Escherichia coli58. Refolded TCRs were purified via anion exchange and size exclusion chromatography. The protein concentration was determined using Bradford assays, and refolding was determined by native and denaturating polyacrylamide gel electrophoresis. Biolayer interferometry (Octet) technology was used to determine the affinity of the refolded TCR toward target pHLA compared to unrelated pHLA. For the measurements, biotinylated PRAME-004 pHLA was immobilized on streptavidin sensors, and the TCRs were applied at concentrations of 0.8 µM, 1.6 µM, 3.1 µM, 6.25 µM, 12.5 µM, 25 µM and 50 µM, respectively, for analysis of association and dissociation parameters. Binding affinity for PRAME-004 pHLA (equilibrium dissociation constant, Kd) was determined at steady state.
Determination of PRAME expression
Patient tumors had to express PRAME, as assessed by an assay based on a reverse transcriptase qPCR analysis of a fresh tumor biopsy specimen stored in RNAlater stabilization solution (Thermo Fisher Scientific). For PRAME, a correlation between mRNA and immunopeptidome levels (both in-house data) was established as demonstrated previously16. From this correlation, a reads per kilobase per million mapped reads threshold was generated and translated into an RT–qPCR assay threshold in which PRAME was considered positive if expression levels were above a target-specific delta cycle threshold (ΔCt). For ΔCt generation, Ct average of three reference genes (RPLP0, OAZ1 and RPL37A) was calculated and subtracted from observed Ct value for PRAME. A threshold (ΔCt of 4.75 for PRAME) was chosen to maximize the sensitivity and specificity of prediction of peptide presentation as described previously16.
Manufacturing of IMA203 products
The IMA203 manufacturing process is a continuous, semi-closed process lasting for 7 d from thawing of starting material to formulation of the drug product. During the dose-escalation phase, process improvements were implemented to ensure successful manufacturing using variable and heterogenous patient material as well as for meeting higher doses. These changes, compared to the original process (version 1.0), included elimination of serum from the transduction step (version 3.0), extension of harvest on day 6 to day 7 (version 3.5), monocyte depletion (version 4.0) and T cell enrichment (version 5.0). Overall, IMA203 manufacturing starts with cryopreservation of the starting material—that is, PBMCs (version 1.0 to version 4.0)—or leukapheresis (version 5.0). In brief, whole (version 1.0 to version 3.5) or monocyte-depleted (version 4.0) PBMCs or T cells selected from thawed leukapheresis (version 5.0) are activated with anti-CD3 and anti-CD28 antibodies. Activated T cells are genetically modified by transducing with a lentiviral vector encoding the PRAME-specific TCR. Transduced cells are expanded until day 7 before being harvested, washed, concentrated and formulated into the drug product. All products undergo in-process and product release testing before infusion and are stable for more than 12 months.
Quantification of IMA203 T cells (exploratory endpoint)
Genomic DNA was extracted from PBMC and/or tissue biopsies using a QIAamp DNA Mini Kit and an AllPrep DNA/RNA Mini Kit (both Qiagen), respectively, according to the manufacturer’s instructions. The concentration of IMA203 T cells was assessed in DNA samples using qPCR specific for the Psi sequence (Applied Biosystems, cat. no. 44-445-56) of the lentiviral construct. The limit of detection for this assay is 40 copies per microgram of genomic DNA.
Phenotypical T cell analysis
For flow cytometry–based ex vivo immunomonitoring, isolated and cryopreserved PBMCs collected at different timepoints before and after infusion were subjected to pHLA multimer and cell surface staining. PBMCs were rested overnight in RPMI 1640 + HEPES + 10% human serum + 1 ng ml−1 IL-15 and 20 U ml−1 IL-2. Between 5 × 105 and 5 × 106 cells were plated and stained with fixable viability stain, followed by multimer staining for 20 min at room temperature (each multimer at a concentration of 0.8 µg ml−1) and subsequent surface staining using antibodies listed in Supplementary Table 6. All washing steps were carried out in PBS, 2% FCS, 2 mM EDTA and 0.01% azide. Stained cells, fixed using PBS with 1% FCS and 1% formaldehyde, were acquired either on a BD LSRII SORP flow cytometer using BD FACSDiva software version 8.0.1 and analyzed using FlowJo software version 10.4 (Tree Star) or on a Cytek Aurora spectral analyzer using SpectroFlo software versions 3.0.1, 3.0.3 and 3.1.0 and analyzed using FlowJo software version 10.8 (Tree Star).
Memory T cell subsets were classified using the markers CD197 (CCR7) and CD45RA, with naive cells being CCR7+CD45RA+, central memory cells being CCR7+CD45RA−, effector memory cells being CCR7−CD45RA− and terminally differentiated effector memory cells being CCR7−CD45RA+. IMA203 cells after infusion were detected as CD3+CD8+ tetramer-positive cells or CD3+CD8+ tetramer-positive Vβ8+ cells.
Statistical methods and sample size used for each comparison are described in the figure legends.
