
Abstract
The grain sorghum inbred line 654 serves as a parent for numerous Chinese commercial hybrids and recombinant inbred lines (RILs), which have played a pivotal role in the cloning of several agronomically important traits. In this study, we present a telomere-to-telomere (T2T) genome assembly of the inbred line 654 (728.81 Mb) using PacBio HiFi, ultra-long Oxford Nanopore Technology, and Hi-C sequencing data. The T2T genome assembly has high integrity (contains all of 10 centromeres and 20 telomeres without any gaps), high contiguity (contig N90: 52.02 Mb), high completeness (98.33% BUSCO completeness, 98.88% k-mer completeness, and LAI 24.38), and extremely low base error (3.37 × 10−7, QV: 64.72). A total of 62.34% sequences were identified as repetitive, and rest region contained 44,399 protein-coding genes, of which 30,245 were functionally annotated. The gap-free T2T genome assembly enables the full picture of the potential translational genomics, and provides the highest resolution genetic map for future studies on genome evolution, structure variation, and the genetic control of agronomic traits in sorghum breeding.
Similar content being viewed by others

Background & Summary
The sorghum cultivar 654, developed by the Chinese National Sorghum Improvement Center at the Liaoning Academy of Agricultural Sciences (LAAS), is a high-yielding grain sorghum variety characterized by its photoperiod insensitivity, dwarf, small grain, compact plant architecture, and early maturity1. The inbred line 654 acts as a fundamental parent for modifying numerous restored lines, which have been extensively utilized to breed elite commercial hybrids in China (Fig. 1a). Furthermore, line 654 was utilized to develop various recombinant inbred lines (RILs) in conjunction with other notable sorghum lines, such as the sweet sorghum LTR1082. These have been instrumental in the cloning of several agriculturally significant traits, including grain size3 and color4, mesocarp thickness5, and polyphenol oxidase6. However, these works were rely on the reference genome BTx623, which can only explore the conserved genome regions, but know little about the diversity variety-specific regions. So, high-quality genome assembly of grain sorghum 654 is urgently needed.

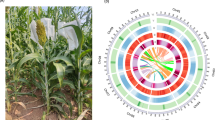
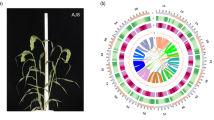
Genome characteristics of T2T assembly of Chinese grain sorghum inbred line 654. (a) Characteristics of the inbred line 654. (b) Circos plot showing genome features including GC content (I), transposable elements (TEs) and satellites (II), and gene density(III). Black triangles represent telomeres and blue lines represent centromeres where containing thousands copies of 137-bp satellites in track II.
The completion of a gapless and telomere to telomere (T2T) genome has always been a long-term goal of genome research. The rapid development of sequencing technologies including PacBio high-fidelity (HiFi) sequencing, ultra-long Oxford Nanopore Technology (ul-ONT) sequencing, and Hi-C sequencing, make the first T2T human genome come true in 2022, taking over twenty years to fix 8% gaps of the original version7,8. The T2T genome of the plant closely followed, maize9, rice10 and soybean11 were completed in 2023. And sorghum kept pace with this progress and the T2T genome assemblies were blowing out in 2024, including baijiu-brewing landraces Hongyingzi12,13 and Huandiaonuo12 released by our group, the reference inbred line BTx62314,15, the red-seeded inbred line Ji205514, and an ancient local landrace “Cuohu Bazi”16. However, no genome assembly of inbred line 654 is available.
