
Abstract
Head and neck squamous cell carcinoma (HNSCC) is notorious for poor prognoses, and effective biomarkers are urgently needed for early diagnosis of HNSCC patients. We investigate the role of alkaline ceramidase 1 (ACER1) and its relationship with immune infiltration in HNSCC. The differential expression and clinical prognostic significance of ACER1 in HNSCC patients are explored using bioinformatics methods and verified in human HNSCC samples. Genetic mutation, DNA methylation and drug sensitivity linked with ACER1 are examined. The potential biological function of ACER1 co-expression genes is assessed, and a series of functional assays are performed on ACER1in vitro. The results comprehensively reveal a relationship between ACER1 and immune infiltration in HNSCC patients. ACER1 expression is significantly downregulated in HNSCC tissues and closely correlated with better prognoses for HNSCC patients, and this prognostic significance is determined by distinct clinical characteristics. Genetic alteration and promoter hypomethylation of ACER1 are involved in progression of HNSCC, and ACER1 expression is significantly related to several drug sensitivities. Functional analysis shows that ACER1 co-expression genes are mainly enriched in the sphingolipid signaling pathway associated with inhibition of tumorigenesis, leading to better prognoses for HNSCC patients. In vitro, ACER1 overexpression inhibits proliferation and migration, induces apoptosis, and promotes adhesion of Fadu and SCC9 cells. In addition, high ACER1 expression is closely linked with infiltration levels of immune cells, and strongly associated with biomarkers of immune cells in HNSCC, suggesting the important role of ACER1 in regulating tumor immunity in HNSCC patients. In summary, ACER1 may be a useful indicator for diagnosis and prognosis, and may regulate immune infiltration in HNSCC patients, thus promising targeted immunotherapy for HNSCC.
Similar content being viewed by others


Introduction
Head and neck cancers is the sixth most common type of cancer in the world, 90% of whom are head and neck squamous cell carcinoma (HNSCC)1,2. HNSCC is a highly aggressive cancer that occurs in the tissues surrounding the head and neck, including the salivary glands and mucosa3,4. Over 900,000 new cases of head and neck cancers are discovered each year, and 64% of patients are diagnosed with locally advanced disease, which leads to more than 500,000 deaths5,6. Despite great advances in diagnosis and therapy, the prognosis of patients with advanced HNSCC remains challenging owing to high rates of recurrence and metastasis, with a 5-year survival rate less than 50%7. Smoking and alcohol abuse are regarded as major risk factors for head and neck squamous cell carcinoma, and it has shown that these unfavorable exposures could contribute to the accumulation of genetic and epigenetic alterations8.While several biological biomarkers have been identified in HNSCC in the past few years, no well-validated prognostic indicators have been discovered. This emphasized the significance of identifying a biomarker for HNSCC diagnosis and treatment.
Molecular targeted therapy is a revolutionary approach to prevent tumor growth, progression, and metastasis by interfering with specific molecules, and it achieves remarkable clinical success in the treatment of different cancer types including lung, breast, head and neck cancers9,10. In our previous research, using the nCounter®Metabolic Pathways, we have determined that metabolic reprogramming occurs and is correlated with cellular stress and immune response in HSCCC. We also find that several metabolism-related genes, such as LDHB, SLC3A2, SLC7A5, and HPRT1, are significantly upregulated and can be unfavorable prognostic markers in HNSCC11. Another study has shown that the CYP4F12 gene may be a potential therapeutic target for HNSCC and restrain the migration of FaDu and SCC-9 cells via EMT pathway12. Furthermore, we have indicated that CCDC60 overexpression obviously inhibits the growth of HNSCC, is linked to a better prognosis for patients with HNSCC, and is implicated in controlling the immune infiltration of HNSCC13. Numerous studies on the possible biological targets and mechanisms of HNSCC have been available in recent years. It has been reported that pleckstrin-2 (PLEK2) is strongly linked to poor overall survival and treatment resistance in HNSCC, and it can stimulate the tumorigenesis via the c-Myc signaling pathway. These findings suggest that PLEK2 may be a promising therapeutic target for HNSCC patients14. The primary treatment difficulty for HNSCC is the response to immunotherapy. According to Cao et al., FAT1 mutations are linked to immunotherapy resistance and engaged in the metabolism-related pathways in HNSCC, which indicates that the FAT1 gene can function as an assistant predictor when employing TMB as a biomarker of drug resistance in HNSCC15. The D. Basu team develop a generalizable procedure for assessing clinically significant targets depending on different cancer types using DepMap. Its application to HNSCC patients reveals new implications for PAK2 inhibition and indicates biomarkers associated with the PAK2 inhibitor responses in HNSCC patients16.
Ceramides are hydrolyzed into sphingosine (SPH) by ceramidases encoded by five different genes, one of which is alkaline ceramidase 1 (ACER1)17. Serving as bioactive lipids, ceramides and their metabolite SPH, as well assphingosine-1-phosphate (S1P), are able to mediate various biological processes18. Initially detected in yeast Saccharomyces cerevisiae, ACER1 is later identified in mammals19,20. Expression of ACER1 is upregulated in the epidermis, and research suggests that ACER1 knockdown may inhibit growth suppression induced by calcium, and mediate differentiation of epidermal keratinocytes21. Lin et al. find that loss of ACER1 promotes cell proliferation by regulating the metabolism of ceramides in the epidermis22. In the past findings, we noticed that compared with the normal tissues, the expression of ACER1 gene was definitely downregulated in the HNSCC samples, but the prognostic significance of ACER1 in HNSCC has not been discussed. Meanwhile, much remains unknown about the underlying roles and mechanisms of ACER1 gene in tumor development and immune engagement with HNSCC.
In our research, we used multiple databases to investigate ACER1 expression and its association with prognoses of HNSCC patients. We explored genomic alterations and DNA methylation of ACER1 in HNSCC, and identified the co-expressed genes of ACER1, as well as their biological function in HNSCC. We used the CellMiner database to examine correlation between ACER1 expression and drug sensitivity, and the TIMER2.0 database for correlation analysis of ACER1 expression and immune cell infiltration in distinct tumor microenvironments. In vitro, we validated ACER1 expression in human HNSCC tissues through the quantitative real-time polymerase chain reaction (qRT-PCR), and further detected the impact of ACER1 expression on proliferation, apoptosis, cell cycle, migration, and adhesion of Fadu and SCC9 cells. Our findings reveal the role of ACER1 in HNSCC and the potential mechanisms between ACER1 and tumor-immune interactions, suggesting that ACER1 may be a promising immunotherapy target for HNSCC.