RNAscope
FFPE tumors were acquired from commercial providers. In brief, 4-μm sections were evaluated using RNAscope from Advanced Cell Diagnostics for the expression of the following probe sets: PRAME (1209068), PPIB (313908) and PRAME scramble (1210868). All tumor stainings were performed using RNAscope 2.5 LS Reagent Kit-RED assays (Advanced Cell Diagnostics, 322150) on the Leica BOND RX platform (6.0.1.14) (Leica Biosystems). Serial sections for FFPE tissues were baked for 90 min at 60 °C before deparaffinization and dehydration on the instrument. Antigen retrieval was performed using Leica Epitope Retrieval Buffer 2 (cat. no. AR9551) for 15 min at 88 °C or 95 °C and subsequent protease treatment for 15 min at 40 °C. Probes were hybridized for 2 h at 42 °C, followed by RNAscope amplification and Fast Red signal detection applying BOND Polymer Refine Red Detection (Leica, DS9390). The slides were counterstained with hematoxylin on the Leica BOND. Slides were scanned at ×40 magnification (Olympus, VS200 ASW 3.4.1). The staining quality was evaluated by checking intact RNA expression (PPIB of ≥4 dots per cell) and minimal background signal (PRAME scramble of ≤1 dots in 10 cells). Artifacts including folding, dust, necrosis or other tissue-specific artifacts were excluded from analysis. Analysis and tissue scoring were performed by a trained pathologist using a semi-quantitative scoring system as previously described59.
Serum cytokine analysis
Invitrogen Cytokine & Chemokine 34-Plex Human ProcartaPlex Panel 1 (cat. no. EPX340-12167-901) was used to perform serum cytokine analysis. This method uses Luminex XMAP technology, where magnetic beads are internally color-coded with fluorescent dyes of varying intensities. The bead sets are coated with a target-specific capture antibody against the analyte, and the assay reagents are free-floating in the solution. This technology combines flow cytometry and sandwich immunoassays. In the first step of the experiment, the analytes from the test samples were captured by the beads coated with a target-specific capture antibody. Next, a biotinylated analyte-specific detection antibody was introduced to detect specific analytes from the test sample. The beads were then incubated with streptavidin-conjugated R-phycoerythrin (SA-PE), the reporter molecule, for analyte quantification. Finally, the magnetic beads were illuminated to determine the different assays by internal bead color, and analyte concentration was measured by measuring the reporter molecule fluorescence. The experiment used a kit that contained all the necessary components for the assay procedure. The kit was used only on serum samples that were thawed on ice on the day of the experiment. The kit measured a variety of cytokines, including granulocyte-macrophage colony-stimulating factor (GM-CSF), INF-γ, IFN-α, IL-1α, IL-1RA, IL-1β, IL-2, Il-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12p70, IL-13, IL-15, IL-17A, IL-18, IL-21, IL-22, IL-23, IL-27, IL-31, TNF-β, eotaxin, Gro-α, IP-10, MCP-1, macrophage inflammatory protein (MIP)-1α, MIP-1β, regulated upon activation, normal T cell expressed and secreted (RANTES) and TNF. The assay was carried out according to the manufacturer’s protocol, which involved adding prepared beads to each well of a 96-well plate, followed by incubation and washing steps. The cytokine lyophilized standard was reconstituted, and different concentration ranges for specific analytes were prepared. After incubation with detection antibody mixture and SA-PE, the beads were resuspended in reading buffer, and the Luminex LX200 plate reader was used to detect the individual beads conjugated with analyte. The median fluorescence intensity was then compared to the standard curve to calculate the cytokine concentration in pg ml−1 using ProcartaPlex Analyst cloud software version 1.0. The five-parameter logistic curve fitting was used to derive the analyte concentrations in each sample.
RNA-seq (exploratory endpoint)
Total RNA was isolated and purified in-house using an AllPrep DNA/RNA miRNA Universal Kit (Qiagen) according to the manufacturer’s protocol. RNA samples with a concentration of ≥25 ng µl−1 and an RNA integrity number or an RNA quality number of ≥6.0 were considered acceptable for downstream library preparation and sequencing. Library preparation, including mRNA selection, RNA fragmentation, cDNA conversion and addition of sequencing adaptors, as well as the sequencing process itself, were performed by GENEWIZ Germany GmbH. Sequencing libraries were generated using an NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England Biolabs) following the manufacturer’s instructions. For sequencing, libraries were multiplexed and loaded onto a NovaSeq 6000 sequencer (Illumina) according to the manufacturer’s instructions, targeting a minimum of 80 million paired-end reads per sample with a read length of 150 base pairs.
The obtained reads were trimmed using BBDuk (BBTools version 38.81, 24 March 2020) to remove adapter sequences and low-quality bases. Subsequently, the trimmed reads were then mapped to the Genome Reference Consortium Human Build 38 patch release 13 (GRCh38.p13) using STAR (version 2.7.3a)60, and the exon expression was quantified using featureCounts (version 2.0.0)61. For quantifying transcript expression, Kallisto (version 0.46.1)62 was used. For the quantifications, the Ensembl 99 reference annotations were used, and the results were normalized using DESeq2 (ref. 63) to a predefined reference sample.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The datasets used and/or analyzed for this publication are not publicly available due to proprietary considerations and to protect the patients and their sensitive data. Regarding patient data, this avoids irregular access to data related to the presence or risk of developing particular diseases or conditions and information regarding family relationships or ancestry. Upon reasonable request and authorization by the corresponding author, and after appropriate data-sharing and transfer agreements have been agreed upon, eligible academic researchers in the field may be provided access to trial-related data after product approval and ending 36 months thereafter. Requesters should submit a proposal outlining the objective, data format and features, hypothesis and specific rationale to [email protected]. Requests will usually be processed within 15 weeks. Sequencing data of tumor biopsies collected during the conduct of this trial have not been made publicly available in a repository to protect the privacy and confidentiality of the participating patients. Source data of Extended Data Figs. 8 and 9 are provided as Supplementary Information to this paper. Access to de-identified gene-limited datasets may be provided to eligible academic researchers after product approval and ending 36 months thereafter and upon request as outlined above for clinical data. A redacted clinical trial protocol is available at https://euclinicaltrials.eu/search-for-clinical-trials/?lang=en&EUCT=2024-515114-41-00. The results shown in Supplementary Table 1 and gray histograms depicted in Extended Data Fig. 1 are, in part, based on data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Change history
29 April 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41591-025-03731-6
References
Norberg, S. M. & Hinrichs, C. S. Engineered T cell therapy for viral and non-viral epithelial cancers. Cancer Cell 41, 58–69 (2023).
