
Abstract
The major crop, soybean, forms root nodules with symbiotic rhizobia, providing energy and carbon to the bacteria in exchange for bioavailable nitrogen. The relationship is host-specific and highly host-regulated to maximize energy efficiency. Symbiotic nitrogen fixation (SNF) is greener than synthetic fertilizer for replenishing soil fertility, contributing to yield increase. Nodulation Outer Protein P (NopP) and NopI of the type 3 secretion system (T3SS) of the rhizobium determine host specificity. Sinorhizobium fredii CCBAU25509 (R2) and CCBAU45436 (R4) have different NopP and NopI variants, affecting their respective symbiotic compatibilities with the cultivated soybean C08 and the wild soybean W05. Swapping the NopP variants between R2 and R4 has been shown to switch their compatibility with C08 with the rj2/Rfg1 genotype. To understand the effects of Nops on host compatibility, analyses on the transcriptomic data of W05 roots and nodules inoculated with S. fredii strains containing Nop variants uncovered many differentially expressed genes related to nodulation and nodule functions, providing important information on the effects of Nops on hosts and nodules.
Similar content being viewed by others

Background & Summary
Soybean (Glycine max) is an important cash crop and leads the oilseed production in the 2022/2023 crop year worldwide. Soybean seeds contain roughly 18% oil and 38% protein1, and are commonly used for human food and animal feed. Soybean establishes symbiosis with soil bacteria (rhizobia) to form root nodules where the rhizobium carries out symbiotic nitrogen fixation (SNF). SNF is an essential biological process and the initial stage of the nitrogen cycle, where nitrogen in the atmosphere is assimilated into ammonia at the expense of sugars supplied by the host. SNF could significantly reduce the usage of synthetic nitrogen fertilizer, the production of which is highly energy intensive with high carbon emission. SNF fulfills 40 to 70% of the plant’s total nitrogen demand, depending on the growing conditions and the compatibility between host plants and rhizobia2,3. Therefore, SNF is critical for soybean growth and yield.
The symbiosis between legumes and rhizobia depends on molecular signals and determining factors produced by both symbiotic partners. Among these factors, rhizobial Nodulation Outer Proteins (Nops) are critical in determining host specificity. Nops are secreted into the host plant cells through the type III secretion system (T3SS)4. T3SS is common to bacterial pathogens of plants that directly delivers effectors into the cytoplasm of host plant cells, thereby helping bacteria evade the host immune response, by altering host signaling pathways and suppressing host defense genes4,5. Nops have dual effects on symbiosis, by promoting symbiosis with one legume while impairing the symbiotic process with another6. For example, NopP from both Bradyrhizobium and Sinorhizobium was responsible for the symbiotic incompatibility with soybean carrying different alleles of Rj2/Rfg14,7. An incompatible Rj2/Rfg1-NopP pair suppressed the formation of the infection thread at two days post-inoculation by activating the host defense gene PR-24. Consequently, the soybean host rejected the rhizobial infection, leading to failure in nodule formation.
In our previous work, the soybean cultivar C08 carrying the rj2/Rfg1 allele was found to be incompatible with Sinorhizobium fredii CCBAU25509 (R2) but compatible with CCBAU45436 (R4)7,8. The incompatibility was due to the sequence variations in NopP between R2 and R4. NopI, the paralog of NopP, also exhibited sequence variations between R2 and R4, although NopI did not appear to contribute to host specificity9.
In this research, to investigate the functions of NopP and NopI in effecting host compatibility and nodule functions, we generated transcriptomic data on a compatible soybean host inoculated with different NopP and NopI variants. The wild-type R2 and R4 strains of S. fredii were used as background and their respective NopP and NopI genes were swapped between them. To be specific, NopP- and NopI-swapped strains of R2 and R4, along with their corresponding wild-type strains and an R2 T3SS mutant (rhcN), were inoculated into the compatible wild soybean W05 carrying the rj2/rfg1 allele. Transcriptomic data from W05 uninfected roots, stripped infected roots and nodules were collected and analyzed. A flowchart of the experimental design is presented in Fig. 1. The purpose of the present study was to analyze the transcriptional changes in soybean nodules attributable to the NopP and NopI variants, to further the investigation into the roles of Nops in mature soybean nodules.