In this study, we presented a gap-free telomere-to-telomere (T2T) genome assembly of grain sorghum inbred line 654 in size of 728.81 Mb using 26.53 Gb PacBio HiFi reads (N50: 14.53 kb), 17.74 Gb ul-ONT reads (N50: 100 kb), and 69.75 Gb Hi-C reads (Figs. 1b, 2 and Tables 1, 2). The genome survey yielded a smaller 626-Mb estimated genome size (Fig. 3) and 2n = 20 chromosomes by Karyotyping (Fig. 4). Using all of three types of reads, we first obtained a 736.98 Mb draft genome assembly (V1) consisting of 162 contigs (N50: 69.96 Mb), of which 8 reached T2T chromosome level (Table 3). Then, the top longest 12 contigs were selected and rearranged as a chromosome-level assembly (V2) by comparing with the reference genome BTx623 (Fig. 5a). Only 2 gaps remained, Gap 1 in Chr01 and Gap2 in Chr09 were fixed with model sequence of 358 copies 45S rRNA arrays, and 690 copies of 5S rRNA units, respectively (Fig. 5b). Finally, we obtained a T2T genome assembly (654-T2T) consisting of 10 chromosomes, and no inter- or intra-chromosomal assembly errors were detected by Hi-C chromatin interaction heat map (Fig. 6), each chromosome with one centromere and two telomeres (Figs. 1a, 7a and Table 4). Genome coverage was universal along whole chromosomes (Fig. 7b). Genome completeness assessment revealed 98.88% k-mer based Merqury completeness, 98.33% BUSCO completeness, over 99% read mapping rate (99.14% for HiFi, 99.98% for ONT), and LTR assembly index (LAI) of 24.38 (Tables 1–2 and Fig. 7c,d). The average genome base error evaluated by Merqury with HiFi reads was extremely low at 3.37 × 10−7 (base accuracy > 99.9999%, QV: 64.72) (Table 1 and Fig. 7c). In short, the quality of the 654-T2T genome assembly is comparable to that of other genomes.

Quality and length distribution of ul-ONT (a) and HiFi reads (b).

Genome size estimated by k-mer based GenomeScope using HiFi reads.

Karyotying shown 2n = 20 chromosomes in sorghum 654.

T2T genome assembly finished by gap filling. (a) Two gaps in draft genome assembly shown by Mummer against reference genome BTx623. (b) Gap1 in chromosome 1 fixed by 358 copies of 45S rRNA array and Gap2 in chromosome 9 fixed by 690 copies of 5S rRNA unit clusters.

Hi-C chromatin interaction heat map.

T2T genome quality assessment. (a) Whole genome alignment shown by Mummer. (b) Genome coverage shown by HiFi and ul-ONT reads. (c) Base accuracy and assembly completeness assessed by Merqury using HiFi reads. (d) Assembly and gene set BUSCO completeness against embryophyta_odb10 (n = 1614) database.
We identified 62.34% repeat sequences in the 654-T2T genome assembly, and the LTR retrotransposon (Gypsy: 38.96% and Copia: 5.66%) is the most abundant, followed by satellites (5.75%) (Table 5). A total of 44,399 protein-coding genes were identified, of which 30,245 were functionally annotated (Table 6 and Supplementary Table 1). Comparing with other T2T genome assemblies using homologous genes clustering, we obtained 22,637 core orthogroups (19,744 single-copy orthogroups), and 1,611 unique genes in 654 (Fig. 8a). The complete 654-T2T genome assembly shed light on the black hole regions, such as telomeres and centromeres, and provided a complete picture of the genetic map, for future studies on sorghum diversity, evolution, and new variety-specific agronomic genes to benefit sorghum breeding.

Pfam function of 654 unique genes. (a) 654 variety-specific gene analysis using homologous gene clustering. (b) Top20 Pfam functional analysis of 654 unique genes.
Methods
Plant materials and sequencing
Three-week young whole plants of Chinese grain sorghum inbred line 654 were collected and immediately frozen in liquid nitrogen, and sent to BIOZERON Biotechnology Company Ltd (Shanghai, China) for whole genome sequencing, including PacBio HiFi, 100-kb ul-ONT and Hi-C, and RNA-seq. DNA extraction and sequencing library construction were conducted following relative protocols of sequencing technology. We obtained 26.53 Gb HiFi reads (~36×), 17.74 Gb ul-ONT reads (~24×), and 69.75 Gb Hi-C reads (~96×) for de novo T2T genome assembly, and 9.84 Gb RNA-seq reads and 0.90 Gb clustered Iso-seq reads (GSA accession: CRR933028) from our previous study12 for gene annotation (Table 2 and Fig. 2).