Leko, V. & Rosenberg, S. A. Identifying and targeting human tumor antigens for T cell-based immunotherapy of solid tumors. Cancer Cell 38, 454–472 (2020).
Klebanoff, C. A., Chandran, S. S., Baker, B. M., Quezada, S. A. & Ribas, A. T cell receptor therapeutics: immunological targeting of the intracellular cancer proteome. Nat. Rev. Drug Discov. 22, 996–1017 (2023).
Johnson, L. A. et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 114, 535–546 (2009).
Moore, T. et al. Clinical and immunologic evaluation of three metastatic melanoma patients treated with autologous melanoma-reactive TCR-transduced T cells. Cancer Immunol. Immunother. 67, 311–325 (2018).
Robbins, P. F. et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1–reactive T-cell receptor: long-term follow-up and correlates with response. Clin. Cancer Res. 21, 1019–1027 (2015).
Hong, D. S. et al. Autologous T cell therapy for MAGE-A4+ solid cancers in HLA-A*02+ patients: a phase 1 trial. Nat. Med. 29, 104–114 (2023).
Nagarsheth, N. B. et al. TCR-engineered T cells targeting E7 for patients with metastatic HPV-associated epithelial cancers. Nat. Med. 27, 419–425 (2021).
D’Angelo, S. P. et al. Antitumor activity associated with prolonged persistence of adoptively transferred NY-ESO-1 c259T cells in synovial sarcoma. Cancer Discov. 8, 944–957 (2018).
Ikeda, H. et al. Characterization of an antigen that is recognized on a melanoma showing partial HLA loss by CTL expressing an NK inhibitory receptor. Immunity 6, 199–208 (1997).
Kaczorowski, M. et al. PRAME expression in cancer. A systematic immunohistochemical study of >5800 epithelial and nonepithelial tumors. Am. J. Surg. Pathol. 46, 1467–1476 (2022).
Lezcano, C., Jungbluth, A. A., Nehal, K. S., Hollmann, T. J. & Busam, K. J. PRAME expression in melanocytic tumors. Am. J. Surg. Pathol. 42, 1456–1465 (2018).
Zhu, H. et al. Downregulation of PRAME suppresses proliferation and promotes apoptosis in hepatocellular carcinoma through the activation of p53 mediated pathway. Cell. Physiol. Biochem. 45, 1121–1135 (2018).
Kessler, J. H. et al. Efficient identification of novel HLA-A*0201–presented cytotoxic T lymphocyte epitopes in the widely expressed tumor antigen PRAME by proteasome-mediated digestion analysis.J. Exp. Med. 193, 73–88 (2001).
Weinschenk, T. et al. Integrated functional genomics approach for the design of patient-individual antitumor vaccines. Cancer Res. 62, 5818–5827 (2002).
Fritsche, J. et al. Translating immunopeptidomics to immunotherapy-decision-making for patient and personalized target selection. Proteomics 18, e1700284 (2018).
Neelapu, S. S. et al. Chimeric antigen receptor T-cell therapy—assessment and management of toxicities. Nat. Rev. Clin. Oncol. 15, 47–62 (2018).
Gyurdieva, A. et al. Biomarker correlates with response to NY-ESO-1 TCR T cells in patients with synovial sarcoma. Nat. Commun. 13, 5296 (2022).
Lei, W. et al. Treatment-related adverse events of chimeric antigen receptor T-cell (CAR T) in clinical trials: a systematic review and meta-analysis. Cancers (Basel) 13, 3912 (2021).
Kawai, A. et al. Safety and efficacy of NY-ESO-1 antigen-specific T-cell receptor gene-transduced T lymphocytes in patients with synovial sarcoma: a phase I/II clinical trial. Clin. Cancer Res. 29, 5069–5078 (2023).
D’Angelo, S. P. et al. Primary efficacy and safety of letetresgene autoleucel (lete-cel; GSK3377794) pilot study in patients with advanced and metastatic myxoid/round cell liposarcoma (MRCLS). J. Clin. Oncol. https://doi.org/10.1200/JCO.2022.40.16_suppl.11500 (2022).
Hong, D. S. et al. Updated safety and efficacy from SURPASS, the phase I trial of ADP-A2M4CD8, a next-generation autologous T-cell receptor T-cell therapy, in previously treated patients with unresectable or metastatic tumors. Ann. Oncol. 33, S879–S880 (2022).