Materials and methods
Clinical samples
We collected 60 pairs of HNSCC samples from patients undergoing surgery at the Qilu Hospital of Shandong University, Jinan, China. The HNSCC samples were drawn from hypopharyngeal squamous cell carcinoma tissues and patients’ clinicopathologic features was shown in Supplementary Material-1. None of the patients had received any chemotherapy prior to surgery. A total of 60 HNSCC tissues and paired adjacent tissues were gained. Samples were snap-frozen in liquid nitrogen and stored at -80 °C until use. All were randomly selected and provided written informed consent. This research was approved by the Medical Ethnical Committee of Qilu Hospital, Shandong University.
RNA extraction and qRT-PCR
Total RNA was obtained by applying 1 ml TRIzol reagent (Sigma-Aldrich) from tissues, then approximately 1 µg of RNA of each sample was reversely transcripted into cDNA using a reverse transcription kit (TaKaRa). ACER1 mRNA expression was detected by qRT-PCR with SYBR Green PCR Master Mixes (Thermo Fisher). The 2−ΔΔCt method was applied to calculate the relative quantification, and β-Actin was selected as the internal control. The sequences of ACER1 were: GATGCCTAGCATCTTCGCCT (forward primer) and AGGGGATATTGGAGAACGTGT (reverse primer). The sequences of ß-actin were: CACCATTGGCAATGAGCGGTTC (forward primer) and AGGTCTTTGCGGATGTCCACGT (reverse primer).
Gene expression analysis of ACER1
Pan-cancer analysis of the expression of ACER1 was carried out using the Cancer Genome Atlas (TCGA) datasets and the TIMER2.0 database23. The Gene Expression Profiling Interactive Analysis (GEPIA) database was then employed to examine differences in ACER1 expression between tumor and normal tissues in the Genotype Tissue Expression (GTEx) dataset24. Violin plots of ACER1 expression were obtained for four TCGA tumors at different pathological stages via the “pathological stage plot” module of GEPIA. Using the University of Alabama at Birmingham Cancer (UALCAN) portal25, we confirmed ACER1 expression between tumor and normal tissues, and datasets of the four tumors were selected. The UALCAN database was used to study the transcription of ACER1 in HNSCC subgroups based on several clinicopathological features, such as cancer stage, tumor grade, lymph node metastasis, and patients’ gender, race and age. We validated expression of ACER1 in HNSCC tissues from the TCGA datasets (n = 546) using the limma package in R. The ROC curve of ACER1 for HNSCC was plotted using the pROCR package26, and the area under the curve (AUC) was calculated to detect the predictive value of the ACER1 gene. Values of AUC > 0.7 and p < 0.05 suggested high predictive ability for HNSCC.
Survival prognosis analysis of ACER1
We obtained overall survival (OS) and disease-free survival (DFS) maps for ACER1 in the four selected TCGA tumors using GEPIA. The Kaplan-Meier (KM) plotter database was used to plot the survival curve and evaluate the impact of 54,000 genes on survival in 21 human cancers27. The correlations of various clinicopathological factors with ACER1 mRNA expression in HNSCC, OS and relapse-free survival (RFS), were analyzed using the KM plotter, including patients’ sex, race, cancer stage, tumor grade and mutation burden. The hazard ratio (HR) was estimated using a Cox regression model, and the log-rank test was used to determine the statistical significance of differences.
Genetic alteration and DNA methylation analysis of ACER1
The cBioPortal was used to perform genetic alteration analysis of ACER1 in HNSCC and alteration-related prognosis28. The mutation types and substitutional mutation types of ACER1 in HNSCC were studied using the COSMIC database29. Correlation analysis of ACER1 expression and DNA methylation levels in HNSCC was carried out using the UALCAN database and the TCGA dataset. Promoter methylation levels of ACER1 in HNSCC were determined based on sample types, cancer stages, tumor grades, lymph node metastasis, TP53 mutation status, tobacco smoking history, and patients’ gender, race and age.
Correlation between ACER1 expression and drug sensitivity
Correlation between ACER1 expression and anticancer drug sensitivity was performed using the CellMiner database30,31. The “impute”, “limma” and “ggpubr” packages in R were used for data processing and graphing. Finally, the Wilcoxon test of these drugs was conducted to predict the level of response to the drug in HNSCC patients, and the difference was statistically significant at p < 0.05.
ACER1 co-expression genes and Gene Set Enrichment Analysis (GSEA)
We predicted ACER1 co-expression genes in HNSCC from the TCGA dataset (n= 520) using the LinkedOmics database32. A Spearman correlation test was applied, with a false discovery rate (FDR) < 0.001 and rho statistic < 1. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and gene ontology (GO) enrichment analysis of ACER1 co-expression genes were conducted using GSEA tools. Biological processes, molecular functions and cellular components were included in the GO biological process. We selected FDR < 0.05 as the rank standard, with a minimum of three genes and 500 simulations. Using GeneMANIA, we constructed a protein-protein interaction (PPI) network and gene-gene interaction network for ACER1-related co-expressed genes33.
Correlation analysis of ACER1 expression and Immune Cell Infiltration
The relationship between ACER1 and tumor-infiltrating immune cells (TILs) was examined using the TIMER2.0 database. The left-most panel shows the effects of gene expression level on tumor purity34. Next, we explored the correlation between ACER1 expression and gene markers of immune cells by examining markers of B cells, CD4+/CD8+ T cells, natural killer (NK) cells, monocytes, neutrophils, tumor-associated macrophages (TAMs), M1/M2 macrophages, dendritic cells (DCs), T cells and related subtypes. The correlation and Spearman estimates were tested for statistical significance, and log2 RSEM was used to detect gene expression levels.
Gene expression data with Immunotherapy
The IMvigor210 cohort was utilized to investigate the predictive value of ACER1 expression35. Under a Creative Commons 3.0 license, the IMvigor210 dataset was downloaded from a freely available, fully documented software and data package. HNSCC samples with entire clinical information treated with immunotherapy were investigated to determine ACER1 expression.
Cell cultures
Human SCC9 cell line was preserved in our laboratory. Fadu cell line was gained from American Type Culture Collection. SCC9 cells were cultured in DMEM medium (BasalMedia) containing 10% fetal bovine serum (FBS). Fadu cells were maintained in MEM medium (BasalMedia) with 10% FBS. All cells were cultured in a humidified atmosphere containing 5% CO2 at 37 °C.
