
Abstract
Quinoa (Chenopodium quinoa) is an important crop for the future challenges of food and nutrient security. Deep characterization of quinoa diversity is needed to support the agronomic improvement and adaptation of quinoa as its worldwide cultivation expands. In this study, we report the construction of chromosome-scale genome assemblies of eight quinoa accessions covering the range of phenotypic and genetic diversity of both lowland and highland quinoas. The assemblies were produced from a combination of PacBio HiFi reads and Bionano Saphyr optical maps, with total assembly sizes averaging 1.28 Gb with a mean N50 of 71.1 Mb. Between 43,733 and 48,564 gene models were predicted for the eight new quinoa genomes, and on average, 66% of each quinoa genome was classified as repetitive sequences. Alignment between the eight genome assemblies allowed the identification of structural rearrangements including inversions, translocations, and duplications. These eight novel quinoa genome assemblies provide a resource for association genetics, comparative genomics, and pan-genome analyses for the discovery of genetic components and variations underlying agriculturally important traits.
Similar content being viewed by others


Background & Summary
Over the past fifty years, quinoa (Chenopodium quinoa Willd.) cultivation transformed from the state of a subsistence crop for local farmers exclusively grown in the Andean region of South America, to being cultivated or experimented with by more than 110 countries across all continents1. The rapid global expansion of quinoa beyond the Andes arose from the international recognition of the high nutritional value of its seeds, resilience to environmental extremes, tolerance to several abiotic stresses, and the large genetic diversity available for this crop. These qualities make quinoa a promising future crop, specifically for diversifying sustainable cropping systems and food markets which in turn will address nutrient and food security in marginal agricultural regions of the world2,3,4. Along with the spread of quinoa cultivation comes the need to develop breeding programs, collect and catalog the available genetic diversity, and identify favorable allelic variation underlying beneficial traits that will allow quinoa to adapt to new environments and agronomic practices5,6. Underlying these breeding program and genetics studies is the need to generate genomic resources specific to quinoa to support its accelerated improvement.
Quinoa germplasm banks are composed of several thousand accessions, both landraces and cultivars, from diverse geographical origins7,8. Diversity studies based on molecular markers classified quinoa into two major groups: (1) highland quinoa, which displays the greatest genetic diversity and which spans the high plains of Bolivia, Peru, and Ecuador near its center of origin by Lake Titicaca (between 3650 and 4200 m above sea level); and (2) lowland quinoa, also named coastal or sea-level quinoa, which defines a unique South-Central Chilean ecotype of lesser diversity and has been used to breed quinoa for lowland regions worldwide2,8,9. Additional studies grouped quinoa according to morphological traits linked with adaptation to specific agro-ecological conditions in major production areas, further classifying quinoa into four smaller genetic groups also referred to as ecotypes (Valley, Northern and Southern Highland, and Sea Level)7,10,11,12,13. Phenotypic diversity and genetic variation for adaptation to various environments and stresses have been described within each regional geographical distribution. However, very little is known of the natural allelic variation underlying these traits since genomic resources are still restricted to a limited representation of the available genetic diversity.
Several assemblies have been produced for the quinoa genome (2n = 4x = 36) in the past decade and have been important for these initial characterizations of quinoa genetic diversity and genetic studies. Draft assemblies have been produced for four quinoa accessions, including Kd14, a Japanese inbred accession; PI 614886 (QQ74)15, a coastal Chilean accession; Real16, a highland accession representing the most commonly cultivated commercial variety; and the Bolivian accession CHEN12517. Though fragmented and incomplete, these resources have enabled transcriptomic analysis and targeted gene family approaches for the identification and mapping of genes involved in adaptation and domestication traits including seed saponin synthesis15, quinoa seed quality18, flowering time19, and the response to abiotic16,20,21,22,23,24 and biotic25 stresses. Recently, we produced an improved version, both in terms of contiguity and completeness, of the coastal quinoa PI 614886 genome assembly (QQ74-V226) which has enabled larger-scale genetic studies27,28,29,30,31,32 (both in terms of population size and allelic variation representation) through quantitative genetics and genome-wide association studies. This new assembly, together with the resequencing of a set of quinoa accessions from both highland and lowland groups, uncovered large genomic rearrangements which may have significant implications for quinoa breeding and improvement. The results of that study further emphasized the need to consider not only nucleotide but also structural polymorphism to better understand quinoa diversity, especially as structural variants can be important components underlying phenotypic variation.