Hong, D. S. et al. Phase 1 clinical trial evaluating the safety and anti-tumor activity of ADP-A2M10 SPEAR T-cells in patients with MAGE-A10+ head and neck, melanoma, or urothelial tumors. Front. Oncol. 12, 818679 (2022).
Blumenschein, G. R. et al. Phase I clinical trial evaluating the safety and efficacy of ADP-A2M10 SPEAR T cells in patients with MAGE-A10+ advanced non-small cell lung cancer. J. Immunother. Cancer 10, e003581 (2022).
Abramson, J. S. et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet 396, 839–852 (2020).
Schuster, S. J. et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 380, 45–56 (2019).
Locke, F. L. et al. Axicabtagene ciloleucel as second-line therapy for large B-cell lymphoma. N. Engl. J. Med. 386, 640–654 (2022).
Pazdur, R., Samson, M. K. & Baker, L. H. Fludarabine phosphate. Phase II evaluation in advanced soft-tissue sarcomas. Am. J. Clin. Oncol. 10, 341–343 (1987).
Von Hoff, D. D. et al. Phase II study of fludarabine phosphate (NSC-312887) in patients with advanced ovarian cancer. A Southwest Oncology Group Study. Am. J. Clin. Oncol. 11, 146–148 (1988).
Lambert, H. E. & Berry, R. J. High dose cisplatin compared with high dose cyclophosphamide in the management of advanced epithelial ovarian cancer (FIGO stages III and IV): report from the North Thames Cooperative Group. Br. Med J. (Clin. Res Ed.) 290, 889–893 (1985).
Bramwell, V. H. et al. Cyclophosphamide versus ifosfamide: final report of a randomized phase II trial in adult soft tissue sarcomas. Eur. J. Cancer Clin. Oncol. 23, 311–321 (1987).
Gogas, H. J., Kirkwood, J. M. & Sondak, V. K. Chemotherapy for metastatic melanoma: time for a change? Cancer 109, 455–464 (2007).
Wermke, M. et al. First-in-human dose escalation trial to evaluate the clinical safety and efficacy of an anti-MAGEA1 autologous TCR-transgenic T cell therapy in relapsed and refractory solid tumors. J. Immunother. Cancer 12, e008668 (2024).
Galon, J. & Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 18, 197–218 (2019).
Lanitis, E., Dangaj, D., Irving, M. & Coukos, G. Mechanisms regulating T-cell infiltration and activity in solid tumors. Ann. Oncol. 28, xii-18–xii32 (2017).
Morotti, M. et al. Promises and challenges of adoptive T-cell therapies for solid tumours. Br. J. Cancer 124, 1759–1776 (2021).
Melero, I., Rouzaut, A., Motz, G. T. & Coukos, G. T-cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discov. 4, 522–526 (2014).
Gajewski, T. F. et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr. Opin. Immunol. 25, 268–276 (2013).
Xie, X., Li, X. & Yao, W. A narrative review: depth of response as a predictor of the long-term outcomes for solid tumors. Transl. Cancer Res. 10, 1119–1130 (2021).
Heinemann, V. et al. Early tumour shrinkage (ETS) and depth of response (DpR) in the treatment of patients with metastatic colorectal cancer (mCRC). Eur. J. Cancer 51, 1927–1936 (2015).
Martin, T. et al. Ciltacabtagene autoleucel, an anti-B-cell maturation antigen chimeric antigen receptor T-cell therapy, for relapsed/refractory multiple myeloma: CARTITUDE-1 2-year follow-up. J. Clin. Oncol. 41, 1265–1274 (2023).
Betof Warner, A. et al. Long-term outcomes and responses to retreatment in patients with melanoma treated with PD-1 blockade. J. Clin. Oncol. 38, 1655–1663 (2020).
D'Angelo, S. P. et al. Identification of response stratification factors from pooled efficacy analyses of afamitresgene autoleucel (‘afami-cel’ [formerly ADP-A2M4]) in metastatic synovial sarcoma and myxoid/round cell liposarcoma phase 1 and phase 2 trials. J. Clin. Oncol. 40, 11562 (2022).
Chesney, J. et al. Efficacy and safety of lifileucel, a one-time autologous tumor-infiltrating lymphocyte (TIL) cell therapy, in patients with advanced melanoma after progression on immune checkpoint inhibitors and targeted therapies: pooled analysis of consecutive cohorts of the C-144-01 study. J. Immunother. Cancer 10, e005755 (2022).
Tawbi, H. A. et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N. Engl. J. Med. 386, 24–34 (2022).
Joseph, R. W. et al. Baseline tumor size is an independent prognostic factor for overall survival in patients with melanoma treated with pembrolizumab. Clin. Cancer Res. 24, 4960–4967 (2018).
Locke, F. L. et al. Tumor burden, inflammation, and product attributes determine outcomes of axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv. 4, 4898–4911 (2020).
Cappell, K. M. & Kochenderfer, J. N. Long-term outcomes following CAR T cell therapy: what we know so far. Nat. Rev. Clin. Oncol. 20, 359–371 (2023).
Davar, D. et al. Phase I dose escalation of IMC-C103C, a CD3×MAGE-A4 T-cell receptor (TCR) bispecific protein. Ann. Oncol. 32, S1411–S1413 (2021).
