
Abstract
Climate change is dramatically affecting vector ecology in extreme environments such as the Arctic. However, little is known about the current status of viruses of arthropod vectors located in such northerly locations. As part of a field survey on the role of wildlife in international movement of zoonotic pathogens, we sampled mammalophilic mosquitoes near the settlement of Kangerlussuaq, Greenland in July 2022 and July 2023 to investigate their virome. The majority of mosquitoes were identified as either Aedes impiger or Aedes nigripes. Metagenomic analysis of RNA extracted from species pools detected a number of novel RNA viruses belonging to a range of different virus families, including Flaviviridae, Orthomyxoviridae, Bunyavirales, Totiviridae and Rhabdoviridae. However, the sequence identities when compared to previously published, were as low as 34% at the amino acid level. Furthermore, a comparison of virome diversity between Aedes species emphasises the uniqueness of both Aedes impiger and Aedes nigripes from this secluded ecosystem. It also highlights the need to better understand the viromes of potential pathogen vectors as the impacts of climate change are experienced in such northerly ecosystems.
Similar content being viewed by others


Introduction
Global changes in climate are causing a shift in the distribution of vectors1 and increasing the likelihood of vector-borne disease outbreaks in regions that have not experienced such disease emergence2,3. Yet, very little is known about the vectorial capacity and virome of mosquitoes in less accessible areas such as the Arctic. In addition to a potential change in the localisation of Arctic insects in the long term, climate change already affects vectors in their current locations. With rapid environmental change, and warming at twice the global average4 the ecology of Arctic insects will be dramatically affected. Arctic insects have uniquely adapted to long, cold winters and short, cool, unpredictable summers5. Despite this, mosquito species have established in Arctic locations and are voracious feeders readily feeding on humans if present6. Due to the different ways a warming climate will affect the microclimate that they inhabit, the interplay of factors impacting Arctic mosquitoes can be complex. How climate change exactly impacts the vector ecology in the Arctic is therefore hard to predict. Nevertheless, we can be sure that all aspects of the Arctic insect life will be affected, including survival, development time, life cycle, host-seeking activity, interactions with other species, and range expansions6. This necessarily comes with implications for the entirety of northern ecosystems as well as the importance of Arctic mosquitoes as disease vectors. Additionally, a warming climate might also favour species introduction into the Arctic through an increase in tourism and travel, causing additional concerns around the implications of the diseases they carry for the local ecosystems and indigenous populations7,8. Newly arriving diseases could disproportionally impact Greenland’s wildlife, because their populations are likely immunologically naïve9.
Metagenomic analysis based on mass sequencing is an established methodology for determining the virome of arthropod species10. This approach has been used extensively for analysis of the viral composition of mosquito species associated with pathogen transmission, particularly those within the genus Aedes11,12,13,14. Understanding the diversity of the mosquito virome is a critical first step that can determine the relationship between known pathogens and the insect-only virus composition of mosquitoes that in turn could lead to novel strategies of control of mosquito-borne disease15. However, studies of the virome of indigenous mosquito species in extreme northernly locations have been limited16,17.
The aim of this study was to explore the virome of the local mosquito population in Greenland, applying a metagenomics approach. The few studies conducted in Greenland so far identified Aedes nigripes (Zetterstedt, 1838) and Aedes impiger (Walker, 1848) as the main native mosquito species18, but did not detect any arthropod-vectored viruses19,20. However, exploring the entirety of the mosquito virome, including both insect specific viruses as well as potential arboviruses will enable us to better understand the risks a change in ___location of Arctic mosquitoes might harbour, but even more importantly, take us one step closer to estimating the vector competence of so far understudied mosquito species for relevant virus families. Our study highlights the importance of exploring the virome in secluded locations such as the Arctic as climate change is already causing dramatic changes in species distribution that is accompanied by a shift in pathogen localisation.