Plasmid and transfection
ACER1 plasmid was purchased from Vigenebio. Transiently transfection of ACER1 plasmid into Fadu and SCC9 cells was performed by the Lipofectamine 3000 (Thermo Fisher) according to the manufacturer’s recommendations. Cells were incubated in 6-well plates at 2.5 × 105 cells /mL for 24 h. Transfection was performed when the cell fusion reached 50-70%. 5 µL Lipofectamine 3000 reagent and 2 µg plasmid were diluted into 125 µL Opti-MEM (Thermo Fisher) medium, respectively. Then, 5 µL P3000 reagent was added to the diluted plasmid solution. After a short rotation, incubate at room temperature for 5 minutes. The diluted plasmid solution was then added to the diluted Lipofectamine 3000 reagent. After incubation at room temperature for 15 min, the plasmid lipid complex was added to the cells. Forty-eight hours after transfection, cells were collected and sub-cultured for subsequent experiments.
Cell proliferation assay
Approximately 5 × 103 transiently transfected cells were planted into 96-well plates in 100 µL medium and cultured for 24, 48, 72 and 96 h, respectively. 10 µL of cell counting kit-8 (CCK-8, Dojindo) reagent was added into each well. One hour after incubation at 37 °C in the dark, a microplate reader (BioTek) was used to measure the optical density (OD) at 450 nm. The values were processed using the GraphPad Prism7 software.
Flow Cytometric Analysis
Apoptosis of cells was determined using an Annexin V-FITC/PI Apoptosis Detection Kit (Bestbio). Approximately 1.5 × 105 transfection cells were seeded into each well of a six-well plate. After 48 h, the cells were harvested by ethylene-diamine-tetraacetic acid (EDTA)-free trypsin and washed with cold phosphate-buffered saline (PBS), and the collected cells were resuspended in 400 µL binding buffer. The cells were then stained with 5 µL of fluorescein isothiocyanate (FITC) annexin V and 5 µL of propidium iodide (PI) for 15 min at 37 °C in the dark. Cell apoptosis was examined using CytEpert v2.0 (Beckman Coulter).
A cell cycle detection kit (BestBio) was applied to detect changes in the cell cycle distribution. Transfected cells were plated in 6-well plates at a concentration of 2 × 105 cells/mL for 48 h. After the cells were digested with trypsin, they were collected and washed twice with PBS, and fixed with 75% ice-cold ethanol at -20 °C for one hour. Add 20µL Rnase A reagent and incubate at 37℃ for 30 min. After the supernatant was discarded, the cells were re-suspended with 0.5 mL PI staining buffer and incubated in the dark at room temperature for 15 min. Finally, the cell cycle was determined with a flow cytometer (Beckman Coulter).
Wound healing assay
After transfection, cells were seeded into six-well plates at a concentration of 3 × 105 cells/mL. After 24 h, scratch with a 10-ml micropipette tip in the center of the wells. After being washed twice by PBS, the cells were incubated in MEM, or DMEM medium with 1% FBS was added to plates. Pictures of the scratches were taken every 6 h to compute the scratch healing area.
Transwell assay
Transwell chambers were applied to determine the invasion of cells., and approximately 1 × 105 transiently transfected cells were added into the upper chamber with serum-free medium, while medium containing 20% FBS was added to the lower chamber. Cells were maintained at 37 °C for 24 h. After being washed by PBS, the cells were fixed with methanol and stained with 0.1% crystal violet (Solarbio). Finally, the stained cells were counted under a microscope, and stained cells were counted.
Cell adhesion assay
A cell adhesion kit (BestBio) was used to conduct a cell adhesion assay, according to the manufacturer’s recommendations. 100 µL coating buffer was added into each well of a 96-well plate, incubated overnight at 4°C, and then washed with a washing solution. Approximately 5 × 104 cells were seeded and incubated for 1.5 h at 37 °C. Each well was washed with medium, then 100 µL of fresh medium was added. Finally, 10 µL of staining solution B was added and incubated for two hours at 37 °C, and then the OD 450 nm was measured.
Statistical analysis
GraphPad Prism7 and R software (version 4.1.2) were used for data visualization and statistical analysis. The presence and strength of relationships between variables were examined by running Spearman and Pearson correlation tests. The KM method was utilized to evaluate survival analyses. A ROC curve was plotted to examine the predictive performance of the ACER1 risk score for diagnosis. The difference was statistically significant at p < 0.05.
Results
Pan-cancer mRNA expression levels of ACER1
Compared with adjacent normal tissues, ACER1 expression is lower in most types of cancer, based on the TIMER2.0 database (Fig. 1A, p < 0.05). The GEPIA tool demonstrated that ACER1 expression is significantly higher in normal tissues than in tissues of esophageal carcinoma (ESCA), HNSCC, skin cutaneous melanoma (SKCM) and testicular germ cell tumors (TGCT) (Figure S1, p < 0.05). For further verification, differences in ACER1 expression between normal and tumor tissues were explored using the UALCAN database, and the results indicate significantly lower expression only in HNSCC (Fig. 1B, p < 0.001). In addition, expression of ACER1 is significantly connected with pathological stages of HNSCC and TGCT (Fig. 1C, p < 0.05).

Expression analysis of ACER1 in diverse cancers. (A) Levels of ACER1 expression in multiple types of human cancer using TIMER2.0 database. (B) Differences in ACER1 expression levels between tumorous and adjacent normal tissues of ESCA, HNSCC, SKCM and TGCT, based on the TCGA dataset. (C) ACER1 expression levels in patients at different pathological stages of the four selected cancers. * p < 0.05, ** p < 0.01, *** p < 0.001.
Prognostic analysis of ACER1 in cancers
We examined the correlation between ACER1 expression and prognosis in the four selected cancers. OS analysis (Fig. 2A) shows that highly expressed ACER1 is linked with longer survival in HNSCC (HR = 0.55, p = 0.014). Upregulated ACER1 expression is also significantly related to better DFS for HNSCC (HR = 0.56, p = 0.047) in the TCGA datasets (Fig. 2B). However, there is no significant difference in OS and DFS probabilities for the ESCA, SKCM and TGCT groups. Considering the significant difference in the expression and survival analysis of ACER1 in HNSCC, we focused further on the role of ACER1 in HNSCC.