Majzner, R. G. & Mackall, C. L. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 8, 1219–1226 (2018).
Da Via, M. C. et al. Homozygous BCMA gene deletion in response to anti-BCMA CAR T cells in a patient with multiple myeloma. Nat. Med. 27, 616–619 (2021).
Gettinger, S. et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. 7, 1420–1435 (2017).
Paschen, A., Melero, I. & Ribas, A. Central role of the antigen-presentation and interferon-γ pathways in resistance to immune checkpoint blockade. Annu. Rev. Cancer Biol. 6, 85–102 (2022).
Lee, D. W. et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 124, 188–195 (2014).
Eisenhauer, E. A. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247 (2009).
Walter, S. et al. Cutting edge: predetermined avidity of human CD8 T cells expanded on calibrated MHC/anti-CD28-coated microspheres. J. Immunol. 171, 4974–4978 (2003).
Kobayashi, E. et al. A new cloning and expression system yields and validates TCRs from blood lymphocytes of patients with cancer within 10 days. Nat. Med. 19, 1542–1546 (2013).
Willcox, B. E. et al. Production of soluble αβ T-cell receptor heterodimers suitable for biophysical analysis of ligand binding. Protein Sci. 8, 2418–2423 (1999).
Kim, G. B. et al. Quantitative immunopeptidomics reveals a tumor stroma-specific target for T cell therapy. Sci. Transl. Med. 14, eabo6135 (2022).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Field, M. G. et al. PRAME as an independent biomarker for metastasis in uveal melanoma. Clin. Cancer Res. 22, 1234–1242 (2016).
Acknowledgements
Immatics US, Inc. is the sponsor of this trial. We thank all patients for their participation and acknowledge the support of their families throughout the trial. Furthermore, we thank A. Alpert and S. Kuttruff-Coqui for contributing to the development of Immatics’ adoptive T cell therapy concept; L. Alten for contributing to the TCR identification, design and characterization; Z. Coughlin for leading the manufacturing of IMA203 products; K. Sieger for quality control oversight; O. Ebert and D. Pankov for the medical monitoring during the trial; J. Ludwig for overseeing IMA203 pharmacovigilance; and H. Bösmüller for pathology consultations. M. Baumeister, B. Chesson, D. Alcoba, E. Farrar, S. Grund-Gröschke, J. Gunesch, F. Hoffgaard, F. Köhler, A. Schmidmeir, A. Marisetty, M. Ruh and our Clinical Trial Sample Management Team supported the scientific work, data analysis, preparation of figures and/or writing of the manuscript. Finally, we thank A. Acs for coordinating the compilation of the manuscript and the Cancer Prevention and Research Institute of Texas for co-funding this work (DP150029). The funders had no role in study design, data collection and analysis, preparation of the manuscript or decision to publish. S.W. has received the funding. Immatics’ work on IMA203 was supported by the Cancer Prevention and Research Institute of Texas.
Author information
Authors and Affiliations
Contributions
Conception or design of the work: A.M.-M., A.M., O.S., C.R., H.S.-J., S.W., C.M.B., H.G.R. and M.W. Acquisition, analysis or interpretation of data: M.W., D.M.A., M.C., A.M.T., T.A.W.H., A.A.J., R.R., C.B., W.A., K.W., P.B., J.J.L., K.A., L.B., J.F., S.G., S.H., M.B.H., J.H., M.K., M.L., R.M., M.A.K. and A.S. Manuscript drafting and revision: S.B., N.H., D.K., M.A.K., D.M., K.P., H.S., C.W., H.R., T.W. and M.W.
Corresponding author
Ethics declarations
Competing interests
M.W.: honoraria: Amgen, Boehringer Ingelheim, Bristol Myers Squibb, Eli Lilly, GWT, Janssen, Merck Novartis, Pfizer, Serono and Synlab; consulting or advisory role: Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Bristol Myers Squibb, Daiichi Sankyo Europe GmbH, Eli Lilly, ImCheck Therapeutics, Immatics, ISA Pharmaceuticals, Novartis, PharmaMar, Regeneron, Tacalyx and Zymeworks; research funding (to institution): Roche; travel, accommodations, expenses: Amgen, AstraZeneca, Bristol Myers Squibb, Daiichi Sankyo Europe GmbH, GEMoaB, Immatics, Iovance Biotherapeutics, Janssen Oncology, Merck/Senoro, Pfizer and Sanofi/Aventis. D.M.A.: research funding: Adaptimmune, GlaxoSmithKline and Immatics. A.M.T.: consulting or advisory role: Avstera, BioEclipse Therapeutics, BrYet, Diaccurate, Macrogenics, Nex-I and Vincerx; research funding: BrYet (to institution), AbbVie, Agenus, Immatics, Macrogenics, Novocure Ltd., OBI Pharma, Orionis, Parker Institute for Cancer Immunotherapy, Tachyon, Tempus, Tvardi Therapeutics and