Materials and methods
Mosquito collection
Mosquitoes were trapped at an open air camp at lake Sanningasoq, approximately 11.5 km northeast from Kangerlussuaq (67°01’N 50°41’W) in central-western Greenland (Fig. 1a), from 05.07. to 25.07.2022 (14 sampling days) and from 04.07. to 22.07.2023 (seven sampling days) using aspirators (Tubular suction aspirator, 7 mm intake, model D-601, Entomopraxis, Barcelona). They were killed by pipe smoke and kept at low temperature by immersing the collection tubes in a lake (estimated temperature between 3 ºC and 6 ºC). To prevent nucleic acid degradation, in 2023 a total of 70 mosquitoes were instantly homogenised individually in DNA/RNA Shield (Zymo) after trapping by shaking them in a 1.5 ml Eppendorf tube with a 5 mm stainless steel bead (Qiagen, Manchester, UK).

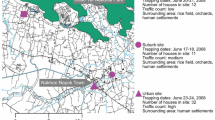
Aedes impiger was the predominant species collected in Kangerlussuaq, Greenland. (a) Map of Greenland depicting the sampling ___location at lake Sanningasoq, approximately 11.5 km northeast from Kangerlussuaq. The map was produced with ggplot2 (version 3.5.1) in R (version 4.4.1). (b) Species identification by morphology, Danks and Corbet, 1973. Image shows the tarsal claw of an Aedes impiger sampled in 2022. (c) Pie charts representing the number of mosquitoes identified for each year and species by DNA barcoding.
Morphological identification of mosquitoes
Mosquito species identification by morphology was carried out under a microscope (Leica M165 C) following the guide by Danks and Corbet18. For Aedes nigripes the tarsal claw curves gradually, while for Aedes (A) impiger the tarsal claw curves abruptly (Fig. 1B).
Molecular identification of mosquitoes
To confirm the morphological species identification and to identify specimens where the tarsal claws were missing, cytochrome c oxidase I (cox1) sequences were obtained.
In the pilot project i.e., mosquitoes trapped in 2022, two legs were used to extract DNA for species identification by DNA barcoding. Total DNA was extracted using DNeasy kits (Qiagen, Manchester, UK) according to the manufacturer’s instructions. Mosquitoes were then homogenised individually in 350 µl tissue culture medium using the Qiagen TissueLyser II with 5 mm stainless steel beads (both Qiagen, Manchester, UK) and centrifuged (10,000 rpm/10 min). Total RNA was extracted from 250 µl of the supernatant using RNeasy kits (Qiagen, Manchester, UK). The precipitated RNA was resuspended in 40 µl nuclease free water and pooled by species and date for next generation sequencing (NGS).
In 2023, to prevent nucleic acid degradation, mosquitoes were homogenised individually in DNA/RNA Shield (Zymo) immediately after trapping. DNA and RNA were then extracted separately using the AllPrep DNA/RNA Mini Kit (Qiagen, Manchester, UK). The precipitated DNA and RNA were resuspended in 40 µl nuclease free water. DNA was used for species identification by DNA barcoding and RNA was later pooled by species and sampling date for further analysis by NGS.
A 658 bp region located at the 5’ end of the cox1 gene was amplified by PCR with the primer pair (LCO1490 and HCO2198) published by Folmer et al.21. PCR products were visualised on a 1.5% agarose gel and samples of the correct band size were submitted for Sanger sequencing using primers LC01490 and HCO2198. Mosquito species were identified following a BLASTN search. Sequence identities were > 99% when compared with published Aedes impiger (Genbank: JN303080) and Aedes nigripes (Genbank: KR395472) sequences.
Next generation sequencing
Extracted mosquito RNA was pooled based on species and date of trapping and subjected to next generation sequencing for metagenomic analysis (Supplementary Tables 1 and 2). Sequencing libraries were prepared using the Nextera XT kit (Illumina, Cambridge, UK) and analysed on a NextSeq sequencer (Illumina, Cambridge, UK) with 2 × 150 base paired-end reads.