Here we present the genome assembly and annotation of a panel of eight quinoa accessions of diverse geographical origins selected to represent the diversity of quinoa phenotypes and to support the genetic improvement and adaptation of quinoa to warm and arid environments. All eight genomes are high-quality, chromosome-scale reference sequences assembled from over 30x genome coverage of PacBio HiFi long-reads, with validation and scaffolding provided by Bionano Saphyr optical maps. We further provide a map of structural rearrangements (inversion, duplication, and translocation) between the eight genomes. These genomes represent important resources for the investigation of the genetic components underlying important agronomic traits for quinoa improvement needed to meet the challenges of adapting to novel environments outside its native cultivation range.
Methods
Plant material
A set of eight quinoa accessions, including three lowland (Regalona and Javi from Chile; D-12282 from Argentina) and five highland genotypes (CHEN-199 and PI-614919 from Bolivia; 03-21-072RM, D-10126 and CHEN-90 from Peru), were primarily selected to represent the diversity of phenotypes (growth habit, panicle architecture, leaf shape, stem and seed colors – Fig. 1) and within the selected diversity for performing relatively well in hot, dry and short-day environments33. Each of these eight accessions is highly homogeneous and exhibits low seed shattering33. Previous analysis of SNPs produced through short-read resequencing indicated that all eight genotypes have high coefficient of inbreeding (F.coef ≥ 0.85; homozygous) and are well distributed across the spread of genetic diversity of quinoa28.

Phenotypic diversity between highland and lowland quinoas. Photos depicting the contrasting phenotypes for plant architecture, leaf shape and panicle between (a) the lowland accession Regalona and (b) the highland accession 03-21-072RM. Photos are from 2019 field trials in Hada Al-Sham, Saudi Arabia.
Reference genome sequencing and assembly
Genome size estimation
We performed genome size estimation for each of the eight quinoa genotypes to inform the genome sequencing and assembly strategy. DNA amounts of each Chenopodium quinoa accession were estimated using CytoFLEX flow cytometer (Beckman Coulter, Brea, USA) equipped with a 488 nm solid-state laser. Samples were prepared following the protocol of Doležel et al.34 using LB01 nuclei isolation buffer and propidium iodide staining. Lycopersicon esculentum cv. Stupicke polni tyckove rane (2 C = 1.96 pg) was used as the internal reference standard35. Five individuals were analyzed per accession, and each plant was analyzed three times on three different days. 2 C nuclear DNA amounts were calculated according to Doležel et al.34. Genome sizes (1 C values) were then determined considering 1 pg DNA equal to 0.978 × 109 bp according to Doležel et al.36. The genome sizes averaged 1.42 Gb with limited variation between accessions, which is in agreement with previous studies37,38. The smallest genome (1.40 Gb) was found for CHEN-199 and the largest (1.43 Gb) for Javi, representing less than 2% genome size difference (Table 1).
DNA extraction and sequencing
The seed stock used for genome sequencing is the second generation of propagation through single-seed descent from the original seeds received from the genebanks. Seeds were sown in pots containing a mixture of 30% sand + 70% soil & rocks (3:1 part), and grown in KAUST greenhouses in short-day conditions (10-hour day length) with 26 °C day / 18 °C night temperatures. Quinoa plants at the bolting stage (50 on the Biologische Bundesanstalt Bundessortenamt und CHemische Industrie (BBCH)-scale39,40) were dark-treated for 48 hours, at which point the youngest leaf material from a single plant for each genotype was collected and immediately frozen in liquid nitrogen and kept at −80 °C until DNA extraction. Samples were ground into powder in liquid nitrogen using mortar and pestle, and high molecular weight (HMW) genomic DNA was isolated using the LeafGo protocol41 for long-read sequencing. DNA was quantified using a Qubit dsDNA HS Assay (Q32851, ThermoFisher Scientific), purity was confirmed using a Nanodrop spectrophotometer by checking that 260/280 and 260/230 ratios and DNA fragments size (>50 kb) was validated using the FemtoPulse system (Agilent, Santa Clara, CA, USA). HiFi libraries were then prepared according to the manual “Procedure & Checklist - Preparing HiFi SMRTbell® Libraries using the SMRTbell Express Template Prep Kit 2.0” (PN 101-853-100, Pacific Biosciences, Menlo Park, CA, USA) with 10 μg DNA sheared using the Megaruptor 2 system (Diagenode, Liège Science Park, Belgium) to obtain an average fragment size of 15–20 kb. Size-selected libraries were sequenced on a PacBio Sequel II system in CCS mode for 30 hours. For each accession, two SMRT cells were sequenced, and an average of 53 Gb PacBio HiFi reads per sample were obtained, corresponding to a ~36-fold coverage (Table 2).