Vividion Therapeutics. T.A.W.H.: honoraria: Amgen, Bristol Myers Squibb, GlaxoSmithKline and Jazz Pharmaceuticals; consulting or advisory role: AbbVie, Amgen, Bristol Myers Squibb, GlaxoSmithKline, Janssen, Jazz Pharmaceuticals, Kite/Gilead, Novartis, Otsuka, Pfizer, Pierre-Fabre and Sanofi; travel, accomodations, expenses: AbbVie, Amgen, Astellas, BeiGene, Bristol Myers Squibb, GlaxoSmithKline, Immatics, Janssen, Jazz Pharmaceuticals, Kite/Gilead, Kyverna Therapeutics, Medac, Neovii, Sanofi and Sobi. A.A.J.: stock or other ownership: Avenge Bio; consulting or advisory role: Avenge Bio, Eisei, GLG, Guidepoint, Macrogenics, Sentinel Bio and Theolytics; research funding: AstraZeneca, Eli Lilly, Immatics, Immunon, Iovance, Macrogenics, Merck and Xencor; travel, accomodations, expenses: Striker. R.R.: consulting or advisory role: Allogene, Autolus, Gilead, Incyte, Orca Bio, Quell Biotherapeutics, Sana Biotechnology and TScan; research funding (all to institution): AbbVie, Atara Biotherapeutics, Bristol Myers Squibb, Cabaletta, CareDx, Genentech, Gilead, Immatics, Incyte, Johnson & Johnson, Precision Biosciences, Sanofi, Synthekine, Takeda, TCR2 and TScan; expert testimony: Bayer; travel, accommodations, expenses: Gilead and Johnson & Johnson. C.B.: consulting fees: AOK Germany, AstraZeneca, Bayer Healthcare, BioNTech, Lindis Biotech and Sanofi Aventis; payment or honoraria for lectures, presentations, speakersʼ bureaus, manuscript writing or educational events: med update, Merck Serono and Roche Pharma; data safety monitoring board or advisory board: German Society of Hematology and Oncology (DGHO), Hamburg Cancer Society (HKG), German Cancer Society (DKG), National Network of German Cancer Centers (CCC)/German Cancer Aid (DKH) and Northern Germany Society of Internal Medicine (NWGIM). W.A.: consulting or advisory role: Janssen; research funding (received through institution): Affimed and BioNTech; travel, accommodation, expenses: BioNTech, Immatics and Janssen; honoraria: Astellas, AstraZeneca, GlaxoSmithKline and Janssen. H.-G.R.: stock or other ownership: CureVac, Immatics and ViferaXS; consulting or advisory role: Immatics; patents, royalities, other intellectual property: CureVac, Immatics and ViferaXS. P.B.: consulting or advisory role: BeiGene, Bristol Myers Squibb and Kite; research funding: Bristol Myers Squibb. J.J.L.: data safety and monitoring board: AbbVie, Agenus, Evaxion, Immutep and Shionogi; scientific advisory board (no stock): 7 Hills, Affivant, BioCytics, Bright Peak, Exo, Fstar, Inzen, RefleXion and Xilio; scientific advisory board (stock): Actym, Alphamab Oncology, Arch Oncology, Duke Street Bio, Elipscience, Kanaph, NeoTx, Onc.AI, OncoNano, physIQ, Pyxis, Saros, Stipe and Tempest; consultancy with compensation: AbbVie, Agenus, Alnylam, AstraZeneca, Askgene, Atomwise, Bayer, Bristol Myers Squibb, Castle, Checkmate, Codiak, Crown, Cugene, Curadev, Day One, Eisai, EMD Serono, Endeavor, Flame, G1 Therapeutics, Genentech, Geneos, Gilead, Glenmark, HotSpot, Kadmon, Ko Bio Labs, Krystal, KSQ, Janssen, Ikena, Inzen, Immatics, Immunocore, Incyte, Instil, IO Biotech, LegoChem, Lyvgen, Macrogenics, Merck, Mersana, Nektar, Novartis, Partner, Pfizer, Pioneering Medicines, PsiOxus, Regeneron, Replimmune, Ribon, Roivant, Servier, STINGthera, Storm, Sumoitomo, Synlogic, Synthekine and Teva; research support (all to institution): AbbVie, Astellas, AstraZeneca, Bristol Myers Squibb, Corvus, Day One, EMD Serono, Fstar, Genmab, Hot Spot, Ikena, Immatics, Imugene, Incyte, Janux, Kadmon, KAHR, Macrogenics, Merck, Synlogic, Takeda, Trishula, Tizona, Tscan, Werewolf and Xencor; patents: US-11638728 (‘Microbiome biomarkers for anti-PD-1/PD-L1 responsiveness: diagnostic, prognostic and therapeutic uses thereof’). K.A., L.B., S.B., J.F., S.G., S.H., N.H., J.H., M.A.K., D.M., A.M.-M., R.M., M.L., H.S., O.S., C.W., C.R., H.S.-J., T.W. and C.M.B. were employees of Immatics Biotechnologies GmbH during the course of this work and may have securities from Immatics. M.B.H., M.K., D.K., A.M., K.P., A.S. and S.W. were employees of Immatics US, Inc. during the course of this work and may have securities from Immatics. The other authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks Christopher Klebanoff, Menggang Yu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Saheli Sadanand and Jean Nakhle, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 PRAME mRNA expression across many tumor indications is unimodal.