Genome survey
The genome size was estimated using k-mer based methods (k-mer number/k-mer depth) by GenomeScope v217 (genomescope2 -i reads.histo -o genomescope -k 21 -p 2), the k-mer count distribution reads.histo was generated by KMC v3.2.418 truncate the histogram at 10,000 using highly accurate HiFi reads. (kmc -fm -k21 -m64 -ci1 -cs10000 654_hifi.fa reads /tmp/; kmc_tools transform reads histogram reads.histo -cx10000). The estimated genome size of sorghum 654 is ~ 626 Mb with 0.15% heterozygous (Fig. 3), much smaller than previously reported T2T genome assemblies including BTx623 (719.90 Mb)14, Hongyingzi (724.37 Mb)12 and Huandiaonuo (726.89 Mb)12, which may affected by the up to 70% repeats in genome.
Karyotyping
Oligonucleotide-based chromosome painting was used for karyotyping. Oligo libraries derived from sugarcane Saccharum officinarum19 were used and labeled by Cy3 (red) and FAM (green). Fluorescence in situ hybridization (FISH) steps following published protocol in sugarcane19 and maize20. Slides were inoculated overnight at 37°C, washed for 5 minutes in 2 × SSC (RT), for 10 minutes in 2 × SSC (RT), and for 3 minutes in 1 × PBS (RT), finally dried, and counterstained with 10 µL of 4, 6-diamidino-2-phenylindole (DAPI). Chromosomes were imaged using an Olympus BX53 microscope. A total of 10 pairs (2n = 10) of sister chromatids were detected in inbred line 654 (Fig. 4).
T2T genome assembling
Hifiasm v0.19.7-r59821 was employed to generate the draft genome assembly (V1) of sorghum 654 using HiFi, ul-ONT, and Hi-C reads with T2T assembly model (hifiasm -t 32 --h1 Hi-C_R1.fq.gz -- h2 Hi-C_R2.fq.gz -- ul ul-ONT.fq.gz HiFi.fq.gz). The V1 draft genome assembly comprises 162 contigs (N50: 69.96 Mb and N90: 52.02 Mb) in size of 736.98 Mb (Table 3). The V1 draft genome assembly was aligned with the reference genome BTx623 using Mummer v4.0.0rc122 (nucmer–mum -t 32 -b 500 -c 100 -l 10000), then the top 12 longest contigs were reordered and rearranged into chromosome-level assemblies (V2) with only 2 gaps (Gap1 in Chromosome 1 and Gap2 in Chromosome 9), according the virtualization of alignments (mummerplot–png–large -f) (Fig. 5a).
Contigs assembled from whole HiFi reads or gap flanking region mapped HiFi reads, were failed to close any gaps. The boundary sequences of Gap1 in Chromosome 1 were nearly identical repeats of 45S rRNA array (consist of 18S, 5.8S, and 28S rRNA subunits) identified by Infernal v1.1.523 using Rfam v14.724 (cmscan -Z 100 –cut_ga --rfam–nohmmonly --fmt 2 --cpu 60 --tblout). And mapped long reads were also identical repeats of 45S rRNA array. So Gap1 was fixed with artificial model sequence of 45S rRNA array with the mean copies estimated using Blastn v2.1425 against mapped HiFi and ONT reads (-task megablast -max_hsps 5000 -max_target_seqs 100000) (Fig. 5b). Similarly, Gap2 in Chr09 was fixed with artificial model sequences of the mean copies of 5S rRNA units (Fig. 5b). Finally, we got a complete T2T genome assemblies of 654 (654-T2T) in size of 724.37 Mb, by filling artificial model sequences of 358 copies of 45S rRNA assays in Gap1 in chromosome 1 (much higher than other genomes including BTx623, Ji2055, and Cuohu Bazi), and 690 copies of 5S rRNA units in Gap1 in chromosome 9 (Figs. 1, 5 and Table 1).