Iso-Seq library preparation and sequencing
Iso-Seq libraries were produced from 5-6 tissues at different developmental stages for the Regalona and 03-21-072RM genotypes (root, shoot, apical meristem, leaves, and flowers for both genotypes, and developing seeds for Regalona only), in order to support the gene annotation of one lowland (Regalona) and one highland (03-21-072RM) reference genome assembly, respectively. Plants issued from the same seed stock as for the genome sequencing were sown and grown in the same soil, temperature and daylength conditions as described in the DNA extraction section. Each sample was made of tissues collected from three plants. Cleaned tissues washed in pure water were flash frozen in liquid nitrogen and kept at −80 °C until processing. RNA extraction was done using RNeasy Mini Kit, Qiagen, Cat. No. 74104, then DNAse treatment was performed using Invitrogen™ DNA-free™ DNA Removal Kit, Catalog No. AM1906. The concentration and purity of the RNA samples were measured on Thermo Scientific™ NanoDrop™ 2000 Spectrophotometer, and RNA integrity was assessed on the Agilent 2100 Bioanalyzer system. Only samples with RNA-integrity (RIN) value above 6 were retained for sequencing. Library preparation following the PacBio Iso-SeqTM Express Template Preparation, and sequencing on Sequel II System was performed at Novogene Co., Ltd.
Bionano optical map
Genome assembly
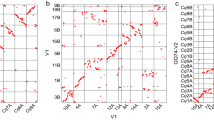
For each of the eight quinoa accessions, PacBio HiFi reads were assembled using hifiasm42 v16.1 with default parameters to generate primary contig assemblies, and hybrid scaffolds were then generated by combining contig assemblies and optical maps using the hybridScaffold pipeline (https://bionano.com/wp-content/uploads/2023/01/30073-Bionano-Solve-Theory-of-Operation-Hybrid-Scaffold.pdf; Bionano Solve version 3.7). Hybrid scaffolds for each quinoa accession were compared to the quinoa reference QQ74-V226 using a dotplot produced by chromeister43. For the accession 03-21-072RM, the dotplot comparison (Fig. 2) showed that the 18 longest (with an average length of 71.2 Mb) hybrid scaffolds out of 21 (with 0.2 Mb average length for the three remaining scaffolds) represented the chromosome-scale assembly, and the orientation and ordering of each pseudomolecule were assigned according to the quinoa reference genome QQ74-V2. Thus, the 03-21-072RM assembly was used as reference to guide the scaffolding of the other quinoa accessions to chromosome-scale level using RagTag44 v2.1.0 with the following parameters: -C -r -u. LTR Assembly Index (LAI) was determined with LTR_retriever45,46 v2.8.7, and BUSCO47 v5.4.5 scores were computed with the eudicots_odb10 lineage dataset to assess the completeness of each genome (Table 3).

Dotplot comparison of the 18 longest hybrid scaffolds of the quinoa accession 03-21-072RM (x-axis) against the quinoa reference genome assembly QQ74-V2 (y-axis).
Genome analysis
Repeat analysis
We performed the annotation, filtering, and consolidation of the repeat elements across the 8 quinoa genomes using default parameters of panEDTA48 with the REPET49,50 curated library provided for QQ74-V226 as input. On average, 66% of each quinoa genome is classified as repetitive sequences (Table 4).