Prognostic analysis of ACER1 in different cancers. (A) Overall survival. (B) Disease-free survival for the four tumors detected by ACER1 gene expression using the GEPIA tool.
To validate the prognostic value of ACER1, we used the KM plotter to detect the effects of sex, race, stage, gender and mutation burden on OS and RFS in HNSCC patients (Table 1). Our results indicate that upregulated ACER1 expression is connected with better OS in male and female, White and Black-African American patients and with two types of mutation burden (p < 0.001). We observe that high expression of ACER1 is linked with longer RFS in stage 2 cancer (DFS, HR = 11.99, p = 0.0046) and better OS in stage 4 (OS, HR = 0.59, p = 0.0037). Specifically, increased ACER1 expression is related to longer OS and RFS of HNSCC patients with grade 2 (OS, HR = 0.68, p = 0.037) and grade 3 tumors (OS, HR = 0.50, p = 0.0095; RFS, HR = 7.52, p = 0.028). These findings reveal that the prognostic significance of ACER1 depends on specific clinicopathological characteristics in HNSCC patients.
ACER1 expression and its relationship with clinical characteristics in HNSCC
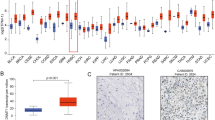
Having confirmed decreased ACER1 expression in HNSCC tissues in the TCGA database (n = 546, p < 0.001; Fig. 3A), we then conducted qRT-PCR to verify ACER1 mRNA expression in HNSCC tissues. This indicated that expression of ACER1 mRNA in 60 pairs of HNSCC tissues was also lower than in paired adjacent healthy tissues (Fig. 3B). In addition, based on the TCGA dataset, analysis of the ROC curve demonstrates that ACER1 has high sensitivity and specificity in HNSCC diagnosis (AUC > 0.74), suggesting that ACER1 may act as a potential biomarker for HNSCC diagnosis (Fig. 3C). Analysis of ACER1 expression according to clinical characteristics of HNSCC patients is illustrated in Fig. 3D–I. Our analysis of cancer stage, tumor grade, lymph node metastasis, gender, race and age reveal that ACER1 expression in different subgroups of HNSCC patients is significantly lower than in the normal group. Interestingly, ACER1 expression in grade 1 tumors is found to be significantly higher than in grades 2, 3 and 4, and significantly lower in grade 3 than in grade 2. With regard to lymph node metastasis classification, ACER1 expression is significantly upregulated in N0 compared with N1, N2 and N3, while expression in N2 is significantly lower than in N1.

ACER1 expression and its relationship with clinical characteristics in HNSCC patients. (A) Decreased ACER1 expression in HNSCC compared with normal tissues, based on the TCGA database (n = 546). (B) ACER1 mRNA expression analyzed by qRT-PCR in 60 pairs of human HNSCC tissues and normal adjacent tissues. (C) ROC curve analysis of ACER1 in HNSCC. Box-whisker plots reveal ACER1 transcription levels in patients with different subtypes of HNSCC, stratifed according to (D) individual cancer stage, (E) tumor grade, (F) node metastasis status, (G) patient gender, (H) race and (I) age. * p < 0.05, ** p < 0.01, *** p < 0.001.
Genetic alteration of ACER1 in HNSCC
Extant research suggests that genetic alterations are closely associated with tumorigenesis and have an impact on cancer prognoses36. We investigated the alteration frequency of ACER1 in 504 HNSCC samples using the cBioPortal database. Our analysis shows that ACER1 altered in 0.99% of samples, including mutation, amplification and deep deletion, with alteration frequencies of 0.4% (2/504), 0.2% (1/504) and 0.4% (2/504) respectively. In exploring the mutation sites of ACER1, we found two missense mutation types occurring in ceramidase S118F and R258Q (Fig. 4A). Further analysis revealed that the most common copy number variations are shallow deletion and diploid in HNSCC patients (Fig. 4B). Between the altered and unaltered groups, the difference in OS probability is not significant, with an overall median of 56.44 months (p = 0.904, Fig. 4C). The results show that overall alteration of ACER1 is not the cause of worse outcomes led by low ACER1 expression. Subsequently, using the COSMIC database to detect mutation types and substitutional mutation types of ACER1 in HNSCC, we found that missense and synonymous substitutions occurred in 38.64% and 12.68% of cases, respectively (Fig. 4D). Substitution mutations occurred mainly in C > T (33.15%) and G > A (32.02%) (Fig. 4E).

Genetic alteration of ACER1 in HNSCC. (A) Summary of alteration types, corresponding frequencies and mutation sites of ACER1 in the cBioPortal database. (B) ACER1 copy number variations in HNSCC. (C) Survival analysis of ACER1 altered and unaltered groups. (D, E) Mutation types and substitutional mutation types of ACER1 using the COSMIC database.
Promoter methylation levels of ACER1 decrease in HNSCC
Hypomethylation has been found to be an important component of epigenetic alterations in tumorigenesis and progression37. Promoter hypomethylation is considered a drug target for lung cancer, as well as a prognostic indicator38. Using the UALCAN database, we observe that, compared with the control, methylation levels of ACER1 promoter are significantly lower in HNSCC tissues (Fig. 5A). We further evaluated the methylation levels of ACER1 promoter in different HNSCC subgroups from the TCGA dataset. Interestingly, levels in stages 3 and 4 cancer groups are lower in HNSCC than in normal tissues (Fig. 5B). Compared with normal tissues, methylation levels of ACER1 promoter for tumors of all grades are lower in HNSCC (Fig. 5C). Our findings reveal that promoter methylation levels of ACER1 in N0 and N2 lymph node metastasis groups are lower than in the control group in HNSCC, and significantly lower in N2 than N1 (Fig. 5D). We find that, compared with the other groups, male HNSCC patients have the lowest methylation, suggesting that ACER1 promoter hypomethylation may be related to gender (Fig. 5E). We also considered tobacco smoking history (Fig. 5F), TP53 mutation (Fig. 5G), and patients’ race (Fig. 5H) and age (Fig. 5I). The results suggest that ACER1 promoter hypomethylation may affect the progress of HNSCC.

Promoter methylation levels of ACER1 in HNSCC. Box-whisker plots show ACER1 promoter methylation levels in patients with different subtypes of HNSCC, stratified according to (A) sample type, (B) individual cancer stage, (C) tumor grade, (D) node metastasis status, (E) patient gender, (F) tobacco smoking history, (G) TP53 mutation, (H) race and (I) age. * p < 0.05, ** p < 0.01, *** p < 0.001.