Evaluation of the genome assembly
The T2T genome assembly quality was assessed by a series of methods. Raw reads were mapping to genome assembly using repeat sensitive long-read mapping algorithm Winnowmap v2.0326 for HiFi (-ax map-pb) and ul-ONT (-ax map-ont) reads. The mapping rate (mapped reads / total reads) was calculated by Samtools v1.16.127 and revealed 99.14% for HiFi reads, 99.98% for ul-ONT reads (Table 2). Hi-C reads were mapped by in 3D-DNA v20100828, then generated a Hi-C chromatin interaction heat map by Juicebox v2.20.0029 to check chromosome integrity and continuity (Fig. 6). The 654-T2T genome assembly was aligned with the reference genome BTx623 using Mummer v4.0.0rc122 to generate a whole-genome plotting and see genome collinearity (Fig. 7a) (nucmer–mum -t 32 -b 500 -c 100 -l 10000; mummerplot–png–large -f). Genome coverage (window = 1 Mb, step = 100 kb) was assessed by Sambamba v1.0.030 (sambamba depth window -t 40 -w 1000000 --overlap 900000) and found universal genome coverage along whole chromosomes (Fig. 7b). Base accuracy was evaluated by reference-free k-mer based assembly evaluation Merqury v1.331 using HiFi reads (meryl count k = 19 654_HiFi.fa.gz output 654.HiFi.meryl; merqury.sh 654.HiFi.meryl 654-T2T.fa 654_merqury). The base quality value score (QV) of 654-T2T genome assembly is 64.72, which means extremely low base error (3.37 × 10−7, <1 bp per 1 Mb), slightly higher than that of BTx623v3, BTx623-CAS and Cuohu Bazi (Table 1 and Fig. 7c). LTR Assembly Index (LAI)32 was calculated by LTR-retriever v2.9.433 based on the intact LTR retrotransposons in assembly. The LAI is 24.38 (>20) suggests 654-T2T assembly touches the highest gold-stand, and does not diff significantly from other T2T genomes (Table 1). BUSCO completeness was performed by BUSCO v5.5.034 using benchmarking universal single-copy orthologs from embryophyta_db10 (n = 1614) in genome model (busco -i 654-T2T.fa -l embryophyta_db10 -m geno for genome assembly or prot for gene annotation). The BUSCO completeness is 98.33% and 98.70%, for 654-T2T assembly and gene set, respectively (Fig. 7d).
The identification of telomeres and centromeres
Telomeres and centromeres were detected by associated simple repeats using TRF v4.0935 (trf 1 1 2 80 5 200 2000 -d -h -r). The chromosome 5′ or 3′ end regions with the 7-bp telomere simple repeat (CCCTAAA / TTTAGGG)n clusters were defined as telomeres (awk ‘$3==7 & & $4>=100). All of the 20 telomeres were identified in the 10 chromosomes of 654-T2T genome assembly, most of them with thousands copies of 7-bp telomeric simple repeats (from 1,011 to 1,967). The 3’ end of chromosome 8 only have 7 copies, which may still have some bug need to fix in future (Table 4).
All of the 10 centromeres were identified by the 137-bp sorghum centromere associated simple repeats36, each one with thousands clustered copies (awk ‘$3==137 & & $4>=100). The 10 centromeres of 654-T2T assembly are in average length of ~7.38 Mb, consist of average ~40,032 copies (28,955 to 48,726) of the 137-bp simple tandem repeats (Table 4).
Repeat sequence identification and masking
de novo transposable element (TE) families in 654-T2T genome assembly were identified and classified by RepeatModeler v2.0437. Then the de novo TE families were used as query library to detect and mask repeats in 654-T2T genome assembly using RepeatMasker v4.1.538. The 654-T2T genome assembly contains a total of 62.34% repeat sequences, of which, the highest abundance repeat is the long terminal repeat (LTR) retrotransposon Gypsy (38.96%), next is satellites (5.75%) and LTR retrotransposon Copia (5.66%) (Table 5).