Data analysis
Reads were analysed using the Chan Zuckerberg Illumina pipeline22, a cloud-based, open-source bioinformatics platform: Reads were aligned against NCBI NT and NR databases using Minimap2 and Diamond, contigs were assembled with SPAdes, reads mapped against contigs using Bowtie2 and contigs aligned against nucleotide and protein databases with BLASTN and BLASTX. According to the Chan Zuckerberg pipeline (for all projects created prior April 19, 2023), the Host Filtering and Quality Control steps included initial host filtration using STAR, trimming of sequencing adapters using Trimmomatic, quality filtering using PriceSeq, identification of duplicate reads using czid-dedup, filtering out of low complexity sequences using LZW, filtering out remaining host sequences using Bowtie2, subsampling to 1 million fragments (reads/read-pairs) if > 1 M remain after step, and filtering out human sequences, regardless of host (using STAR, Bowtie2, and GSNAP). Details of the total reads per sample, the percentage that passed QC, duplicate compression ratio (DCR) and the number of reads that remained after host filtering can be found in Supplementary Table 3.
Hits that produced a minimum of one contig were investigated further. If multiple contigs were recovered from the same virus, the longest contig was investigated further.
The map depicting the trapping ___location was produced with ggplot2 (version 3.5.1) in R (version 4.4.1).
The bubble blots in Fig. 2 were made with ggplot2 (version 3.5.1) in R (version 4.4.1).
Sequence alignments (Fig. 3) were produced using MAFFT v7.471 and the resulting alignment was imported into BEAST (v1.10.4). A Bayesian phylogenetic tree was produced using the Blosum62 amino acid substitution model and 10,000,000 Markov chain Monte Carlo generations. Log files were analysed in Tracer v1.7.1 to check the effective sample size and a 10% burn-in was included (TreeAnnotator v.1.10.4) before being visualised and annotated in FigTree v1.4.4.

Distribution of non-host reads. (a) Distribution of non-host reads across the 2023 mosquito pools for each indicated sampling date and species. (b) Bubble plots representing the top15 viral hits (genus-level) of mosquito pools sampled in 2023 in percentage by species and date. The asterix marks non-genus specific reads in a virus family. (c) Overlap of viral hits (genus level) between the species across the top 15 reads only (left) or all viral reads (right).

Phylogenetic analysis. Bayesian phylogenetic relationships of virus sequences identified in this study (labelled red and blue) with published sequences. The analysies were based on polyprotein (Flaviviridae, Totiviridae), nucleocapsid (Bunyavirales) or nucleoprotein (Orthomyxoviridae, Rhabdoviridae) amino acid sequences. Node labels represent posterior probabilities and accession numbers are shown.
For the heatmap in Fig. 4 a list of viruses for all assigned reads (BLASTX) for both A. nigripes and A. impiger was added to the database assembled by Moonen et al.23. Detections versus non-detections for all viruses and Aedes species in the database were calculated with Tidyverse (version 1.3.1) in R (version 4.4.1). A heatmap was created with GraphPad Prism (version 8.4.2).

The virome of Aedes impiger and Aedes nigripes is distinct from other Aedes species. (a) Heatmap comparing all assigned virus reads in both Aedes impiger and Aedes nigripes (n = 94) with a list of viruses (n = 685) published to infect Aedes spp. as summarized by Moonen et al. 2023. Two-way Anova with ***P < 0.001 (b) Overlap of viruses found between indicated species.
Results
Aedes impiger was the dominant species collected in Kangerlussuaq
Previous surveys in western Greenland suggested that A. nigripes was the only mosquito species present19. In July 2022 and July 2023, we trapped a total of 75 and 70 specimen, respectively, near Kangerlussuaq (Fig. 1a and b). The mosquitoes trapped in 2022 served as a pilot study to assess the best methods of preservation, transport, species identification, RNA extraction and metagenomic analysis. Species identification by morphology proved to be challenging due to the poor sample conditions after transport and the minor visible differences between the most common species described for Greenland (Fig. 1c). Species identification using the cox1 partial sequence, effectively discriminated the species present provided the quality of extracted nucleic acids was high. This identified the majority of mosquitoes collected in 2022 as Aedes impiger. Of the 75 Diptera trapped, 4 were excluded (3 midges, 1 fly), 37 were identified as Aedes impiger (52%), 5 as Aedes nigripes (7%), with the remaining 29 samples unidentifiable, due to poor DNA quality and failure to amplify the cox1 amplicon. Consequently, all mosquitoes sampled in 2023, (n = 70) were individually homogenised in DNA/RNA Shield immediately after trapping. This improved the quality of the nucleic acid extracted after transport, leading to 93% of samples identifiable by DNA barcoding (Fig. 1c). 49 mosquitoes were identified as A. impiger (70%) and 16 as A. nigripes (23%). No other mosquito species were identified in either field survey.