Correlation between ACER1 expression and drug sensitivity
We examined the correlation between expression of ACER1 and drug sensitivity using the CellMiner database, and found 36 anticancer drugs significantly relating to ACER1 (Supplementary Material-2, p < 0.05). Figure S2 shows the top 10 results. ACER1 expression is positively connected with drug response in patients treated with Etoposide, Fulvestrant, Isotretinoin, Quizartinib and SR16157. In addition, there are negative connections between ACER1 expression and anticancer drugs KX-01, PD-0325901, TAK-733, Trametinib and Varbulin.
ACER1 co-expression genes and Pathway Enrichment in HNSCC
We identified ACER1 co-expression genes and examined their biological function in HNSCC using the LinkedOmics database. As shown in a volcano plot (Fig. 6A), 2,125 genes are positively correlated and 2,845 genes negatively correlated with ACER1 in HNSCC (FDR < 0.001). Figure 6B, C indicate the top 50 genes positively or negatively correlated with ACER1 in HNSCC. Analysis of GO enrichment suggests that ACER1-related co-expressed genes in HNSCC are mainly involved in DNA replication, protein-DNA complex subunit organization, chromosomal region and single-stranded DNA binding (Fig. 6D–6F). KEGG pathway analysis reveals enrichment in the sphingolipid signaling pathway and the Ras signaling pathway, linked with cell cycle and apoptosis (Fig. 6G). Using the GeneMANIA database, we constructed a PPI network to describe the biological functions among genes relating to ACER1. Gene sets enriched with ACER1 are involved in the sphingolipid catabolic process and the sphingoid and sphingosine metabolic processes, as well as the sphingoid biosynthetic process (Figure S3A). These results further validate that ACER1 may regulate cell cycle and induce apoptosis of HNSCC cells through the sphingolipid signaling pathway. Figure S3B shows the KEGG pathway annotation for the sphingolipid signaling pathway.

ACER1 co-expression genes and their functional analysis. (A) Volcano plot of ACER1 co-expression genes. Red dots represent genes positively related to ACER1, and green dots represent negatively related genes. (B) and (C) Top 50 significantly positive and negative genes linked to ACER1 in HNSCC. Functional analysis of ACER1 co-expression genes in HNSCC includes (D) biological processes, (E) cellular components, (F) molecular functions and (G) KEGG pathway using GSEA.
ACER1 Overexpression Inhibits Proliferation, Induces Apoptosis, Suppresses Migration, and Promotes Adhesion of Fadu andSCC9 Cells
To analyze the functional relevance of ACER1, cell proliferation of ACER1 was investigated using CCK-8 assay. Compared with the controls, we observed an apparent decrease in cell viability in both Fadu and SCC9 cells after ACER1 transfection at 24, 48, 72 and 96 h (Fig. 7A, p < 0.05). In the cell apoptosis assay, compared with the control group, overexpression of ACER1 significantly induces apoptosis of Fadu and SCC9 cells (Fig. 7B, P < 0.001); however, the results of the cell cycle assay reveal no significant difference between these groups (Fig. 7C). Next, we detected the role of ACER1 in migration. Twenty-four hours after scratch, we observed that the migration area of Fadu and SCC9 cells transferred with ACER1 was obviously smaller than in the controls (Fig. 8A, p < 0.01). The number of migration cells also decreased after ACER1 overexpression (Fig. 8B, P < 0.05). The assay of cell adhesion demonstrates that the number of adhesion cells with ACER1 plasmid increases significantly in Fadu cells (p < 0.05), but there is no significant difference in SCC9 cells (Fig. 8C, p < 0.05).

ACER1 inhibits growth, induces apoptosis, and regulates cell cycle progression of Fadu and SCC9 cells. (A) Growth curves for Fadu and SCC9 cells transfected with the indicated plasmid, measured by CCK-8 assay. (B) Apoptotic rate of Fadu and SCC9 cells with indicated treatment, measured via flow cytometry from triplicate experiments. (C) Distribution of cell cycle examined by flow cytometry analysis in Fadu and SCC9 cells after overexpressing ACER1.

ACER1 suppresses migration, and promotes adhesion of Fadu and SCC9 cells. (A) Wound-healing assay and related analysis after ACER1 overexpression in Fadu and SCC9 cells. (B) Transwell assay and related analysis after ACER1 overexpression in Fadu and SCC9 cells. (C) Cell adhesion assay in Fadu and SCC9 cells after ACER1 overexpression and related analysis. * p < 0.05, ** p < 0.01, *** p < 0.001.
ACER1 expression linked with Immune Cell Infiltration in HNSCC
Tumor immune microenvironment (TIME) is a complex regulatory network composed of various immune cells, which plays a crucial role in tumor biological process. We employed the TIMER2.0 database to examine correlations between ACER1 and TILs in HNSCC. As shown in Fig. 9 (p < 0.05), we observe that ACER1 expression is adversely associated with the purity of HNSCC (Rho = -0.155, p = 5.52e-04). ACER1 expression is positively linked with levels of immune infiltration of CD8+ T cells (rho = 0.268, p = 1.44e-09), B cells (rho = 0.224, p = 5.36e-07), neutrophils (rho = 0.236, p = 1.11e-07) and mast cells (rho = 0.264, p = 2.59e-09) in HNSCC. Furthermore, ACER1 expression is negatively correlated with infiltration levels of CD4+ T cells (rho = -0.235, p = 1.27e-07), regulatory T cells (rho = -0.103, p = 2.25e-02), monocytes (rho = -0.145, p = 1.30e-03), macrophages (rho = -0.116, p = 1.00e-02) and NK cells (rho = -0.324, p = 1.71e-13) in HNSCC.

Relationship between ACER1 expression and infiltration levels of immune cells in HNSCC using TIMER2.0 database.
Correlation between ACER1 expression and Immune marker sets in HNSCC
In addition, we find that ACER1 expression is closely associated with marker sets of most immune cells in HNSCC (Table 2). ACER1 expression has negative correlations with TAMs markers (CCL2), monocyte markers (CD86 and CSF1R) and M2 macrophage markers (CD163, VSIG4 and MS4A4A) \(Fig. 10A–C). Expression levels of marker sets of DCs (HLA-DPB1, HLA-DQB1, HLA-DRA, HLA-DPA1, NRP1 and ITGAX) are significantly related to ACER1 expression (Fig. 10D). Moreover, expression of ACER1 is negatively linked with T cell exhaustion markers such as PDCD1, PDCDLG2, CTLA4, LAG3, HAVCR2 and GZMB (Fig. 10E).