Gene annotation
Protein-coding genes in 654-T2T genome assembly was identified by BRAKER v3.0339 using both of ab initio prediction and evidence-based prediction (braker.pl–genome = 654-T2T.fa–prot_seq = homo_prot.fa–bam = RNA.bam). Both of RNA sequencing data (Table 2) and homologous proteins from crop genome database Gramene (http://gramene.org/), including maize (B73 AGPv4), sorghum (NCBIv3), rice (IRGSP1.0), and Arabidopsis (TAIR10), were used and obtained 44,399 protein-coding genes. Gene functional annotation were performed by eggNOG-mapper v2.1.940 and revealed 30,245 functional annotated genes, including 11,494 genes with GO41 terms, 8,333 genes with KEGG42 terms, 28,839 genes with Pfam43 terms, 28,931 genes with COG44 terms, and 686 Carbohydrate-Active Enzymes (CAZys)45, (Table 6 and Table).
Unique genes in 654-T2T assembly
Whole-genome representative proteins were selected (one gene one protein) and clustered with other varieties (including BTx623-AGI, Ji2055, Cuohu Bazi, Hongyingzi and Huandiaonuo) using OrthoFinder v2.5.446 (orthofinder -f./Proteins -M msa). We obtained 22,637 core orthogroups (19,744 single-copy orthogroups) present in all of the six varieties, 465 variety-specific orthogroups, and 9,042 singletons not assigned in any orthogroups (Fig. 8a). A total of 1,611 genes were unique in the variety 654, and revealed top5 Pfam enrichment terms including putative gypsy type transposon (85 genes), integrase core ___domain (47 genes), zinc knuckle (45 genes), integrase zinc binding ___domain (45 genes), RNA-dependent DNA polymerase (42 genes)(Fig. 8b).
Data Records
The T2T genome assembly with gene annotations have been deposited in the Genome Warehouse database (GWH, accession: GWHFFNS00000000.147) in the China National Center for Bioinformation (CNCB) and also in the NCBI GenBank (accession: JBLVXU000000000.148). The raw sequence data including HiFi, ul-ONT, Hi-C, and RNA-seq reads have been deposited in the Genome Sequence Archive (GSA, accession: CRA01955449) and co-deposited in the NCBI Sequence Read Archive (SRA, accession: SRP56483750). The genome assembly data and annotation data have also been shared on the Figshare database51.
Technical Validation
The 654-T2T genome assembly quality was assessed in completeness, contiguity and correctness. For completeness, we revealed 99.14% HiFi and 99.98% ul-ONT reads mapping rate, 98.25% Merqury completeness, 98.33% BUSCO completeness (1587 Complete, 11 fragment and 16 missing BUSCOs from embryophyta_odb10, n = 1614) and LAI of 24.38. For contiguity, the 654-T2T genome assembly consists of 10 gap-free chromosomes with all of 20 telomeres and 10 centromeres, has well whole genome collinearity with reference genome BTx623, and no chromosome assembly error detected by uniform genome coverage along chromosomes or Hi-C chromatin interaction heat map. For correctness, the average base error of 654-T2T genome assembly evaluated by Merqury is 3.37 × 10−7 (QV: 64.72).
Code availability
No custom script was used in this work. All analyses were using publicly available software according to the corresponding manual and protocols. The Methods section provides detailed information about the versions and specific parameters of each software. The default parameters were applied if no specific parameters mentioned.
References
Zou, G. H. et al. Genetic variability and correlation of stalk yield-related traits and sugar concentration of stalk juice in a sweet sorghum (Sorghum bicolor L. Moench) population. Aust. J. Crop Sci 5, 1232–1238 (2011).
Zou, G. H. et al. Identification of QTLs for eight agronomically important traits using an ultra-high-density map based on SNPs generated from high-throughput sequencing in sorghum under contrasting photoperiods. J Exp Bot 63, 5451–5462, https://doi.org/10.1093/jxb/ers205 (2012).