PRAME target expression distribution (dark gray histograms) based on TCGA RNAseq data where available, patient data (dots, colors indicate BOR according to RECIST1.1) based on in-house qPCR testing of screening biopsies. Numbers in parentheses indicate the number of TCGA patient samples used for TCGA prevalence calculation. 1PRAME target prevalence is based on TCGA RNAseq data combined with a MS-guided RNA expression threshold. 2PRAME target prevalence in uveal melanoma based on in-house qPCR testing of screening biopsies from clinical trial patients (n=33) demonstrates substantial higher prevalence of 91% compared to prevalence based on TCGA data of 50%, TCGA: early & late-stage primary tumor samples; Immatics clinical trials: late-stage/metastatic tumor samples, indicated elevated expression of PRAME in late-stage uveal melanoma patients64. BOR: best overall response; cPR: confirmed partial response; HNSCC: head and neck squamous cell carcinoma; MPNST: malignant peripheral nerve sheath tumor; MS: mass spectrometry; NA: not applicable; PD: progressive disease; PR: partial response; RNAseq: RNA sequencing; SD: stable disease; TCGA: The Cancer Genome Atlas.
Extended Data Fig. 2 PRAME expression across many tumor tissues is homogenous.
Spatial mRNA PRAME expression was analyzed using RNAScope in situ hybridization assay. For tumor tissues, the tumor and stroma regions within the tissue were determined by analyzing hematoxylin and eosin staining. Corresponding serial sections were stained for PRAME expression (lower panel). PRAME expression is seen as red punctate dots homogenously distributed throughout the tumor regions. The staining was quantified using immunoreactivity score depicted in the adjacent table. H&E: hematoxylin and eosin; IRS: immunoreactivity score.
Extended Data Fig. 3 Manufacturing process delivers cells enriched in effector memory phenotype with favorable co-stimulation phenotype.
Phenotypic analysis of collected apheresis (n=40) material and final products (n=40) of 40 patients treated with IMA203. Flow cytometric quantification of a) naïve, b) CM, c) TEM, d) TEMRA, e) CD62L, f) CD27, g) CD28, h) CD45RO, i) CD57, j) PD-1, k) TIM-3, l) LAG-3, and m) TIGIT expression on CD3+CD8+-gated lymphocytes. Statistical analysis was performed using two-tailed Wilcoxon matched pairs test with 95% confidence interval. Each dot represent an individual patient. Gating strategy is depicted in Supplementary Fig. 20. CM: central memory T-cells; TEM: effector memory T-cells; TEMRA: effector memory T-cells expressing CD45RA.
Extended Data Fig. 4 Shrinkage of metastatic lesions throughout the body after IMA203 application.
Lesion shrinkage under IMA203 treatment in different locations throughout the body. a) Best percent change in diameter of target lesions from baseline of all measured target lesions (one to five per patient) in IMA203 monotherapy population (n=21 in lung, n=31 in liver, n=10 in pleura, n=6 in abdomen/peritoneum, n=5 in skin, n=21 in lymph node and n=31 in other); ‘Other’ includes the following non-exhaustive list of organs: adrenal gland, bladder, kidney, spleen, pelvis, bone, brain and muscle. Box plots represent box bounds as the first and third quartiles, with horizontal lines inside the boxes depicting the median. Whiskers reach the minimum and maximum value of the data and each point represents the best change of one individual target lesion. Colors indicate the BOR according to RECIST 1.1 of the patient. b) Case study: CT scan and target lesion measurement (baseline and post-treatment) of patient 38 (DL5; cutaneous melanoma, post-baseline scan ~9 months post-T-cell infusion), patient 34 (DL5; uveal melanoma, post baseline scan ~12 months post-T-cell infusion), and patient 14 (DL2; synovial sarcoma, post-baseline scan ~24 months post-T-cell infusion). Red circles mark target lesion and green circles mark non-target lesion. BL: baseline; BOR: best overall response; cPR: confirmed partial response; PD: progressive disease; PR: partial response; RECIST: Response Evaluation Criteria in Solid Tumors; SD: stable disease.
Extended Data Fig. 5 Higher TCR T dose, dose:tumor burden ratio, PRAME expression and lower tumor burden are associated with durable responses.
a) IMA203 T-cell dose, tumor burden and TB ratio is associated with durable responses. Dose, that is, number of infused TCR T-cells (transduced viable CD8+ T-cells), tumor burden (sum of diameters of target lesions at baseline), and TCR T dose:TB ratio (approximation by dividing infused TCR T-cells by TB), are depicted for non-responders/short term responders (PD/SD/PR, n=27) and confirmed durable responders (cPR, n=11). Two patients (39 [DL5] and 36 [DL5]) were not included in this analysis since second post-treatment scan was pending at data cut-off and final BOR is not yet determined. Group comparisons were performed using two-sided Mann-Whitney U statistical test. Box plots depict median as horizontal lines within boxes, with box bounds as the first and third quartiles. Whiskers range from minimum to maximum values. b) PRAME mRNA expression from patient tumor biopsies (cPR n=11, PD/SD/PR n=27) was measured by qPCR. Delta cycle threshold (DCt) values meeting the predefined threshold criteria (DCt threshold) were used as predictive for target peptide presentation. Measured DCt is normalized to the DCt threshold (2^-(dCT-Threshold)). Two-sided Mann-Whitney U statistical test was used. Box plots depict median as horizontal lines within boxes, with box bounds as the first and third quartiles. Lower whisker is the minimum value of the data within 1.5 times the interquartile range below the 25th percentile. Upper whisker is the maximum value of the data within 1.5 times the interquartile range above the 75th percentile. c) Impact of pre-treatment PRAME mRNA expression (relative to threshold) on reduction in tumor size of target lesions and impact of pre-treatment PRAME mRNA expression (relative to threshold) on PFS for patients treated with IMA203 T-cells (n=39). The p-value was determined by two-sided Spearman correlation. The p-values and correlation coefficients are depicted in the respective graphs. Each point represents one patient and the respective BOR (color-coded) according to RECIST1.1. For PFS analysis, censored patients were indicated with triangle shapes. BOR: best overall response; cPR: confirmed partial response; NR: non-responder; PD: progressive disease; PFS: progression-free survival; PR: partial response; R: responder; RECIST: Response Evaluation Criteria in Solid Tumors; SD: stable disease; TB: tumor burden.