Subsequently, we conducted correlation analysis between immunotherapy and ACER1 using IMvigor210. The results show that ACER1 expression is closely associated with the prognoses of HNSCC patients treated with anti-PD-1/PD-L1 (Figure S4A, p < 0.05), and that ACER1 may be effectively used to identify immunotherapy responses in patients (Figure S4B, AUC > 0.73).

Relationship between ACER1 expression and biomarkers of immune cells in HNSCC using TIMER2.0 database. Scatterplots of correlations between ACER1 expression and biomarkers of (A) TAM, (B) monocyte, (C) M2 macrophage, (D) dendritic cell and (E) T-cell exhaustion in HNSCC.
Discussion
Early diagnosis of HNSCC patients is extremely challenging due to the absence of clear clinical symptoms39. The death rate for HNSCC remains above 50% even with significant advancements in treatment40. In order to enhance prognoses and minimize the risk of metastasis and recurrence in HNSCC, early disease detection is definitely important41. Therefore, it is crucial to find a promising gene target as a more sensitive biomarker for HNSCC prognosis, and elucidate the molecular mechanism behind it. The rapid advancement of proteomics and RNA sequencing in recent years has made significant contributions to the discovery of biomarkers which are helpful to tumor classification and targeted therapy42. In particular, an in-depth study of multi-omics data can help us comprehend the molecular mechanisms of carcinogenesis even better43. Our study comprehensively examines the key functions of ACER1 in HNSCC, and we identify that ACER1 gene can act as a prognosis biomarker and be involved in regulating immune infiltration in patients with HNSCC via a series of analyses of RNA-seq data, and this is further confirmed through in vitro experiments.
Firstly, pan-cancer analysis has revealed the lower ACER1 mRNA expression in cancer tissues compared to normal tissues. In the TCGA-HNSCC dataset, ACER1 expression is downregulated in HNSCC tissues relative to normal tissues, which is verified by the qRT-PCR results. Additionally, ACER1 expression has a close association with several clinicopathological characteristics of patients with HNSCC, such as cancer stage, tumor grade, lymph node metastasis, gender, race, and age. Afterwards, we substantiate that ACER1 gene exhibits the capacity to significantly differentiate the prognostic outcomes of HNSCC patients in the TCGA cohorts. Our study has found that increased ACER1 expression is related to better prognoses for HNSCC patients. High ACER1 expression is significantly linked with longer OS for stage 4 cancer and grade 2 and 3 tumors, and with better RFS for stage 2 and grade 3. Together, these observations support our hypothesis that ACER1 is a prognostic biomarker in HNSCC patients, and its prognostic significance is determined by distinct clinical characteristics. Our research strongly suggests that ACER1 may be a prognostic indicator, and may participate in inhibiting tumorigenesis in HNSCC.
It is worth noting that our research also confirms the relationship between ACER1 expression and proliferation, apoptosis, cell cycle, migration and adhesion of Fadu and SCC9 cells. CCK8 analysis indicates that ACER1 overexpression causes a significant reduction in Fadu and SCC9 cells proliferation. Flow cytometric analysis suggests that ACER1 gene significantly induces apoptosis of Fadu and SCC9 cells. Wound healing assay shows that overexpression of ACER1 results in weakened migration capacity compared with empty vector cells. It is inferred that compared to the group with low ACER1 expression, the group with high ACER1 expression has less tumor progression and better prognosis. Our results are perfectly consistent with this inference. Moreover, GSEA results indicates that the ACER1 high-expression group is closely connected with the sphingolipid signaling and Ras signaling pathways. Studies of the sphingolipid signaling pathway are offering promising developments in regulating tumor growth and responses to anticancer therapy44. Li et al. show that Tricin may effectively restrain the growth of lung carcinoma cells by suppressing sphingolipid signaling45. Therapies targeting the RAS signaling pathway are also showing promise in the clinic, with positive effects on inhibition of tumor growth, survival and spread46. Through the Ras/Raf/MEK/ERK signaling pathway, the tumor progression of hepatocellular carcinoma is obviously inhibited47. Thus, these findings provide additional substantiation for our assertion that elevated ACER1 expression group demonstrates a subdued tumor aggressiveness, while low ACER1 expression corresponds to an augmented tumor aggressiveness. Our analyses indicate that ACER1 may be involved in the sphingolipid and Ras signaling pathways, which can suppress tumorigenesis and improve the prognoses of HNSCC patients.
The components of the tumor microenvironment (TME) include immune cells, cytokines, stromal cells, tumor cells, and others. The tumor-infiltrating lymphocytes (TILs) play essential roles in the tumorigenesis, prognosis and immunotherapy of different cancers48. There is increasing evidences that the efficiency of immunotherapy can be influenced by the pattern of immune cell infiltration and that the distinct degree of tumor-infiltrating immune cell subsets is closely related to patient outcomes49. For instance, Garon et al. has suggested that pembrolizumab was more beneficial for lung cancer patients with a high number of infiltrating CD8 T cells50. Our study shows that ACER1 expression is negatively correlated with CD4 + T cells, regulatory T cells, monocytes, macrophages, and NK cells, while positively connected to CD8 + T cells, B cells, neutrophils, and mast cells in HNSCC. These interactions can point to a possible mechanism by which the ACER1 gene regulates immune function in HNSCC, contributing to the development of immune-related therapies for patients with HNSCC. Furthermore, the relationships between gene markers of immune cells and ACER1 expression indicate the key role of ACER1 in mediating tumor immunity in patients with HNSCC. It has been reported that the polarization state of TAMs and its proportion in TME can effectively promote tumor growth, invasion and metastasis51,52. Our results show that immune marker sets of TAMs (CCL2), monocytes (CD86 and CSF1R), and M2 macrophages (CD163, VSIG4 and MS4A4A) are strongly negatively correlated with ACER1 expression, suggesting that ACER1 gene may regulate HNSCC progression by influencing macrophage polarization. DCs can modulate tumor metastasis by improving Tregs responses and reducing CD8 + T cell cytotoxicity53. The expression of ACER1 has a close correlation with the DCs markers (HLA-DPB1, HLA-DQB1, HLA-DRA, HLA-DPA1, NRP1 and ITGAX), which reminds us that ACER1 may be involved in the HNSCC metastasis via regulating DCs infiltration and Tregs responses. Immune checkpoint blocking is a promising immunotherapy approach. For example, checkpoint blockade therapy for CTLA-4 and PD1/PDL1 is commonly used for a variety of malignant cancers54,55. The association between checkpoint blockade therapy and HNSCC patients hasn’t been thoroughly investigated, yet. According to our research, T cell exhaustion markers (PDCD1, PDCDLG2, CTLA4, LAG3, HAVCR2, and GZMB) are also substantially correlated with ACER1 expression. It reflects that ACER1 gene is associated with stronger immunotherapy responses in patients with HNSCC, and targeting the ACER1 gene may be a useful strategy to boost the effectiveness of immunotherapy. In a word, ACER1 plays a crucial role in the recruitment and regulation of tumor-infiltrating lymphocytes in HNSCC, and shows a close connection with the TIL marker genes, which may be the molecular pathways of ACER1 gene in modulating tumor microenvironment in HNSCC patients.