Zou, G. H. et al. Sorghum encodes a G-protein subunit and acts as a negative regulator of grain size. J Exp Bot 71, 5389–5401, https://doi.org/10.1093/jxb/eraa277 (2020).
Zhang, L. Y. et al. GWAS of grain color and tannin content in Chinese based on whole-genome sequencing. Theor Appl Genet 136, 77, https://doi.org/10.1007/s00122-023-04307-z (2023).
Feng, Z. et al. Mapping QTL for sorghum physical properties using ultra-high-density bin map. J Plant Genet Resour 23, 1746–1755, https://doi.org/10.13430/j.cnki.jpgr.20220707001 (2022).
Yan, S. et al. Molecular cloning and expression analysis of duplicated polyphenol oxidase genes reveal their functional differentiations in sorghum. Plant Sci 263, 23–30, https://doi.org/10.1016/j.plantsci.2017.07.002 (2017).
Nurk, S. et al. The complete sequence of a human genome. Science 376, 44–53, https://doi.org/10.1126/science.abj6987 (2022).
Rhie, A. et al. The complete sequence of a human Y chromosome. Nature 621, 344–354, https://doi.org/10.1038/s41586-023-06457-y (2023).
Chen, J. et al. A complete telomere-to-telomere assembly of the maize genome. Nat Genet 55, 1221–1231, https://doi.org/10.1038/s41588-023-01419-6 (2023).
Shang, L. et al. A complete assembly of the rice Nipponbare reference genome. Mol Plant 16, 1232–1236, https://doi.org/10.1016/j.molp.2023.08.003 (2023).
Zhang, C. et al. The T2T genome assembly of soybean cultivar ZH13 and its epigenetic landscapes. Mol Plant 16, 1715–1718, https://doi.org/10.1016/j.molp.2023.10.003 (2023).
Bao, J. D. et al. Telomere-to-telomere genome assemblies of two Chinese Baijiu-brewing sorghum landraces. Plant Communications 5, 100933, https://doi.org/10.1016/j.xplc.2024.100933 (2024).
Ding, Y. Q. et al. A telomere-to-telomere genome assembly of Hongyingzi, a sorghum cultivar used for Chinese Baijiu production. Crop J 12, 635–640, https://doi.org/10.1016/j.cj.2024.02.0112214-5141 (2024).
Wei, C. Z. et al. Complete telomere-to-telomere assemblies of two sorghum genomes to guide biological discovery. Imeta 3, e193, https://doi.org/10.1002/imt2.193 (2024).
Deng, Y. et al. A complete assembly of the sorghum BTx623 reference genome. Plant Communications 5, 100977, https://doi.org/10.1016/j.xplc.2024.100977 (2024).
Li, M. et al. Telomere-to-telomere genome assembly of sorghum. Sci Data 11, 835, https://doi.org/10.1038/s41597-024-03664-8 (2024).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nature Communications 11, 1432, https://doi.org/10.1038/s41467-020-14998-3 (2020).
Kokot, M., Dlugosz, M. & Deorowicz, S. KMC 3: counting and manipulating-mer statistics. Bioinformatics 33, 2759–2761, https://doi.org/10.1093/bioinformatics/btx304 (2017).
Yu, F. et al. Chromosome-specific painting unveils chromosomal fusions and distinct allopolyploid species in the complex. New Phytol 233, 1953–1965, https://doi.org/10.1111/nph.17905 (2022).
Braz, G. T. et al. A universal chromosome identification system for maize and wild species. Chromosome Res 28, 183–194, https://doi.org/10.1007/s10577-020-09630-5 (2020).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Marcais, G. et al. MUMmer4: A fast and versatile genome alignment system. PLoS Comput Biol 14, e1005944, https://doi.org/10.1002/cpz1.323 (2018).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935, https://doi.org/10.1093/bioinformatics/btt509 (2013).
Kalvari, I. et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res 49, D192–D200, https://doi.org/10.1093/nar/gkaa1047 (2021).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421, https://doi.org/10.1186/1471-2105-10-421 (2009).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008, https://doi.org/10.1093/gigascience/giab008 (2021).