Extended Data Fig. 6 Rapid T-cell engraftment and long-term persistence observed in pharmacokinetic studies.
IMA203 product persisted up to 743 days post-infusion which was determined using qPCR-based assay. Genomic DNA was isolated from post-infusion PBMC and IMA203 T-cells were detected by using absolute qPCR assay with lentiviral Psi sequence of vector. Number of T-cells is reported per µg of gDNA used in the assay.
Extended Data Fig. 7 Trends emerge with post-infusion activation/differentiation state of IMA203 T-cells association with responses and depth of responses.
a) Memory phenotype associates with responses 2 weeks post-infusion. Changes in central memory phenotype (CCR7+ CD45RA-) from post-infusion (week 2) to FP were compared between non-responders (n=19) vs. responders (n=19). b) Changes in activation/differentiation markers post-infusion at week 2 or week 2 compared to FP between non-responders (n=19) vs. responders (n=20). Week 2 %CD27 expression in tetramer-positive CD8+ T-cells and the difference between week 2 to FP for %CD45RO-expressing tetramer-positive CD8+ T-cell ratio. c) Changes in activation/exhaustion markers post-infusion (week 2) compared to FP associates with clinical responses. Changes in TIGIT and PD-1-expressing tetramer-positive CD8+ T-cell ratio from product level to week 2 post-infusion in non-responders (n=19) vs. responders (n=19). Group comparisons were performed using a two-tailed Mann-Whitney test with 95% confidence interval. A two-tailed Spearman r correlation with 95% confidence interval was performed for correlation analysis. The p-values and correlation coefficients are depicted in the respective graphs. The bar inside the violin plots represents the median value and each point represents one patient and the respective BOR (color-coded) according to RECIST1.1. Gating strategy is depicted in Supplementary Fig. 20. BOR: best overall response; CM: central memory T-cells; cPR: confirmed partial response; FP: final product; NR: non-responder; PD: progressive disease; PFS: progression-free survival; PR: partial response; R: responder; RECIST: Response Evaluation Criteria in Solid Tumors; SD: stable disease.
Extended Data Fig. 8 Antigen processing and presentation machinery is upregulated in tumor microenvironment in responders.
Gene expression analysis for the antigen processing and presentation machinery gene set was performed via RNAseq obtained from matching pre-treatment (Pre) and post-treatment day 42 (D42) tissue biopsies from five responder and three non-responder patients. Gene expression was provided as log2(transcript per million [TPM]) and a gene specific pseudocount. Gene products upregulated more than twofold difference were marked with green while the ones downregulated more than twofold marked with red. The antigen processing and presentation machinery gene set contains the following genes: ABCB9, ACE, AZGP1, B2M, CALR, CLEC4A, ERAP1, ERAP2, FCER1G, FCGR1A, HFE, HLA-A, IDE, IFI30, IKBKB, LNPEP, MFSD6, MR1, PDIA3, SAR1B, TAP1, TAP2, TAPBP, and TAPBPL. NR: non-responder; P: patient; R: responder.
Extended Data Fig. 9 Signs of adaptive resistance observed in the tumor microenvironment.
Gene expression analysis for the antigen processing and presentation machinery gene set was performed via RNAseq obtained from matching pre-treatment (Pre) and post-treatment day 42 (D42) tissue biopsies from five responder and three non-responder patients. Gene expression was provided as log2(transcript per million [TPM]) and a gene specific pseudocount. Gene products upregulated more than twofold difference were marked with green while the ones downregulated more than twofold marked with red. The gene set to find signs of adaptive resistance contained: ARG1, BTLA, CD160, CD244, CD274, CEACAM1, CLEC4G, CTLA4, FCGR2B, FGL1, FOXP3, HAVCR2, HLA-G, HMGB1, IFNA2, IFNB1, KLRC1, LAG3, LGALS3, LGALS9, LILRB1, LILRB4, NECTIN2, NECTIN3, PDCD1, PDCD1LG2, PVR, TBX21, TIGIT, TNFRSF14, and VSIR. NR: non-responder; P: patient; R: responder.
Supplementary information
Supplementary Information
Supplementary Figs. 1–20 and Supplementary Tables 1–6.
Supplementary Data 1
Source data for Extended Figs. 8 and 9.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wermke, M., Araujo, D.M., Chatterjee, M. et al. Autologous T cell therapy for PRAME+ advanced solid tumors in HLA-A*02+ patients: a phase 1 trial. Nat Med (2025). https://doi.org/10.1038/s41591-025-03650-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41591-025-03650-6