However, our study has several limitations. First, there are relatively few clinical HNSC samples, more patients must be collected for further examination. Additionally, our study lacks of data regarding patient complications, treatment option, and other related topics. At last, we only conduct one series of functional assays in vitro, and more research is needed to determine the possible immune regulatory mechanism linked to the ACER1 gene in HNSCC patients in the future. Conclusion.
In conclusion, ACER1 expression is significantly decreased in HNSCC patients and indicates a longer prognosis. Overexpression of ACER1 can induce the apoptosis, suppress the proliferation and migration, as well as promote the cell adhesion in HNSCC. ACER1 gene is involved in regulating the immune cell infiltration and closely associated with immune checkpoint expression. Therefore, ACER1 may function as a tumor suppressor gene and an indicator of the immunotherapy response in patients with HNSCC.
Data availability
The data used and/or analyzed of this study are available from the corresponding author upon request.
6. References
Siegel, R. L. et al. Cancer Stat. ;73(1):17–48. (2023).
Kocarnik, J. M. et al. Cancer incidence, mortality, years of life lost, years lived with disability, and disabilityadjusted life years for 29 cancer groups from 2010 to 2019: a systematic analysis for the global burden of Disease Study 2019. JAMA Oncol. 8, 420–444 (2022).
Lim, A. M. et al. Assessment of endpoint definitions in curative-intent trials for mucosal head and neck squamous cell carcinomas: Head and Neck Cancer International Group consensus recommendations. Lancet Oncol. 25 (7), e318–e330 (2024).
Muijlwijk, T. et al. Immune cell topography of head and neck cancer.J. Immunother Cancer ;12(7). (2024).
Ren, S. et al. Intratumoral CD103 + CD8 + T cells predict response to neoadjuvant chemoimmunotherapy in advanced head and neck squamous cell carcinoma. Cancer Commun. 43 (10), 1143–1163 (2023).
Sung, H. et al. Global Cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J. Clin. 71 (3), 209–249 (2021).
Chaturvedi, A. K. et al. Human papillomavirus and rising Oropharyngeal Cancer incidence in the United States. J. Clin. Oncol. 41 (17), 3081–3088 (2023).
Colevas, A. D. et al. NCCN guidelines insights: head and neck cancers, version 1.2022. J. Natl. Compr. Canc Netw. 20 (3), 224–234 (2022).
Lee, Y. T., Tan, Y. J. & Oon, C. E. Molecular targeted therapy: treating cancer with specificity. Eur. J. Pharmacol. 834, 188–196 (2018).
Malone, E. R. et al. Molecular profiling for precision cancer therapies. Genome Med. 12 (1), 8 (2020).
Li, C. et al. Identify metabolism-related genes IDO1, ALDH2, NCOA2, SLC7A5, SLC3A2, LDHB, and HPRT1 as potential prognostic markers and correlate with immune infiltrates in head and neck squamous cell carcinoma. Front. Immunol. 13, 955614 (2022).
Jia, W. et al. CYP4F12 is a potential biomarker and inhibits cell migration of head and neck squamous cell carcinoma via EMT pathway. Sci. Rep. 13 (1), 10956 (2023).
Liu, Z. et al. Comprehensive analysis reveals CCDC60 as a potential biomarker correlated with prognosis and immune infiltration of head and neck squamous cell carcinoma. Front. Oncol. 13, 1113781 (2023).
Zhao, X. et al. PLEK2 promotes cancer stemness and tumorigenesis of head and neck squamous cell carcinoma via the c-Myc-mediated positive feedback loop. Cancer Commun. 42 (10), 987–1007 (2022).
Cao, H. et al. FAT1 as a tumor mutation burden specific gene affects the immunotherapy effect in head and neck squamous cell cancer. Drug Resist. Updat. 76, 101095 (2024).
Sannigrahi, M. K. et al. A novel pipeline for prioritizing cancer type-specific therapeutic vulnerabilities using DepMap identifies PAK2 as a target in head and neck squamous cell carcinomas. Mol. Oncol. 18 (2), 336–349 (2024).
Mao, C. & Obeid, L. M. Ceramidases: regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochim. Biophys. Acta. 1781 (9), 424–434 (2008).
Hannun, Y. A. & Obeid, L. M. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat. Rev. Mol. Cell. Biol. 9 (2), 139–150 (2008).
Houben, E. et al. Differentiation-associated expression of ceramidase isoforms in cultured keratinocytes and epidermis. J. Lipid Res. 47 (5), 1063–1070 (2006).
Mao, C. et al. Cloning and characterization of a mouse endoplasmic reticulum alkaline ceramidase: an enzyme that preferentially regulates metabolism of very long chain ceramides. J. Biol. Chem. 278 (33), 31184–31191 (2003).
Sun, W. et al. Upregulation of the human alkaline ceramidase 1 and acid ceramidase mediates calcium-induced differentiation of epidermal keratinocytes. J. Invest. Dermatol. 128 (2), 389–397 (2008).
Lin, C. et al. Alkaline ceramidase 1 protects mice from premature hair loss by maintaining the homeostasis of hair follicle stem cells. Stem Cell. Rep. 9 (5), 1488–1500 (2017).
Li, T. et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 48 (W1), W509–W514 (2020).