Virus diversity in Arctic mosquitoes
Mosquitoes were pooled based on sampling date and species (Supplementary Tables 1 and 2) and submitted to NGS. The majority of reads from specimens trapped in 2022 mapped to host and bacterial genomes, due to poor nucleic acid quality caused by the challenging storage and transport conditions. For mosquitoes trapped in 2023, the majority of reads mapped to eukaryotes and bacteria (Fig. 2a). However, a proportion of reads mapped to a variety of virus families representing positive and negative single-stranded RNA viruses, double-stranded RNA viruses as well as retroviruses (Fig. 2b). Many of the sequences mapped to viruses that have no assigned order or family, others to assigned virus families but with no ascribed genus. The highest percentage of virus reads for both A. impiger and A. nigripes mapped to non-genus specific reads in the Totiviridae, Chrysoviridae and Flaviviridae families. Most virus families were detected consistently through the sampling period. However, certain virus families, for example Orthophasmavirus, were only detected in a single mosquito pool. Although the identified virus families significantly overlapped between A. nigripes and A. impiger, especially in the top 15 virus families detected (including 90% and 99% of total viral reads, depending on sampling date and species), they also encompassed their own individual virome footprint (Fig. 2c).
Phylogenetic analysis reveals the identification of novel viruses
Sequences mapping to those virus families comprising virus species with known zoonotic potential were analysed phylogenetically (Fig. 3, Supplementary Table 4). For all contigs analysed, we aimed to always phylogenetically represent the homologous sequence obtained from both Aedes species. In few cases this led to contigs of sizes < 1000 bp or supported by only few reads being included in the analysis (details see Supplementary Table 4). Within the family of Flaviviridae, two novel flavi-like virus sequences were identified, with their polyproteins displaying 35% similarity to the nearest published polyprotein (Fig. 3). This was a flavi-like virus derived from Culex tritaeniorhynchus (Protein Accession Number: BBQ05092). Similarly, two novel orthomyxo-like sequences were identified within the family of Orthomoyxoviridae (Fig. 3). Their nucleoprotein comprised only 36% identity to that of Byreska virus (Protein Accession Number: UQS95351), the nearest published sequence. Within the order of Bunyavirales, we discovered two novel phasiviruses as well as two novel phasmaviruses (Fig. 3). The Phasivirus sequences shared 36% and 66% identity with nucleocapsid sequences belonging to Guadeloupe mosquito phasivirus (Protein Accession Number: YP_010839970) and Coredo virus (Protein Accession Number: YP_010840334), respectively. Within the family Totiviridae, we discovered novel nucleocapsid sequences (Fig. 3) with 68% shared amino acid sequence to Vaasa toti-like viruses (Protein Accession Number: OP019898 and OP019899) and RNA-dependent RNA polymerase with 89% shared amino acid sequence. Within the family of Rhabdoviridae, we also discovered three distinct nucleoprotein sequences, two of which were present in both A. nigripes and A. impiger (Fig. 3).