Experimental design of this study. To detect the transcriptome profile changes in wild soybean (W05) roots and nodules inoculated with different strains of Sinorhizobium fredii versus uninfected roots, stripped roots and nodules were harvested 28 days after inoculation. For each strain, there were three biological replicates, each consisting of three individual plants harvested together as one sample. Three biological replicates per treatment were used for transcriptome sequencing. Only the clean reads were used for analyses. R2 and R4 are two wildtype strains of S. fredii with different host compatibilities. R2p4, R2 with NopP from R4 swapped in; R2i4, R2 with NopI from R4 swapped in; R4p2, R4 with NopP from R2 swapped in; R4i2, R4 with NopI from R2 swapped in; rhcN, R2 T3SS mutant.
Methods
T3SS, NopP- and NopI-swapped mutants of Sinorhizobium fredii CCBAU25509 (R2) and CCBAU45436 (R4)
The R2 T3SS mutant (rhcN::Tn5, rhcN thereafter) and NopP-swapped mutant of R2 (R2p4) and R4 (R4p2) were constructed in our previous work7. For the NopI mutants, the NopI coding sequences with ~1 kb upstream/downstream were amplified from the genomic DNA of R2 and R4, respectively, and inserted into pK18mobsacB (ATCC [American Type Culture Collection], Cat.# 87097). The protocol for swapping of NopI genes was described in our previous study7. The primers for generating and screening the mutants can be found in Supplementary Table 1.
Cultivation of bacteria and plants
Seeds of the wild soybean (Glycine soja) accession W0510 were surface-sterilized with chlorine gas for 16 h and germinated in sterilized vermiculite in the greenhouse in total darkness11. Four days after sowing, seedlings were transferred into individual pots supplemented with 1X B & D solution according to a previous report12.
The Sinorhizobium fredii strains (R2, R2p4 [R2 with NopP from R4], R2i4 [R2 with NopI from R4], rhcN [an R2 T3SS mutant], R4, R4p2 [R4 with NopP from R2] and R4i2 [R4 with NopI from R2]) were cultured in TY medium containing 5 g L−1 tryptone, 3 g L−1 yeast extract and 0.88 g L−1 CaCl2 at 28 °C with shaking at 180 rpm for 40 h13. Before inoculation, the rhizobium cultures were harvested by centrifugation at 1,500 rpm and resuspended in sterile saline solution (0.9% NaCl w/v) to a final OD600 = 0.2. One milliliter of the bacterial suspension was inoculated onto the root of each seedling in the vermiculite. Control plants were prepared following the same procedure but inoculated with sterile saline only. At 28 days after inoculation, nodules and roots were harvested separately and frozen immediately in liquid nitrogen. The samples were then stored at −80 °C for RNA extraction later.
RNA extraction and RNA-Seq
Twenty-four root samples (three uninfected roots and 21 stripped roots) and 21 mature nodule samples, with three biological replicates per treatment/strain-Nop combination per tissue, were used for total RNA extraction using RNAiso Plus (Takara, Cat.# 9108/9109). Total RNAs were sent to Novogene Co, Ltd. for RNA sequencing. Strand-specific polyA-enriched libraries were sequenced on an Illumina NovaSeq6000 platform to generate at least 50 million PE150 reads for each library.
Analysis of differential gene expression and quantification of gene expressions
The nf-core RNAseq version 3.3 pipeline (https://github.com/nf-core/rnaseq) was employed for the processing and analysis of raw RNA sequencing data. This pipeline utilizes STAR, DESeq2, and Salmon to generate gene counts and conducts comprehensive quality control assessments. Raw RNA sequencing data, including 45 sequencing data files, and the wild soybean W05 reference genome10, were utilized. Within the nf-core/rnaseq pipeline, quality control procedures involved the utilization of FastQC version 0.11.9 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) for adapter trimming, filtering of low-quality sequences, and the removal of bases with a Phred quality score below 20. Subsequently, alignment was performed using STAR version 2.7.6a14 to map the sequencing reads to the reference genome. The expression levels were estimated using Salmon version 1.4.015, and the gene expression matrix was generated. To evaluate sample similarities based on gene expression profiles, a sample similarity clustering heatmap was generated using the expression data processed through the nf-core/rnaseq pipelineIn R language. Euclidean distance was used to calculate the similarity between samples, and R package pheatmap was used to visualize the clustering heatmap.
DESeq2 version 1.28.016 was employed to detect differential gene expression. The negative binomial distribution model was utilized to characterize the distribution properties of RNA-seq data and assess gene expression changes under varying conditions. Significantly altered gene expression levels were identified using the criteria of log2(fold change) ≥ 1 and FDR (False Discovery Rate) ≤ 0.05.