Tang, Z. et al. GEPIA: a web server for Cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 45 (W1), W98–W102 (2017).
Chandrashekar, D. S. et al. UALCAN: a portal for facilitating Tumor Subgroup Gene expression and survival analyses. Neoplasia. 19 (8), 649–658 (2017).
Robin, X. et al. pROC: an open-source package for R and S + to analyze and compare ROC curves. BMC Bioinform. 12, 77 (2011).
Lánczky, A. et al. miRpower: a web-Tool to Validate Survival-Associated miRNAs utilizing expression data from 2178 breast Cancer patients. Breast Cancer Res. Treat. 160 (3), 439–446 (2016).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6 (269), pl1 (2013).
Forbes, S. A. et al. COSMIC: somatic cancer genetics at high- resolution. Nucleic Acids Res. 45 (D1), D777–D783 (2017).
Shankavaram, U. T. et al. CellMiner: a relational database and query tool for the NCI-60 cancer cell lines. BMC Genom. 10 (1), 277 (2009).
Reinhold, W. C. et al. CellMiner: a web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set. Cancer Res. 72 (14), 3499–3511 (2012).
Vasaikar, S. V. et al. LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 46 (D1), D956–D963 (2018).
Warde-Farley, D. et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 38 (suppl_2), W214–W220 (2010).
Aran, D., Sirota, M. & Butte, A. J. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 6, 8971 (2015).
Lu, M. et al. N6-methyladenosine-related non-coding RNAs are potential prognostic and immunotherapeutic responsiveness biomarkers for bladder cancer. EPMA J. 12 (4), 589–604 (2021).
Li, Y. & Yuan, J. Role of deubiquitinating enzymes in DNA double-strand break repair. J. Zhejiang Univ. Sci. B. 22 (1), 63–72 (2021).
Imperatori, A. et al. LINE-1 hypomethylation is associated to specific clinico-pathological features in Stage I non-small cell lung cancer. Lung Cancer. 108, 83–89 (2017).
Noguera-Uclés, J. F. et al. The roles of imprinted SLC22A18 and SLC22A18AS gene overexpression caused by promoter cpg island hypomethylation as diagnostic and prognostic biomarkers for non-small cell lung cancer patients. Cancers (Basel). 12 (8), 2075 (2020).
Tanadini-Lang, S. et al. Radiomic biomarkers for head and neck squamous cell carcinoma. Strahlenther Onkol. 196 (10), 868–878 (2020).
Moskovitz, J., Moy, J. & Ferris, R. L. Immunotherapy for head and neck squamous cell carcinoma. Curr. Oncol. Reps. 20 (2), 22 (2018).
Arantes, L. et al. Serum, plasma and saliva biomarkers for head and neck cancer. Expert Rev. Mol. Diagn. 18 (1), 85–112 (2018).
Dienstmann, R. et al. (ed Vermeulen, L.) Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 17 4 268 (2017).
DuW,ElementoO,etal.Cancer systems biology: embracing complexity to develop better anticancer therapeutic strategies.Oncogene.2015;3215–3225.
Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer. 18 (1), 33–50 (2018).
Li, J. et al. Metabolomics and integrated network pharmacology analysis reveal Tricin as the active anti-cancer component of Weijing decoction by suppression of PRKCA and sphingolipid signaling. Pharmacol. Res. 171, 105574 (2021).
Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer. 3 (1), 11–22 (2003).
Zhou, K. et al. MicroRNA-30a suppresses tumor progression by blocking Ras/Raf/MEK/ERK signaling pathway in hepatocellular carcinoma. Biomed. Pharmacother. 93, 1025–1032 (2017).
Klauschen, F. et al. Scoring of Tumor-infiltrating lymphocytes: from visual estimation to machine learning. Semin Cancer Biol. 52 (Pt 2), 151–157 (2018).
Zheng, X. et al. Progression on the roles and mechanisms of Tumor-infiltrating T lymphocytes in patients with Hepatocellular Carcinoma. Front. Immunol. 12, 729705 (2021).
GaronEB,HellmannMD,RizviNA,etal.Five-year overall survival for patients with advanced non–small-cell lung cancer treated with pembrolizumab: results from the phase I KEYNOTE-001 study.J Clin Oncol.2019;2518–2527.
Song, W., Mazzieri, R., Yang, T. & Gobe, G. C. Translational significance for tumor metastasis of tumor-associated macrophages and epithelial-mesenchymal transition. Front. Immunol. 8, 1106 (2017).
Zhang, D. et al. TGF-b secreted by tumor-associated macrophages promotes proliferation and invasion of colorectal cancer via miR-34a-VEGF axis. Cell. Cycle. 17 (24), 2766–2778 (2018).
Lee, Y. S. & Radford, K. J. The role of dendritic cells in cancer. Int. Rev. Cell. Mol. Biol. 348, 123–178 (2019).
Topalian, S. L. et al. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer. 16 (5), 275–287 (2016).
Topalian, S. L. Targeting Immune checkpoints in Cancer Therapy. JAMA. 318 (17), 1647–1648 (2017).
Acknowledgements
The authors expressed their sincere thanks to all databases used in this study.
Funding
This project was supported by China Postdoctoral Science Foundation Special Funded Project, Grant/Award Number: 2017T100498; Science and Technology Project of Jinan City, Grant/Award Number: 201805053; Shandong Provincial Natural Science Foundation, China, Grant/Award Number: ZR2020MH280.
Author information
Authors and Affiliations
Contributions
Liu Heng: Conceptualization, Writing-Review & Editing; Yang Hui: Supervision, Project administration, Funding acquisition; Liu Zhixin: Formal analysis, Writing-Original Draft; Yang Xiaoqi: Methodology, Validation; Chen Shuai: Software; Jia Wenming: Investigation; Qian Ye: Resources; Zhang Minfa: Data Curation; Fang Tianhe: Visualization.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This project was approved by the Institutional Ethics Committee at Qilu Hospital of Shandong University (Ethics approval number: KYLL-2019-2-095). The employment and processing of materials were permitted and obtained informed consent from each patient. All methods involved in human subjects were performed in accordance with the Declaration of Helsinki.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, Z., Yang, X., Chen, S. et al. Tumor suppressor ACER1 correlates with prognosis and Immune Infiltration in head and neck squamous cell carcinoma. Sci Rep 14, 28039 (2024). https://doi.org/10.1038/s41598-024-78663-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-78663-1