Mosquitoes in Greenland host a unique Virome
To emphasize the uniqueness of the virome of A. impiger and A. nigripes, we compared our findings to a database of viruses published for other Aedes species (compiled by Moonen et al.23). The heatmap (Fig. 4a) reveals that the majority of sequences derived from Greenland mosquitoes were unique to A. impiger and A. nigripes, with only 36 (of a total of 94, 38%) assigned viruses overlapping with viruses published for other Aedes spp. (Fig. 4b). Only 22 (23%) overlapped with Aedes aegypti, the Aedes mosquito with the best characterized virome (Fig. 4b). A Two-way Anova revealed a p-value < 0.0001 for the comparison of both A. impiger and A.nigripes with all other Aedes species, with the exception of the comparison of A. impiger with A. cantans (p = 0.0008). This makes the virome composition of A. cantans the most similar published virome composition to that of the mosquitoes we sampled near Kangerlussuaq. This is additionally interesting, as A. cantans is geographically distributed across the Palaearctic, suggesting that mosquitoes from similar ecological habitats might share similar viruses.
Discussion
Aedes nigripes is the most abundant and most widely distributed mosquito in the Arctic24. Despite this, where a definitive identification could be made, A. impiger was the most frequently sampled mosquito at the Kangerlussuaq site. The circumpolar distribution of A. nigripes makes it the most widespread and northernmost mosquito species in the Arctic region25. However, surprisingly little is known about the viruses harboured by this and other indigenous mosquito species. Our study is the first metagenomic exploration of the viromes of A. impiger and A. nigripes in Greenland. In our study, the majority of identified virus sequences were found in both A. impiger and A. nigripes. They also harboured a set of sequence reads assigned to viruses that were unique to each species, implying a distinct virome, although this varied over the sampling period. Due to our limited sample size, future studies are needed to explore the differences and similarities with the virome of other Aedes species in more depth, and to determine how significant the virome difference between A. impiger and A. nigripes is. Larger datasets, including metagenomic data from other locations in Greenland will be crucial to answer remaining questions. Since insect-specific viruses can affect the replication and transmission efficiency of zoonotic viurses, future studies should also investigate if the virome differences observed cause functional consequences for the vector competence of A. impiger and A. nigripes.
A consistent finding in almost all mosquito pools was the presence of sequences with high identity with viruses detected in Finland16. These were reported to be derived from A. excrucians, also referred to a Ochlerotatus excrucians. Totiviridae sequences have also been detected in mosquitoes in extreme northerly locations such as Western Siberia17. However, this family of double-stranded RNA viruses has been associated with a range of arthopods including tabanid flies26 and Culicoides midges27. We also found that the A. impiger and A. nigripes virome comprises of sequences belonging to many of the major virus families described for other mosquito species. However, sequence identity of our assembled contigs with sequences published for other mosquito species was low, sometimes below 35% identity at the on amino acid level. This may explain why the few studies that have previously attempted to characterise the virome of Arctic mosquitoes by PCR often failed to detect viruses19,20.
Even less is known about the vector competence of A. impiger and A. nigripes and the potential impact of novel viruses arriving in Arctic ecosystems or a potential change in global distribution due to a changing climate. Aedes nigripes has been suggested to transmit Getah virus in Siberia28, and there was evidence of infection with Anadyr virus and Chatanga viruses19. Aedes impiger is known to feed on humans and can productively produce eggs and oviposit after a human blood meal29. These observations indicate a realistic risk that these Arctic mosquitoes are capable of transmitting viruses with zoonotic potential. Our findings further support the capability of A. nigripes and A. impiger to replicate viruses belonging to a range of different virus families, including (-)ssRNA, (+)ssRNA, dsRNA and retroviruses. However, none of the viruses detected are closely related to known viruses with zoonotic potential and are likely insect-specific. Further studies will need to functionally characterise the viruses we detected, with regard to their replicative capabilities, potential to cause disease and impact on the replication and transmission of other, better characterized arboviruses.