Gene model prediction
For Regalona and 03-21-072RM accessions, the Circular Consensus Sequencing (CCS) application v6.4.0 (https://github.com/PacificBiosciences/ccs) using the continuous long reads subread dataset and default parameters was used to produce HiFi reads. Iso-Seq tool v4.0.0 (https://github.com/PacificBiosciences/IsoSeq) was used to trim primers and polyadenylation (polyA) tails and produce de novo Full Length Non Chimeric (FLNC) transcripts for downstream mapping and annotation.
Then, the FLNC transcripts from the six and five developmental stages were mapped to their corresponding reference assembly using minimap251 v2.17-r941 (parameter: -ax splice -uf –secondary = no -C5 -O6,24 -B4), and the redundant isoforms were further collapsed into transcript loci using cDNA_Cupcake (http://github.com/Magdoll/cDNA_Cupcake; parameter:–dun-merge-5-shorter -C 0.95). All the gff3 files from each sample were merged into a single gtf file for both Regalona and 03-21-072RM accessions using StringTie52 v2.1.7. The Transdecoder.LongOrfs script (https://github.com/TransDecoder/TransDecoder/blob/master/TransDecoder.LongOrfs) was used to identify open reading frames (ORF) of at least 100 amino acids from the merged gtf file. The predicted protein sequences were compared to the UniProt (2021_03) and Pfam35 databases using BLASTP53 (parameters: -max_target_seqs. 1 -outfmt 6 -evalue 1e−5) and hmmer356 v3.3.2 (parameters: hmmsearch -E 1e-10). The Transdecoder.Predict script (https://github.com/TransDecoder/TransDecoder/blob/master/TransDecoder.Predict) was used with the BLASTP and hmmer results to select the best translation per transcript. Finally, the annotation gff3 file was computed using the perl script “cdna_alignment_orf_to_genome_orf.pl” provided in the Transdecoder package.
In addition, a lifting approach using liftoff54 v1.6.3 was combined with the genome-guided approach to predict gene models for Regalona and 03-21-072RM accessions. For the gene lifting, the annotations of QQ74-V2 and Beta vulgaris cv EL10_v1 were independently transferred using liftoff54 (parameters: -a 0.9 -s 0.9 -copies -exclude_partial -polish).
All the output gff files from the lifting and genome-guided approaches were merged into a single file using AGAT (https://github.com/NBISweden/AGAT; perl script “agat_sp_merge_annotations.pl”). The merged file was then post-processed using gffread tool v0.11.7 (parameters:–keep-genes -N -J) to retain transcripts with a start and stop codon and to discard transcripts containing premature stop codons and/or introns with non-canonical splice sites. In total, 46,730 and 43,733 genes were predicted for Regalona and 03-21-072RM, respectively.
For the six others genomes (CHEN-90, CHEN-199, D-10126, D-12282, Javi, and PI-614919), a gene lifting approach using QQ74-V2, Regalona and 03-21-072RM gene models as reference was performed and merged into a comprehensive gff3 file using the same methods described above (using AGAT (https://github.com/NBISweden/AGAT) and gffread55 v0.11.7). Between 43,733 and 48,564 gene models were predicted for the eight new quinoa genomes (Table 5), and the BUSCO47 score was calculated with the protein mode (Table 6). Finally, the putative functional annotations were assigned using a protein comparison against the UniProt database (2021_03) using DIAMOND56,57 (parameters: -f 6 -k1 -e 1e-6). PFAM ___domain signatures and GO terms were assigned using InterproScan58 v5.55-88.039.
Genome visualization
The eight quinoa genomes were uploaded into the Persephone® multi-genome browser (https://web.persephonesoft.com/). The data tracks available are the DNA sequence and gene model prediction. Among other functions provided by the platform, a BLAST34 search and synteny analysis tool within all quinoa genomes is also available (Fig. 3).

Visualization of quinoa genomes using Persephone® genome browser. Example of visualization of the synteny between (a) all chromosomes and (b) Cq7A chromosome of Regalona and 03-21-072RM genomes, and (c) gene and related sequence search function.