Data Records
The raw data used in this work was submitted to the Sequence Read Archive (SRA) at the National Center for Biotechnology Information (NCBI) database with the accession number PRJNA1112908.
This data set consists of 45 sample files, which are classified into the following three main categories according to the Sinorhizobium fredii strains used (R2, R4 and uninfected control):
-
1.
Soybean W05 infected with R2, R2p4, R2i4 and rhcN, including stripped root and nodule tissues, each with three biological replicates, named as R2/R2p4/R2i4/rhcN_root/Nod _replicate_number_r1/r2/r317.
-
2.
Soybean W05 infected with R4, R4p2 and R4i2, including stripped root and nodule tissues, each with three biological replicates, named as R4/R4p2/R4i2_root/Nod_r1/2/317.
-
3.
Soybean W05 mock-infected with saline, including uninfected root tissues only, with three biological replicates, named as UNino_root_r1/2/317.
The processed data files, gene_counts.tsv and gene_tpm.tsv, were deposited on Gene Expression Omnibus (GEO) at the NCBI database with the accession number GSE27476818.
Technical Validation
Data quality assessment
The symbiosis between soybean and rhizobium can significantly enhance soybean growth under low nitrogen conditions and improve the plant nitrogen content, making leaves greener. To investigate the effect of swapping NopP and NopI between R2 and R4, we inoculated wild soybean W05 with saline (uninoculated), R2, R2p4, R2i4, rhcN, R4, R4p2 and R4i2. The root and nodule samples were harvested to do transcriptome sequencing.
The quality of the RNA-seq data for each sample is shown in Fig. 2. Briefly, 9.93 Gb of data on average were obtained for each sample (Supplementary Table 2). FastQC was used to confirm the base quality of the sequencing reads. The average high-quality score, Q30 (base error < 0.1%), was 95.27% (Fig. 2A).

Technical validation of the transcriptomic data. (A) Overview of the RNA-seq data. Raw reads (top panel), raw data information (second panel), Q30 (%) (third panel) and GC (%) (bottom panel) for each RNA-seq library were plotted. (B) A heatmap showing the correlation coefficients of the scaled expression levels among all sample pairs. R2 and R4 are two wildtype strains of S. fredii with different host compatibilities. R2p4, R2 with NopP from R4 swapped in; R2i4, R2 with NopI from R4 swapped in; R4p2, R4 with NopP from R2 swapped in; R4i2, R4 with NopI from R2 swapped in; rhcN, R2 T3SS mutant; Unino, uninoculated; Nod, nodule; r1, r2 and r3, replicates 1, 2 and 3.
To assess the reproducibility of the biological replicates, we used DEseq2 to calculate the correlation of the expression profile from each sample. In general, gene expressions were highly correlated among biological replicates of the same kind of tissue samples (i.e., roots or nodules) (Fig. 2B).
Differentially expressed gene (DEG) analysis
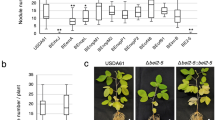
Differential expression analyses between samples/treatments were performed with DEseq2 based on the following criteria: log2(Fold Change) ≥1 and false discovery rate (FDR) <0.05. Summaries of the comparisons are presented in Table 1, Fig. 3 and Supplementary Figure 1. The differentially expressed genes (DEGs) in nodules are shown in Fig. 3 and those from other tissues in Supplementary Figure 1. To assess the data variation caused by Nops, we compared the samples infected with R2 mutants to those infected with wild-type R2 and uninfected roots (Table 1). Similar comparisons were also made with respect to wild-type and mutated R4 strains (Table 1). Among all the R2 mutant-related samples, infection with R2p4 resulted in the lowest number of DEGs in both roots and nodules, with 71 DEGs in roots and 32 in nodules. On the other hand, infection with R2i4 was associated with the highest number of DEGs (1,083 in stripped roots and 2,386 in nodules). rhcN infection had 15 DEGs in roots and 1,468 DEGs in nodules. R4p2 infection resulted in 333 DEGs in roots and 2,325 in nodules. R4i2 infection was associated with 1,100 DEGs in roots and 2,861 in nodules.