Recent publications suggest that a mosquito virome is less driven by ___location, but by species30. In our study, we describe the virome of two uncharacterized mosquito species in a remote ___location. Consequently, we cannot know whether the unique virome we observed is driven by the isolated ___location or the species assemblage, although we note that many virus sequences were present in both species. Mosquitoes currently inhabiting secluded locations, such as the Arctic, might be susceptible to infection and transmit different viruses described in similar species if pathogens or mosquitoes are expanding or changing their distribution3. Understanding and being able to predict which viruses will most likely be able to be transmitted by which mosquito species, based on their described virome, would help to estimate the risk of zoonotic disease transmission and inform policies. As our heatmap shows, there are still numerous gaps in our knowledge about the virome of different Aedine mosquitoes, while most studies concentrate on only very few species. Although it is unclear what risk the viruses assigned to the sequences we observed represent to plants, animals, or the human population, our results highlight that Arctic mosquitoes have a distinct virome and could support replication of viruses with zoonotic potential. As a changing climate will increase the likelihood of novel viruses arriving in remote locations and mosquitoes currently restrained to remote locations migrating into different ecosystems, this will most likely impact animal and public health. In light of the predicted expansion of both vectors and their viruses in a changing climate, this should be considered in future risk management plans.
Data availability
All virus contigs shown in the phylogenetic analysis have been deposited in GenBank under accession numbers: PQ667683 - PQ667701. COX-1 sequences of Aedes impiger and A. nigripes have been deposited in GenBank under accession numbers: PQ645069 - PQ645072. The raw sequence reads generated in this study are available at the NCBI Sequence Read Archive (SRA) database under BioProject PRJNA1230858; BioSamples SAMN47195350 - SAMN47195360.
References
Ryan, S. J., Carlson, C. J., Mordecai, E. A. & Johnson, L. R. Global expansion and redistribution of Aedes-borne virus transmission risk with climate change. PLoS Negl. Trop. Dis. 13 (3), e0007213 (2019).
Paz, S. Climate change impacts on vector-borne diseases in Europe: risks, predictions and actions. Lancet Reg. Health – Europe ;1. (2021).
de Souza, W. M. & Weaver, S. C. Effects of climate change and human activities on vector-borne diseases. Nat. Rev. Microbiol. (2024).
Post, E. et al. The Polar regions in a 2°C warmer world. Sci. Adv. 5 (12), eaaw9883 (2019).
Danks, H. V. Seasonal adaptations in Arctic insects. Integr. Comp. Biol. 44 (2), 85–94 (2004).
Koltz, A. M. & Culler, L. E. Biting insects in a rapidly changing Arctic. Curr. Opin. Insect Sci. 47, 75–81 (2021).
Chown, S. L., McGeoch, M. A. & Marshall, D. J. Diversity and conservation of invertebrates on the sub-Antarctic Prince Edward Islands. Afr. Entomol. 10 (1), 67–82 (2002).
Gabriel, A. G. A. et al. Biological invasions of Southern ocean Islands: the Collembola of Marion Island as a test of generalities. Ecography 24 (4), 421–430 (2001).
Pauchard, A. et al. Non-native and native organisms moving into high elevation and high latitude ecosystems in an era of climate change: new challenges for ecology and conservation. Biol. Invasions. 18 (2), 345–353 (2016).
Li, C-X. et al. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife 4, e05378 (2015).
Xiao, P. et al. Metagenomic sequencing from mosquitoes in China reveals a variety of insect and human viruses. Front. Cell. Infect. Microbiol. ;8. (2018).
Ramos-Nino, M. E. et al. High prevalence of Phasi Charoen-like virus from wild-caught Aedes aegypti in Grenada, W.I. As revealed by metagenomic analysis. PLoS One. 15 (1), e0227998 (2020).
Nebbak, A. et al. Virome diversity among mosquito populations in a Sub-Urban region of Marseille, France. Viruses ;13(5). (2021).
Calle-Tobón, A. et al. Local-scale Virome depiction in Medellín, Colombia, supports significant differences between Aedes aegypti and Aedes albopictus. PLOS ONE. 17 (7), e0263143 (2022).
Patterson, E. I., Villinger, J., Muthoni, J. N., Dobel-Ober, L. & Hughes, G. L. Exploiting insect-specific viruses as a novel strategy to control vector-borne disease. Curr. Opin. Insect Sci. 39, 50–56 (2020).
Truong Nguyen, P. T. et al. Characterisation of the RNA Virome of nine Ochlerotatus species in Finland. Viruses ;14(7). (2022).