Collinearity and structural variation detection
The collinearity and major structural variations (inversions, duplications, and translocations) between QQ74 and the eight new quinoa genomes were assessed using minimap251 v2.26-r1175 (parameters: -ax asm5–eqx) and SyRI59 v1.6.3 with defaults parameters. The results were visualized with plotsr60 v1.1.1 (Fig. 4).

Collinearity and structural rearrangements between QQ74-V2 and the eight quinoa genome assemblies identified using SyRI and visualized with plotsr. On the left and right panels are shown alignments for each chromosome of the A and B sub-genomes, respectively. For each chromosome, the different genomes are ordered per similarity from top to bottom as follows: QQ74-V2, Javi, D-12282, and Regalona for the lowland accessions; 03-21-072RM, PI-614919, CHEN-199, D-10126, and CHEN-90 for the highland accessions. Positions along the chromosomes are given on the x-axes in Mb. Syntenic regions between genomes are shown in grey, inversions in yellow, translocations in green, and duplications in blue.
Data Records
The HiFi and Iso-Seq reads and the final chromosome assemblies were deposited in the Sequence Read Archive at NCBI under BioProject number PRJNA101854861. The raw Bionano optical maps were deposited under EMBL project number PRJEB6627462,63,64,65,66,67,68,69. The final chromosome assembly was deposited at NCBI70,71,72,73,74,75,76,77.
The quinoa assemblies, gene model prediction and the repeat annotations are available on DRYAD Digital Repository (https://doi.org/10.5061/dryad.zkh1893jj)78.
Technical Validation
Assessment of genome assembly and annotation
The average BUSCO47 v5.4.5 (using embryophyta_odb10) score for the eight quinoa genomes is 98.1% at the genome level, indicating a high completeness of the assemblies. In comparison, the BUSCO47 score for the previous quinoa genome QQ74-V2 is 97.9%. The quality of the eight genome assemblies was also assessed with Merqury based on the PacBio HiFi reads using 19-mers. The QV (consensus quality value) scores are in the range of 64.7 and 69 for CHEN-199 and PI-614919, respectively. The k-mer completeness scores were between 98.25% and 99.03 for Regalona and D-10126 genomes, respectively (Table 3).
We validated the concordance of the assembly by re-mapping the optical map onto the pseudomolecules of each of the eight quinoa genomes. Each pseudomolecule was visually inspected using Bionano Access software, and no major discrepancies were found.
Telomeric repeats (TTTAGGG)n26 were screened using tidk79 v0.2.31 (parameter: find -c Caryophyllales) (https://github.com/tolkit/telomeric-identifier). Telomeric repeats are present at the extremities of 35 out of 36 pseudomolecules for the assemblies of 03-21-072RM, CHEN-90, D-10126 and Regalona. Interestingly, telomeric repeat sequences are absent from the short arm of Cq1B in all genome assemblies. Telomeric repeat sequences are missing for one additional chromosome for CHEN-199 (Cq2BL), D-12282 (Cq2BS) and PI-614919 (Cq8AS), while Javi has missing telomere for two additional chromosomes (Cq2AS and Cq7AS).
Completeness of the gene model prediction was evaluated using BUSCO47 v5.4.5 (using embryophyta_odb10, Table 6) and OMark80 v0.3.0 using default settings and the Pentapetalae lineage database (Supplementary Table 1).
Code availability
All software and pipelines were executed according to the manual and protocol of published tools. No custom code was generated for these analyses.
References
Alandia, G., Rodriguez, J., Jacobsen, S.-E., Bazile, D. & Condori, B. Global expansion of quinoa and challenges for the Andean region. Global Food Security 26, 100429 (2020).
Vavilov, N. I. & Dorofeev, V. F. Origin and geography of cultivated plants. Cambridge University Press (1992).
Jacobsen, S.-E. The worldwide potential for quinoa (Chenopodium quinoa Willd.). Food reviews international 19, 167–177 (2003).
Rojas, W., Alandia, G., Irigoyen, J., Blajos, J. & Santivañez, T. Quinoa, an ancient crop to contribute to world food security. Santiago, Chile: FAO, Oficina Regional para America Latina y el Caribe (2011).
Zurita-Silva, A., Fuentes, F., Zamora, P., Jacobsen, S.-E. & Schwember, A. R. Breeding quinoa (Chenopodium quinoa Willd.): potential and perspectives. Molecular Breeding 34, 13–30 (2014).