Jain, C., Rhie, A., Hansen, N. F., Koren, S. & Phillippy, A. M. Long-read mapping to repetitive reference sequences using Winnowmap2. Nat Methods 19, 705–710, https://doi.org/10.1038/s41592-022-01457-8 (2022).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Robinson, J. T. et al. Juicebox.js provides a cloud-based visualization system for Hi-C data. Cell Syst 6, 256–258.e1, https://doi.org/10.1016/j.cels.2018.01.001 (2018).
Tarasov, A., Vilella, A. J., Cuppen, E., Nijman, I. J. & Prins, P. Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034, https://doi.org/10.1093/bioinformatics/btv098 (2015).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol 21, 245, https://doi.org/10.1186/s13059-020-02134-9 (2020).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res 46, e126, https://doi.org/10.1093/nar/gky730 (2018).
Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol 176, 1410–1422, https://doi.org/10.1104/pp.17.01310 (2018).
Manni, M., Berkeley, M. R., Seppey, M. & Zdobnov, E. M. BUSCO: assessing genomic data quality and beyond. Curr Protoc 1, e323, https://doi.org/10.1002/cpz1.323 (2021).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580, https://doi.org/10.1093/nar/27.2.573 (1999).
Melters, D. P. et al. Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol 14, R10, https://doi.org/10.1186/gb-2013-14-1-r10 (2013).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl Acad. Sci. USA 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics Chapter 4, 4.10.1–4.10.14, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Gabriel, L. et al. BRAKER3: Fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS, and TSEBRA. Genome Res 34, 769–777, https://doi.org/10.1101/gr.278090.123 (2024).
Cantalapiedra, C. P., Hernandez-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: functional annotation, orthology assignments, and ___domain prediction at the metagenomic scale. Mol Biol Evol 38, 5825–5829, https://doi.org/10.1093/molbev/msab293 (2021).
Aleksander, S. A. et al. The Gene Ontology knowledgebase in 2023. Genetics 224, iyad031, https://doi.org/10.1093/genetics/iyad031 (2023).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res 53, D672–D677, https://doi.org/10.1093/nar/gkae909 (2025).
Paysan-Lafosse, T. et al. The Pfam protein families database: embracing AI/ML. Nucleic Acids Res 53, D523–D534, https://doi.org/10.1093/nar/gkae997 (2025).
Galperin, M. Y. et al. COG database update 2024. Nucleic Acids Res 53, D356–D363, https://doi.org/10.1093/nar/gkae983 (2024).
Drula, E. et al. The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res 50, D571–D577, https://doi.org/10.1093/nar/gkab1045 (2022).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol 20, 238, https://doi.org/10.1186/s13059-019-1832-y (2019).
CNCB Genome Warehouse database (GWH) https://ngdc.cncb.ac.cn/gwh/Assembly/86159/show (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc:JBLVXU000000000.1 (2025).
CNCB Genome Sequence Archive (GSA) https://ngdc.cncb.ac.cn/gsa/browse/CRA019554 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP564837 (2025).
Wang F. et al. A telomere-to-telomere genome assembly of Chinese grain sorghum 654. figshare https://doi.org/10.6084/m9.figshare.27999161 (2025).
Acknowledgements
This work was supported by the Zhejiang Science and Technology Major Program on Agricultural New Variety Breeding (2021C02064-6).
Author information
Authors and Affiliations
Contributions
Y.Z. and G.Z. conceived and designed the research. J.B. performed bioinformatic analyses and submitted the genome data, F.W. and H.Z. prepared samples, handled sequencing, and wrote the draft manuscript, F.Y. did karyotyping, G.Z., T.S., Z.L., Y.H. discussed and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, F., Bao, J., Zhang, H. et al. A telomere-to-telomere genome assembly of Chinese grain sorghum 654. Sci Data 12, 460 (2025). https://doi.org/10.1038/s41597-025-04791-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-04791-6