Ternovoi, V. A. et al. The Viromes of mosquitoes from the natural landscapes of Western Siberia. Viruses ;15(9). (2023).
Danks HVaCPS. A key to all stages of Aedes nigripes and A. impiger (Diptera: Culicidae) with a description of first-instar larvae and pupae. Can. Entomol. 105/3, 367–376 (1973).
Müllerová, J. et al. No indication of arthropod-vectored viruses in mosquitoes (Diptera: Culicidae) collected on Greenland and Svalbard. Polar Biol. 41 (8), 1581–1586 (2018).
Reeves, W. K., Breidenbaugh, M. S., Thomas, E. E. & Glowacki, M. N. Mosquitoes of thule air base, Greenland. J. Am. Mosq. Control Assoc. 29 (4), 383–384 (2013).
Folmer, O., Black, M., Hoeh, W., Lutz, R. & Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome C oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3 (5), 294–299 (1994).
Kalantar, K. L. et al. IDseq—An open source cloud-based pipeline and analysis service for metagenomic pathogen detection and monitoring. GigaScience 9 (10), giaa111 (2020).
Moonen, J. P., Schinkel, M., van der Most, T., Miesen, P. & van Rij, R. P. Composition and global distribution of the mosquito virome - A comprehensive database of insect-specific viruses. One Health. 16, 100490 (2023).
Vockeroth, J. R. Notes on the identities and distributions of Aedes species of Northern Canada, with a key to the females (Diptera: Culicidae). Can. Entomol. 86 (6), 241–255 (1954).
Coulson, S. & Refseth, S. The terrestrial and freshwater invertebrate fauna of Svalbard (and Jan Mayen). In Prestrud Pål SHaGHV, Editor. A Catalogue of the Terrestrial and Marine Animals of Svalbard (Norwegian Polar Institute, Polar Environmental Centre, NO-9296 Tromsø, 2004).
Litov, A. G. et al. Viromes of Tabanids from Russia. Viruses ;15(12). (2023).
Laredo-Tiscareño, S. V. et al. Discovery of novel viruses in Culicoides biting midges in Chihuahua, Mexico. Viruses ;16(7). (2024).
L’Vov, S. D. et al. [Mosquito-borne arboviruses in the Baikal region]. Vopr Virusol. 40 (4), 170–172 (1995).
Sommermann, K. M. Blood meals and egg production of four species of Alaskan Aedes in captivity (Diptera: Culicidae). Mosq. News. 29 (4), 654–662 (1969).
Thongsripong, P. et al. Metagenomic shotgun sequencing reveals host species as an important driver of Virome composition in mosquitoes. Sci. Rep. 11 (1), 8448 (2021).
Acknowledgements
We would like to thank Luis Hernandez-Triana for his advice on the DNA barcoding. Mirjam would like to thank her friends Laura and Tim for their enthusiasm around this project and for naming her favourite Aedes impiger collected in 2022 Helga. Conference talks without the personal touch of Helga would not have been the same.
Funding
This study was supported by grant SV3045 from Department for Environment, Food & Rural Affairs (Defra), Scottish Government and Welsh Government, the Versatile Emerging Infectious Disease Observatory (VEO) [European Union’s Horizon 2020 research and innovation programme] (Grant No.874735) and Department for Environment, Food & Rural Affairs (Defra) and UK Research and Innovation (UKRI) (EXSE0574).
Author information
Authors and Affiliations
Contributions
Manuscript first draft: MS; Investigations: MS, MJ, HT; Study design and analysis: MS, NJ; Project management/supervision: HT, NJ; Manuscript review and edit: all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
The authors confirm that the ethical policies of the journal, as noted on the journal’s author guidelines page, have been adhered to.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schilling, M., Jagdev, M., Thomas, H. et al. Metagenomic analysis of mosquitoes from Kangerlussuaq, Greenland reveals a unique virome. Sci Rep 15, 17141 (2025). https://doi.org/10.1038/s41598-025-01086-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-01086-z