Murphy, K. M. et al. Quinoa breeding and genomics. Plant breeding reviews 42, 257–320 (2018).
Christensen, S. A. et al. Assessment of genetic diversity in the USDA and CIP-FAO international nursery collections of quinoa (Chenopodium quinoa Willd.) using microsatellite markers. Plant Genetic Resources 5, 82–95 (2007).
Rojas, W. et al. State of the Art Report on Quinoa around the World in 2013. Food and Agriculture Organization of the United Nations, 56–82 (2015).
Gandarillas, H. La quinua (Chenopodium quinoa Willd.): Botánica. La Quinua y la Kañiwa cultivos andinos. Bogota: CIID-IICA, 20–44 (1979).
Tapia, M., Mujica, S. & Canahua, A. A1–A8. Puno, Peru: Proyecto PISCA/UNTA/IBTA/IICA/CIID (1980).
Bertero, H. D., De la Vega, A., Correa, G., Jacobsen, S. & Mujica, A. Genotype and genotype-by-environment interaction effects for grain yield and grain size of quinoa (Chenopodium quinoa Willd.) as revealed by pattern analysis of international multi-environment trials. Field crops research 89, 299–318 (2004).
Curti, R. N. & Bertero, H. D. Botanical context for domestication in South America. The Quinoa Genome, 13–31 (2021).
Wilson, H. D. Quinua biosystematics I: domesticated populations. Economic Botany 42, 461–477 (1988).
Yasui, Y. et al. Draft genome sequence of an inbred line of Chenopodium quinoa, an allotetraploid crop with great environmental adaptability and outstanding nutritional properties. DNA Research 23, 535–546 (2016).
Jarvis, D. E. et al. The genome of Chenopodium quinoa. Nature 542, 307–312 (2017).
Zou, C. et al. A high-quality genome assembly of quinoa provides insights into the molecular basis of salt bladder-based salinity tolerance and the exceptional nutritional value. Cell Research 27, 1327–1340 (2017).
Bodrug-Schepers, A., Stralis-Pavese, N., Buerstmayr, H., Dohm, J. C. & Himmelbauer, H. Quinoa genome assembly employing genomic variation for guided scaffolding. Theoretical and Applied Genetics 134, 3577–3594 (2021).
Grimberg, Å. et al. Transcriptional Regulation of Quinoa Seed Quality: Identification of Novel Candidate Genetic Markers for Increased Protein Content. Frontiers in Plant Science 13 (2022).
Golicz, A. A., Steinfort, U., Arya, H., Singh, M. B. & Bhalla, P. L. Analysis of the quinoa genome reveals conservation and divergence of the flowering pathways. Functional & Integrative Genomics 20, 245–258 (2020).
Mizuno, N. et al. The genotype-dependent phenotypic landscape of quinoa in salt tolerance and key growth traits. DNA Research 27 (2020).
Li, K. et al. Genome-wide identification, phylogenetic analysis, and expression profiles of trihelix transcription factor family genes in quinoa (Chenopodium quinoa Willd.) under abiotic stress conditions. BMC Genomics 23, 499 (2022).
Shi, P. & Gu, M. Transcriptome analysis and differential gene expression profiling of two contrasting quinoa genotypes in response to salt stress. BMC Plant Biology 20, 568 (2020).
Ren, Y. et al. Genome-wide identification and expression analysis of the SPL transcription factor family and its response to abiotic stress in Quinoa (Chenopodium quinoa). BMC Genomics 23, 773 (2022).
Zhu, X., Wang, B., Wang, X. & Wei, X. Genome-wide identification, structural analysis and expression profiles of short internodes related sequence gene family in quinoa. Frontiers in Genetics 13 (2022).
Colque-Little, C. et al. Genetic variation for tolerance to the downy mildew pathogen Peronospora variabilis in genetic resources of quinoa (Chenopodium quinoa). BMC Plant Biology 21, 41 (2021).
Rey, E. et al. A chromosome-scale assembly of the quinoa genome provides insights into the structure and dynamics of its subgenomes. Commun Biol 6 (2023).