The expression profiles of differentially expressed genes in nodules inoculated with different strains of Sinorhizobium fredii carrying variants of NopI and NopP. R2 and R4 are two wildtype strains of S. fredii with different host compatibilities. R2p4, R2 with NopP from R4 swapped in; R2i4, R2 with NopI from R4 swapped in; R4p2, R4 with NopP from R2 swapped in; R4i2, R4 with NopI from R2 swapped in; rhcN, R2 T3SS mutant; nod, nodule; r1, r2, r3, replicates 1, 2 and 3. The expression levels of genes [Log2(FPKM + 1)] were presented in different colors according to the color key.
References
Hartman, G. L., West, E. D. & Herman, T. K. Crops that feed the World 2. Soybean-worldwide production, use, and constraints caused by pathogens and pests. Food Secur. 3, 5–17 (2011).
Ciampitti, I. A. et al. Revisiting biological nitrogen fixation dynamics in soybeans. Front. Plant Sci. 12, 727021 (2021).
Santachiara, G., Borrás, L., Salvagiotti, F., Gerde, J. A. & Rotundo, J. L. Relative importance of biological nitrogen fixation and mineral uptake in high yielding soybean cultivars. Plant Soil 418, 191–203 (2017).
Sugawara, M. et al. Variation in bradyrhizobial NopP effector determines symbiotic incompatibility with Rj2-soybeans via effector-triggered immunity. Nat. Commun. 2018 91 9, 1–12 (2018).
Okazaki, S., Kaneko, T., Sato, S. & Saeki, K. Hijacking of leguminous nodulation signaling by the rhizobial type III secretion system. Proc. Natl. Acad. Sci. 110, 17131–17136 (2013).
Staehelin, C. & Krishnan, H. B. Nodulation outer proteins: double-edged swords of symbiotic rhizobia. Biochem. J. 470, 263–274 (2015).
Rehman, H. M. et al. High-throughput mass spectrometric analysis of the whole proteome and secretome from Sinorhizobium fredii strains CCBAU25509 and CCBAU45436. Front. Microbiol. 10, 2569 (2019).
Jiao, J. et al. Coordinated regulation of core and accessory genes in the multipartite genome of Sinorhizobium fredii. PLoS Genet. 14 (2018).
Jiménez-Guerrero, I., Pérez-Montaño, F., Medina, C., Ollero, F. J. & López-Baena, F. J. The Sinorhizobium (Ensifer) fredii HH103 nodulation outer protein NopI Is a determinant for efficient nodulation of soybean and cowpea plants. Appl. Environ. Microbiol. 83, (2017).
Xie, M. et al. A reference-grade wild soybean genome. Nat. Commun. 10, 1–12 (2019).
Fan, K. et al. MicroRNA 4407 modulates nodulation in soybean by repressing a root-specific ISOPENTENYLTRANSFERASE (GmIPT3). New Phytol. https://doi.org/10.1111/NPH.19222 (2023).
Freiberg, C. et al. Molecular basis of symbiosis between Rhizobium and legumes. Nature 387, 394–401 (1997).
Fan, K. et al. Differentially expressed microRNAs that target functional genes in mature soybean nodules. Plant Genome 14, e20103 (2021).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017 144 14, 417–419 (2017).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, (2014).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP508427 (2024).
NCBI Gene Expression Omnibus https://identifiers.org/geo/GSE274768 (2024).
Ewels, P. A. et al. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 2020 383 38, 276–278 (2020).
Acknowledgements
The work was supported by the Hong Kong Research Grants Council Area of Excellence Scheme (AoE/M-403/16). Any opinions, findings, conclusions, or recommendations expressed in this publication do not reflect the views of the Government of the Hong Kong Special Administrative Region or the Innovation and Technology Commission.
Author information
Authors and Affiliations
Contributions
H.-M.L. planned and coordinated this research. H.-M.L., K.F. and M.-W.L. designed the experimental details. W.-L.C. conducted the swapping of NopP and NopI between the S. fredii strains. K.F., F.-L.W., F.Z. and L.W. collected the root and nodule samples and extracted the total RNA. K.F., Z.X. and L.W. performed the RNA-seq analyses. K.F., M.-W.L. and H.-M.L. wrote the first draft of the manuscript. All authors contributed to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fan, K., Xiao, Z., Wang, L. et al. Transcriptomes of soybean roots and nodules inoculated with Sinorhizobium fredii with NopP and NopI variants. Sci Data 11, 1146 (2024). https://doi.org/10.1038/s41597-024-03964-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03964-z