Maldonado-Taipe, N., Barbier, F., Schmid, K., Jung, C. & Emrani, N. High-density mapping of quantitative trait loci controlling agronomically important traits in quinoa (Chenopodium quinoa willd.). Frontiers in plant science 13, 916067 (2022).
Patiranage, D. S. et al. Genome-wide association study in quinoa reveals selection pattern typical for crops with a short breeding history. Elife 11, e66873 (2022).
Patiranage, D. S. et al. Haplotype variations of major flowering time genes in quinoa unveil their role in the adaptation to different environmental conditions. Plant, Cell & Environment 44, 2565–2579 (2021).
Emrani, N. et al. An efficient method to produce segregating populations in quinoa (Chenopodium quinoa). Plant Breeding 139, 1190–1200 (2020).
Maldonado‐Taipe, N., Rey, E., Tester, M., Jung, C. & Emrani, N. Leaf and shoot apical meristem transcriptomes of quinoa (Chenopodium quinoa Willd.) in response to photoperiod and plant development. Plant, Cell & Environment (2024).
Rahman, H. et al. Mining genomic regions associated with agronomic and biochemical traits in quinoa through GWAS. Scientific Reports 14, 9205 (2024).
Stanschewski, C. S. Domestication and adaptation of Chenopodium quinoa for marginal environments Doctoral dissertation thesis, King Abdullah University of Science and Technology (2023).
Dolezel, J., Greilhuber, J. & Suda, J. Estimation of nuclear DNA content in plants using flow cytometry. Nat Protoc 2, 2233–2244 (2007).
Doležel, J., Sgorbati, S. & Lucretti, S. Comparison of three DNA fluorochromes for flow cytometric estimation of nuclear DNA content in plants. Physiologia plantarum 85, 625–631 (1992).
Dolezel, J., Bartos, J., Voglmayr, H. & Greilhuber, J. Nuclear DNA content and genome size of trout and human. Cytometry A 51, 127–128, https://doi.org/10.1002/cyto.a.10013 (2003).
Kolano, B., Siwinska, D., Gomez Pando, L., Szymanowska-Pulka, J. & Maluszynska, J. Genome size variation in Chenopodium quinoa (Chenopodiaceae). Plant Systematics and Evolution 298, 251–255 (2012).
Palomino, G., Hernández, L. T. & de la Cruz Torres, E. Nuclear genome size and chromosome analysis in Chenopodium quinoa and C. berlandieri subsp. nuttalliae. Euphytica 164, 221–230 (2008).
Sosa‐Zuniga, V., Brito, V., Fuentes, F. & Steinfort, U. Phenological growth stages of quinoa (Chenopodium quinoa) based on the BBCH scale. Annals of Applied Biology 171, 117–124 (2017).
Stanschewski, C. S. et al. Quinoa phenotyping methodologies: An international consensus. Plants 10, 1759 (2021).
Driguez, P. et al. LeafGo: Leaf to Genome, a quick workflow to produce high-quality de novo plant genomes using long-read sequencing technology. Genome Biology 22, 256 (2021).
Cheng, H. et al. Haplotype-resolved assembly of diploid genomes without parental data. Nature Biotechnology 40, 1332–1335 (2022).
Pérez-Wohlfeil, E., Diaz-del-Pino, S. & Trelles, O. Ultra-fast genome comparison for large-scale genomic experiments. Scientific Reports 9, 10274 (2019).
Alonge, M. et al. Automated assembly scaffolding using RagTag elevates a new tomato system for high-throughput genome editing. Genome Biology 23, 258 (2022).
Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant physiology 176, 1410–1422 (2018).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic acids research 46, e126–e126 (2018).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness. Gene prediction: methods and protocols, 227-245 (2019).
Ou, S. et al. Differences in activity and stability drive transposable element variation in tropical and temperate maize. bioRxiv, 2022.2010. 2009.511471 (2022).
Flutre, T., Duprat, E., Feuillet, C. & Quesneville, H. Considering transposable element diversification in de novo annotation approaches. PloS one 6, e16526 (2011).
Quesneville, H. et al. Combined evidence annotation of transposable elements in genome sequences. PLoS computational biology 1, e22 (2005).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biology 20, 278 (2019).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J Mol Biol 215, 403–410 (1990).
Shumate, A. & Salzberg, S. L. Liftoff: accurate mapping of gene annotations. Bioinformatics 37, 1639–1643 (2021).
Pertea, G. & Pertea, M. GFF Utilities: GffRead and GffCompare. F1000Res 9 (2020).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nature Methods 12, 59–60 (2015).
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nature Methods 18, 366–368 (2021).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Goel, M., Sun, H., Jiao, W.-B. & Schneeberger, K. SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biology 20, 277 (2019).
Goel, M. & Schneeberger, K. plotsr: visualizing structural similarities and rearrangements between multiple genomes. Bioinformatics 38, 2922–2926 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP461962 (2024).
European Nucleotide Archive https://identifiers.org/ebi/biosample:SAMEA114426149 (2024).
European Nucleotide Archive https://identifiers.org/ebi/biosample:SAMEA114426156 (2024).
European Nucleotide Archive https://identifiers.org/ebi/biosample:SAMEA114426151 (2024).
European Nucleotide Archive https://identifiers.org/ebi/biosample:SAMEA114426154 (2024).
European Nucleotide Archive https://identifiers.org/ebi/biosample:SAMEA114426155 (2024).
European Nucleotide Archive https://identifiers.org/ebi/biosample:SAMEA114426150 (2024).
European Nucleotide Archive https://identifiers.org/ebi/biosample:SAMEA114426152 (2024).
European Nucleotide Archive https://identifiers.org/ebi/biosample:SAMEA114426153 (2024).
NCBI Assembly https://identifiers.org/ncbi/insdc.gca:GCA_040571405.1 (2024).
NCBI Assembly https://identifiers.org/ncbi/insdc.gca:GCA_040571485.1 (2024).
NCBI Assembly https://identifiers.org/ncbi/insdc.gca:GCA_040571585.1 (2024).
NCBI Assembly https://identifiers.org/ncbi/insdc.gca:GCA_040571465.1 (2024).
NCBI Assembly https://identifiers.org/ncbi/insdc.gca:GCA_040571445.1 (2024).
NCBI Assembly https://identifiers.org/ncbi/insdc.gca:GCA_040571505.1 (2024).
NCBI Assembly https://identifiers.org/ncbi/insdc.gca:GCA_040571545.1 (2024).
NCBI Assembly https://identifiers.org/ncbi/insdc.gca:GCA_040571565.1 (2024).
Rey, E. et al. Data from: Genome assembly of a diversity panel of Chenopodium quinoa. Dryad Digital Repository. https://doi.org/10.5061/dryad.zkh1893jj (2024).
Brown, M., González De la Rosa, P. M. & Mark, B. A Telomere Identification Toolkit. (Zenodo, 2023).
Nevers, Y. et al. Quality assessment of gene repertoire annotations with OMArk. Nature Biotechnology, 1–10 (2024).
Acknowledgements
We would like to thank Ingrid von Baer for enabling the use of Regalona quinoa in this study, and the public release of all related data to the scientific community. We thank the KAUST Bioscience Core Laboratory for sequencing support, the KAUST Plant Growth Facility Core Lab for greenhouse support, and the KAUST Supercomputing Facilities (https://www.hpc.kaust.edu.sa/) for providing computing resources. This research was supported by the King Abdullah University of Science and Technology (KAUST).
Author information
Authors and Affiliations
Contributions
E.R. and M.Tester conceived the project and planned the analyses. E.R., G.F., C.S., D.E.J., E.N.J., P.J.M. and M. Tester contributed to the germplasm selection. E.R. performed the gDNA extractions, sequence assemblies and validations. J.C. and E.H. performed the genome size estimation analyses. N.R., I.D. and S.C. produced the optical maps and hybrid scaffold assemblies. N.S. performed the RNA extractions. M.A. performed the annotations and variants identification. M.T. and M.K. managed the visualization platform. E.R., M.A. and M. Tester wrote the initial manuscript with input from all authors. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rey, E., Abrouk, M., Dufau, I. et al. Genome assembly of a diversity panel of Chenopodium quinoa. Sci Data 11, 1366 (2024). https://doi.org/10.1038/s41597-024-04200-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-04200-4
